1. Introduction
Malignant tumors often feature inadequate
angiogenesis, structure and function as well as over-proliferation
and increased energy demand when growing in microenvironments with
insufficient blood supply (1). When
cancer cells proliferate in these harsh conditions, they must adapt
by regulating their cell cycles to improve blood flow and adjust
the balance of energy metabolism to maintain proliferation. This is
a hypoxic reaction and various molecules are involved in this
process (2). Cancer cell response
in hypoxic conditions has been extensively studied and hypoxia is
widely accepted as a specific marker indicating poor cancer
prognosis (3).
AMPK-related protein kinase 5 (ARK5) is a
serine/threonine kinase that has been identified as the fifth
member of the AMP-activated protein kinase (AMPK) family (4). ARK5 serves a role in the metastasis
and invasion of colorectal (CRC) cancer, pancreatic cancer (PC),
gastric cancer, hepatic cancer and squamous cell carcinoma
(5-8).
Akt, the most important ARK5 upstream regulator, phosphorylates
ARK5 at the Ser600 residue (a C-terminal site outside the catalytic
domain) ans activates 74 kDa kinases (9). Akt is also an important mediator of
cancer proliferation, survival and oncogenesis (9,10).
ARK5-mediated Akt was established as a key element
that functions as a survival factor in the complex tumorigenesis
network (9). ARK5 prevents cell
death under hypoxic and glucose-starved conditions by avoiding
death receptor (RAS) activation in cells and inhibiting caspase-8
activation by inducing cellular Fas-associated protein with death
domain-like interleukin (IL) 1β-converting enzyme-inhibitory
protein (c-FLIP) in cancer cells (9,11).
Previous studies have revealed that ARK5 is involved in
hypoxia-induced cancer cell tolerance to glucose starvation by
regulating the transforming growth factor-β (TGF-β) signaling
pathway (12,13). Furthermore, ARK5 causes drug
resistance by inducing certain cellular morphology transformations,
including myosin filament reorganization (13).
As an important and recently investigated
intermediate molecule, ARK5 has promising long-term research value.
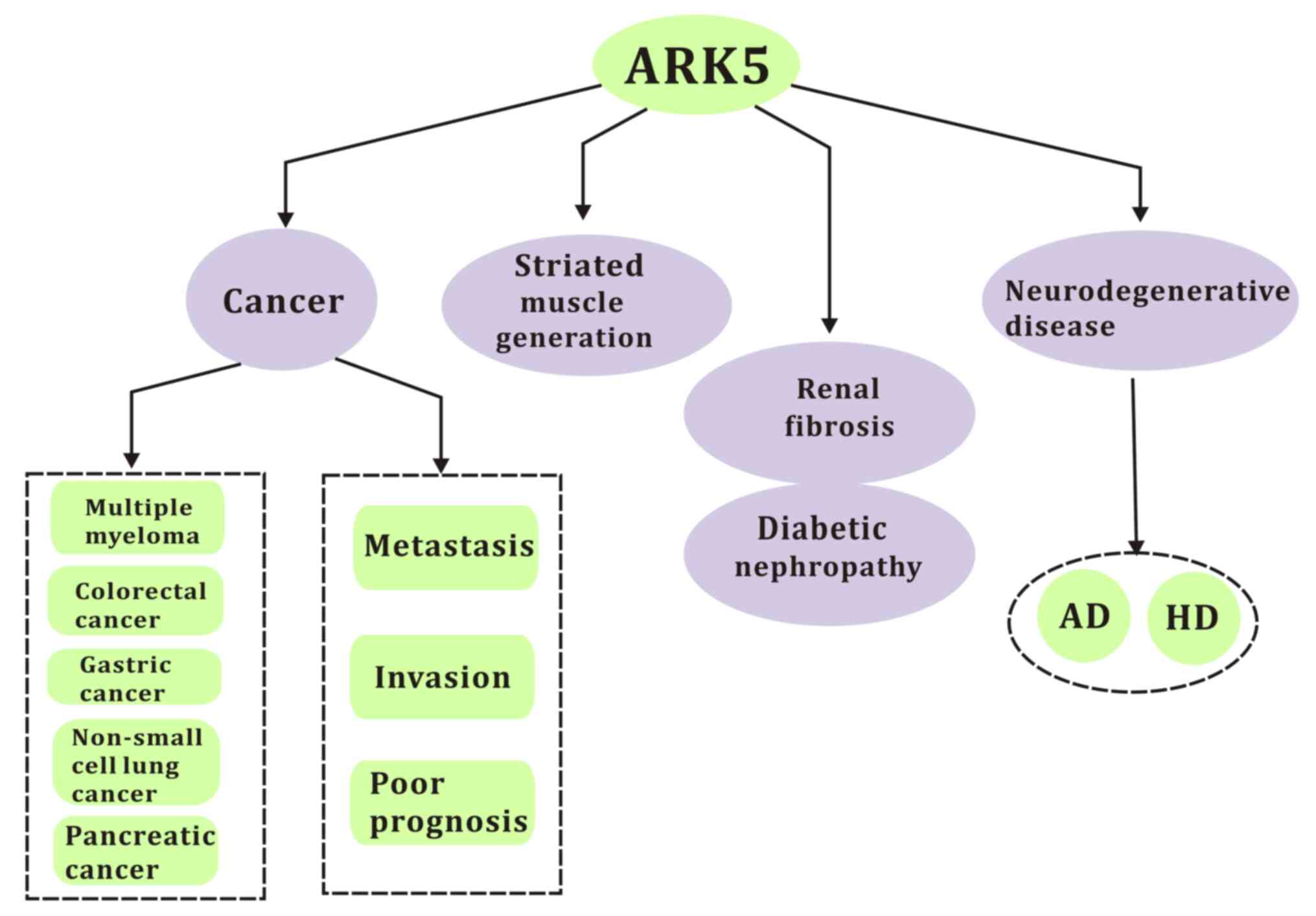
In the current review, the molecular interactions, physical
progress and different functions of ARK5 in cancer and other
diseases, as well as potential therapeutic strategies, are
discussed (Fig. 1).
2. Relevance of ARK5 in cancer
Multiple myeloma
Multiple myeloma is a common cancer where ~20,000
new cases are diagnosed annually in the United States (14,15).
The introduction of autologous stem cell transplantation and novel
drugs with various mechanisms of action (including proteasome
inhibition and immunomodulation) have fundamentally changed the
treatment strategy for multiple myeloma and have significantly
prolonged the overall survival of patients (16-19).
However, despite these advances, patients only survive for 7-8
years after diagnosis due to drug resistance and minimal residual
disease (20).
c-Musculoaponeurotic fibrosarcoma (MAF), which is
transcription factor that involved in immune responses and works as
a T cell stimulator that induces IL-4 and IL-10 release to control
T cell, was revealed to participate in the regulation of fiber cell
differentiation (21-23).
c-MAF translocation and overexpression have been reported in
numerous cases of multiple myeloma (24-26).
In various clinical trials, 351 clinical specimens exhibited clear
ARK5 and c-MAF overexpression in multiple myeloma-derived cell
lines. Sequence analysis of the ARK5 gene promoter further revealed
that the gene contained two putative MAF-recognition elements and
that ARK5 mRNA acts as a regulator in c-MAF-induced multiple
myeloma (8). These results
suggested that ARK5 may be a transcriptional target of the large
MAF family.
A previous in vitro study has demonstrated
that ARK5 expression is a key element in multiple myeloma invasion
and metastasis and participates in these processes by modulating
insulin-like growth factor (IGF)-1 expression (8). Schiller et al and Perumal et
al (27,28) demonstrated that the ratio of AMP/ATP
increased during energy deficiency, leading to phosphoric
acid-dependent AMPK activation. Eventually, the level of ATP
returned to normal due to the inhibition of energy-consuming
pathways.
The overexpression of ARK5 has been previously found
to exert negative effects on the apoptosis regulation of multiple
myeloma by inducing glucose starvation tolerance (29). ARK5 is directly stimulated by Akt,
which regulates cancer cell survival and proliferation (9-11,30).
Inhibition of ARK5 can lead to the reduction in ATP levels in cells
that abnormally express MYC, leading to a variety of pro-apoptotic
reactions (31). MYC is widely
known as a strong factor in tumorigenesis (29,32).
In conclusion, AKR5 has become a more promising
choice to inhibit malignant tumors and prolong patient survival due
to high reversal of drug resistance. ARK5 is closely associated
with multiple myeloma and may be an effective therapeutic
target.
CRC
CRC is one of the most lethal cancers worldwide
(15). An available treatment
method for all stages of CRC is tumor resection, with chemotherapy
and radiotherapy commonly used as neoadjuvants in locally advanced
CRC (33). However, the prognosis
of patients with CRC remain poor (33). A total of 241 pairs of cDNA from
normal tissues and 13 different tumor specimens from patients with
CRC were sequenced via DNA array analysis. The results revealed
that ARK5 was overexpressed in CRC. Additionally, a total of 56
clinical specimens of primary CRC and liver metastasis demonstrated
high ARK5 expression (31).
Poor clinical prognosis caused by ARK5 is primarily
associated with a hypoxic microenvironment (34). Hypoxia is very common in tumors and
is associated with proliferation, invasiveness, metastasis and drug
resistance (35). Hypoxia-inducible
factor (HIF)1 serves a regulatory role and can be used as a key
prognostic indicator of tumor hypoxia in CRC (36). HIF-1 is a dimer composed of
HIF1-α and HIF1-β subunits (37). HIF1-α is a regulator that is
triggered by hypoxia and subsequently regulates the activity of the
entire complex (37). The
expression and absence of HIF1-α is associated with poor prognosis
in patients with CRC (38).
Kusakai et al (31) revealed that ARK5 was overexpressed
in malignant CRC tumors and that this expression dynamically
increased in hypoxic conditions. Additionally, it was determined
that ARK5 and HIF1-α were overexpressed in CRC, demonstrating a
clear linear correlation. ARK5 is regulated by HIF1-α, which
amplifies the ARK5 signal and promotes cancer cell survival in
hypoxic conditions (37). Due to
this, ARK5 is highly expressed in hypoxic solid tumor-associated
blood vessels, which is an another important factor in cancer cell
angiogenesis and metastasis (37).
HIF1-α expression downstream of ARK5 is associated
with tumor stage, tumor grade, lymph node metastasis and liver
metastasis (35). However, further
research is required to assess the degree of malignancy in solid
tumors.
PC
PC is the most fatal cancer and is the eighth
leading cause of cancer-related death worldwide (39). Prognosis is often poor due to high
invasiveness, rapid proliferation and limited treatment (15,39).
By analyzing a rank-based meta-analysis of
individual histological features associated with pancreatic ductal
adenocarcinoma (40), 8 genes
associated with PC progression were identified: ARK5, E2F
transcription factor 3, high mobility group AT-Hook 2, RAS P21
protein activator 1, insulin receptor substrate 1, actinin α1,
Sloan-Kettering oncogene and Δ-like protein 1 pre-cursor. ARK5 is
one of the potential regulators that was significantly
differentially expressed (40).
These results may indicate that AKR5 serves a significant role in
PC.
Gemcitabine (GEM) is the sole first-line drug for
advanced PC, although it only prolongs patient survival for a few
months due to clinical multi-drug resistance (41). Further research into this resistance
mechanism is required to improve PC treatment (15,42,43).
Studies have demonstrated that hypoxia increases PC cell resistance
to GEM-induced apoptosis (44,45).
Additionally, under hypoxic conditions, ARK5 inhibition
significantly increases the sensitivity of PC cells to GEM
(44). In a previous study, the
inhibition of ARK5 reversed the effects of hypoxia on E-cadherin
and vimentin expression, whichh are markers of
epithelial-mesenchymal transition (EMT) (46). Therefore, ARK5 regulates GEM
resistance under hypoxic conditions through the EMT process
(47).
Furthermore, in vivo and in vitro ARK5
overexpression was demonstrated in PC metastasis and invasion
models through EMT (9,10). The detection and intervention of
ARK5 expression may therefore improve drug sensitivity and improve
prognosis and survival time in patients with PC.
Other cancers
ARK5 expression was confirmed in various types of
cancer, including osteosarcoma, ovarian, hepatic, CRC, gastric,
breast and non-small cell lung cancer (NSCLC). NSCLC is one of the
most common malignant tumors worldwide and accounts for ~1/3 of all
cancer-related deaths (48,49). Currently, chemotherapy is one of the
most effective treatments for NSCLC, with cisplatin being the
standard first-line drug, though its long-term therapeutic efficacy
reduced by drug resistance (49).
Although various factors are known to induce
chemical resistance, the mechanism of cisplatin resistance remains
unclear (50). A report has
previously indicated that epithelial phenotype NSCLC is more
sensitive to chemotherapy compared with a mesenchymal phenotype
(51). Mesenchymal tumors that
express E-cadherin regain chemical sensitivity (51). In ARK5-knockdown cells, sensitivity
to cisplatin increased significantly, suggestomg that it may serve
as a potential strategy to improve NSCLC drug resistance.
Furthermore, in head and neck squamous cell carcinoma (HNSCC), a
study has demonstrated that the expression of micro RNA (miRNA or
miR) associated with the invasion and metastasis of melanoma. This
was also observed in epithelial ovarian cancer (52).
ARK5 overexpression is associatied with poor
prognosis (53-55)
and has been identified in various solid tumors. Numerous studies
have also demonstrated that ARK5 activation can induce the survival
of cancer cells during nutritional deficiency (8,56).
3. Emerging role of ARK5 in cancer genesis
and progression
ARK5 and Akt
ARK5 is a member of the AMPK family and its highly
conserved T loop is phosphorylated by two molecules: Akt kinase,
which acts on serine600 and liver kinase (LK) B1, which acts on
threonine200(57). Research has
revealed that ARK5 transcription is regulated by sp1
transcription-activated protein, metabolic pressure and cofactors
required at the sp-1 site (58,59).
This covers almost all the pathways that involve ARK5.
Akt is a serine-threonine protein kinase that
functions as a key regulator of cell survival and serves an
important role in tumor genesis, cell survival, proliferation and
differentiation (60). Accelerated
Akt activation in malignant tumors (including invasion and
metastasis) is associated with gene amplification in various types
of cancer, including CRC, PC, gastric and ovarian cancer (61-63).
Therefore, Akt is essential to malignant tumors.
Numerous studies have confirmed the role of Akt in
promoting. tumor invasion and metastasis (31,64)
during nutritional starvation. However, the downstream factors of
Akt in these processes have not been determined.
A previous study has indicated that Akt-1 and Akt-2
expression in CRC and liver metastasis is higher compared with
normal tissue and that ARK5 is expressed in highly malignant
clinical specimens, especially in those with invasive morphology
(29). Since it has been
demostrated that Akt is the most direct upstream pathway of ARK5
and ARK5 is the only known protein of the AMPK familiy assocuated
with the Akt pathway, this would indicate a close regulatory effect
between Akt and ARK5(55).
Furthermore, Akt mediates several cellular responses
induced by insulin and IGF, such as glycogen synthesis via the
phosphorylation of glycogen synthase kinase 3(65). In a previous study, ARK5 was
revealed to mediate the invasion of PC and CRC and to promote
cancer cell survival by activating the Akt signaling pathway via
IGF-1(66). Furthermore, nuclear
DBF-related kinase 2 (NDR2) was revealed to be activated by IGF-1
treatment, phosphorylating threonine211 on the active T loop of
ARK5(56). ARK5 was also
demonstrated to be downstream of NDR2 during activation of IGF-1
signaling (67). Therefore, ARK5
promotes cancer cell survival under the regulation of Akt and ARK5
serves an important role in hypoxia and the Akt pathway in
apoptosis in various types of cancer.
The RAF-MEK-ERK and PI3K-Akt-HIF-α pathways serve an
important role in cancer development as they are downstream of RAS
(68), a protein that regulates
cancer cell survival. Akt, as the most direct downstream regulator
of RAS, regulates the downstream factor HIF1-α (69). Previous studies have determined a
close association between the HIF1-α-mediated RAS pathway and ARK5
(68-70).
A linear correlation was reported between ARK5 and
HIF1-α expression (68,70). In a previous study, short
interfering RNA suppressed HIF1-α expression under hypoxic
conditions. The results revealed that the protein and mRNA levels
of ARK5 were significantly decreased indicating that ARK is
regulated by HIF1-α and that ARK5 lie downstream of HIF1-α under
hypoxic conditions where HIF1-α amplifies the role of ARK5 in
hypoxia (57). In conclusion, ARK5
is the key gene of HIF1-α-mediated cancer proliferation and
migration under hypoxic conditions.
ARK5 overexpression was demonstrated to
significantly stimulate the invasiveness and metastasis of PC cells
in vitro and in vivo by activating matrix
metallopeptidase (MMP) and matrix metalloproteinase 1 (MT1)-MMP
(7). MMPs, especially MMP-2 and
MMP-9, participate in tumor metastasis and MT1-MMP is the most
common activating agent of these (34,71).
Research has demonstrated that ARK5 increased MMP-2 and MMP-9
production levels and induced their activation via MT1-MMP
production (7).
Davis et al (58) previously hypothesized that ARK5
activation served as a metabolic checkpoint in the regulation of
apoptosis, cell cycle progression and arrest, which confirmed
ARK5-modulated ‘glucose metabolism’ as the most significantly
aberrantly affected cellular signaling pathway in a model system
for highly metastatic tumors.
ARK5 and EMT
EMT is involved in numerous biological and
pathological processes, is associated with chemotherapeutic
resistance and has an invasive and anti-apoptotic role in cancer
tissues (72). EMT is the process
by which polar epithelial cells with firm cell-cell adhesion
transform into mesenchymal cells with highly invasive capacity
(73). At the molecular level, the
gene expression of these cells undergoes numerous changes: The
expression of epithelial genes (E-cadherin, tight junction protein
1 and occludin) decreases and the expression of mesenchymal genes
(N-cadherin, vimentin and fibronectin) increases (74).
In a study on epithelial ovarian cancer, ARK5 was
highly expressed in cancer cells compared with normal tissue and
was revealed to be strongly associated with EMT (51). Furthermore, ARK5 was reported to
regulate the progression of EMT in various solid tumors (74-78).
When ARK5 expression was decreased in cells, the resultant
inhibition of E-cadherin expression and the downregulation of
vimentin expression were related to ARK5 activation (79).
Since the recurrence of E-cadherin expression was
demonstrated to increase the sensitivity of cancer tissues to
chemotherapeutic agents in various studies, reducing the expression
of ARK5 may increase sensitivity to drugs such as doxorubicin (dox)
and cisplatin (75,78).
TGF-β1 is a key induction factor of the EMT pathway
in tumors (80) and induces EMT in
NSCLC (81). ARK5 knockdown was
reported to decrease TGF-β1-induced EMT and invasion and metastasis
of cells under hypoxic conditions (82). The results indicated that ARK5 may
be involved in the hypoxia-induced TGF-β1 pathway in cancer cells,
which contributes to glucose starvation tolerance (82). In a liver study, ARK5 expressed
lipid fibers and ultimately induced hepatic cell necrosis (52,83).
This process is also observed in PC (84). ARK5 inhibits cancer cell death
stimulation and promotes normal tissue necrosis. Additionally,
another study reported that the suppression of ARK5 reversed
hypoxia-induced EMT in hepatic cancer cells (81). The results suggested that ARK5
overexpression is indicative of hypoxia. An additional protein,
zinc finger E-box-binding homeobox 1 (ZEB1), also acts as an
activator of EMT in mantle cell lymphoma cells and determines
resistance to different chemotherapeutic drugs (81). ARK5 was demonstrated to suppress
ZEB1 to improve drug sensitivity (81,85).
In conclusion, ARK5 is associated with drug
resistance in solid tumors and mediates the EMT pathway and
numerous EMT-related molecules such as TGF-β1 and ZEB1. Since ARK5
knockdown improves resistance to clinical drugs, ARK5 may serve as
a new target to reverse drug resistance.
ARK5 and Fas
Fas is a member of the tumor necrosis factor
receptor family (86). As a
transmembrane protein, it transmits apoptotic signals in cells and
induces apoptosis when Fas ligands bind to the Fas receptor
(87). Fas is widely expressed in
normal tissues and tumors.
Fas-cell mediated apoptosis serves an important role
in various biological processes as it triggers a series of
downstream pathways (86,87), suggesting that ARK5 inhibits the
activation of caspase-8 and the expression of caspase-6 containing
two putative ARK5 phosphorylation sites: Ser80 and Ser257(29), and ultimately inhibits the
phosphorylation induced by Fas ligand and Fas. This could promote
the survival of cancer cells under conditions of nutritional
starvation (12,86).
In further studies, ARK5 was revealed to inhibit
caspase-8 activation by preserving c-FLIP in response to Akt
stimulation (4,88), preventing cell death caused by
glucose starvation and RAS activation in cancer cells (8,86).
RAS-induced apoptosis was demonstrated to be inhibited by increased
ARK5 expression (11). These
results suggested that ARK5 is associated with energy metabolism
and cell apoptosis.
ARK5 and MYC
MYC is an important inducer protein in cancer that
interferes with cell cycle metabolism and ribosome synthesis
(89). When combined with
MYC-associated factor X, MYC regulates transcription and oncogenic
activity (90). MYC overexpression
has been demonstrated to induce cell apoptosis as it cannot
maintain an adequate ratio of ATP/ADP (91,92).
Kusakai et al and Cox and Der (67,68)
previously demonstrated that, using synthetic lethal RNAi
screening, ARK5 kinases regulated protein expression by activating
various pathways that ultimately maintained or triggered cancer
cell survival, particularly when MYC was overexpressed.
Activation of ARK5 and AMPK in response to metabolic
stress combats apoptosis in cancer cells and they are markers for
most solid tumors (93,94). AMPK is activated by the tumor
inhibitor LKB1(95). In the absence
of LKB1, AMPK responds to calcium ions by phosphorylating
calcium/calmodulin-dependent protein kinase kinase 2 (95,96).
As a member of the AMPK family, ARK5 is also primarily activated by
LKB1(55).
However, Ciccarese et al (97) revealed a second pathway that
maintains ARK5 activity in the absence of LKB1: ARK5 responds to
calcium signaling through protein kinase C α (PKCα) to regulate
AMPK activation (98) and
mTORC1-dependent protein translation, protecting cells from
MYC-driven apoptosis.
The Calcium-AMPK-mTORC1 metabolic
checkpoint-dependent activation requires PKCα and ARK5 while in the
absence of ARK5, activated mTOR increases ATP consumption and
impairs MYC response to AMPK (98).
Therefore, in the presence of ARK5, ATP synthesis is enhanced
through the MYC pathway. The depletion of PKCα and ARK5 leads to
apoptosis, which suggests that this pathway serves an active role
in tumor maintenance (29). The
results indicated a novel role for calcium ions in supporting
cancer cell viability and elucidated the synthetic lethal
interaction between ARK5 and MYC (98). Similarly, PKCα and -β have been
demonstrated to phosphorylate Akt (99,100),
preventing typical MYC-induced apoptosis by inhibiting the
expression and function of apoptotic Bcl-2 homology protein
(100-102).
Therefore, ARK5 expression is necessary for MYC overexpression,
even if Akt is overexpressed (29),
suggesting that ARK5 may be a potential target for treating
MYC-driven cancer (4,103).
Overall, ARK5 and PKCα may control multiple pathways
that promote cancer cell survival. The targeted suppression of
these pathways may therefore have potential therapeutic benefits in
numerous types of cancer in which MYC is deregulated (103).
At the organelle level, ARK5 is involved in energy
regulation by maintaining mitochondrial adaptability and stability
and increases the expression of proteins in the respiratory chain
by activating MYC, which enhances respiratory capacity (55). When ARK5 is depleted, this
phenomenon is eliminated (55). As
an important upstream regulator of MYC, ARK5 serves an important
role in tumor regulation at the organelle level.
ARK5 and aneuploidy
The AMPK family has 13 different sub-groups with
different regulatory modes (104).
However, all of them are activated by LKB1 under conditions of
metabolic pressure, when ATP levels are low (105). Tumor protein P53 is a downstream
protein of AMPK and its phosphorylation serves an important role in
apoptosis and cell aging (105).
ARK5 is part of the AMPK subfamily. It was
demonstrated experimentally that ARK5 regulates P53 phosphorylation
in vivo and in vitro, directly interacting with the
P53 nucleus under the regulation of LKB1(106). Additionally, ARK5 activation via
P21 and weak acid resistance protein 1 prevents cells from entering
the S phase from the G1 phase (106).
ARK5 was demonstrated to induce premature cell
aging, which is closely associated with genetic aneuploidy, which
had been previously demonstated in fibroblast (89). ARK5 regulates ploidy and senescence.
Decreased ARK5 prevents aneuploidy in cells and enhance their
replicative lifespan, while increased ARK5 induces gross
aneuploidies and senescence (89).
This ARK5-induced aneuploidy increased the genomic
instability of cancer cells and allowed them to overgrow, invade
and metastasize, demonstrating the role of ARK5 in tumor regulation
(89). Similarly to how cancer
develops and exacerbates, genomic instability tends to induce cell
senescence (107-109).
Genomic instability often manifests as an increase in aneuploidy
and a decrease in large tumor suppressor kinase 1 (LATS1)
expression, a kinase involved in mitotic exit (109). Decreased LATS1 levels block cell
division, affect genomic stability and increase the amount of
abnormal DNA in each cell (109).
AMPK accelerates P53-mediated cell senescence and ARK5 also leads
to genomic aneuploidy changes without the involvement of
P53(110). However, experimental
knockdown or overexpression of ARK5 did not affect cellular P53
activity (110).
In summary, in terms of cellular senescence
regulation, ARK5 knockdown extends the lifespan of cells,
metabolism and slows fibroblast senescence in normal individuals,
even in the absence of LKB1. LATS1 is a regulator of stable gene
expression and the overexpression of ARK5 weakens the expression of
LATS1(89). Such changes are
independent of P53, which highlights the potential role of
aneuploidy in ARK5-mediated senescence (89), and suggests a difference between the
ARK5 and AMPK families at downstream action sites.
Furthermore, ARK5-induced aneuploidy leads directly
to the death of MCF10a immortal cells rather than to senescence in
other types of cells (89),
indicating that aneuploidy serves a different role in different
cells.
ARK5 and miRNA
miRNAs are non-coding RNAs 18-22 nucleotides in
length that regulate various physiological activities through
specific binding to the 3'-untranslated regions (3'UTR) of mRNA
(111). miRNAs are involved in
cell proliferation, invasion and metastasis (111). Currently, different miRNA families
serve different roles in malignant tumors (112). For example, the miR-200 family
(miR-200a, -200b, -200c, -141 and -429) slowed EMT in cancer cells
by lowering ZEB1/ZEB2 expression (111,113).
It has been demonstrated that ARK5 was regulated by
different miRNAs in different cancers and that ARK5 was negatively
associated with the expression of certain miRNAs (112). In a liver cancer study, miR-204
reduced ARK5 expression and invasion, and reversed drug resistance
(114). Similarly, miR-145 acted
as a negative regulator of intrahepatic cholangiocarcinoma via the
regulation of ARK5. miR-145 also reduced MT1-MMP, MMP-2 and MMP-9
expression to alter cell metastasis (115). miR-211 is associated with cell
invasion and response to melanoma adhesion (116). In HNSCC, miR-203 regulated EMT by
targeting ARK5 and miR-96 regulated PC malignancy in the same
manner (117). These three RNAs
function upstream of ARK5 to block cell invasion (112). Overall, ARK5-associated miRNA
could be regarded as a potential therapeutic target in the future
treatment of cancer.
4. ARK5 as a potential therapeutic
target
While chemotherapy is still a major strategy in
cancer treatment, drug resistance has become a novel limitation
that has led to its failure in long-term use (118). Therefore, there is an urgent
requirement to elucidate novel strategies to increase drug
efficacy.
Salinomycin
It has been established that ARK5 is highly
expressed after treatment with certain clinical therapeutic drugs,
including dox, 5-fluorouracil and cisplatin (50). Since ARK5 is associated with cancer
cell drug resistance, its downregulation may serve as a potential
therapeutic strategy to increase drug sensitivity (50). To increase drug sensitivity,
salinomycin may be used as it targets ARK5(50).
Salinomycin is an ionophore antibiotic that kills
cancer stem cells and reverses EMT (50). A previous study on lung cancer
demonstrated that salinomycin increased dox sensitivity and
demonstrated a synergistic effect (combination index, 0.430 with
dox) (50). Furthermore, it was
revealed that salinomycin suppresses ZEB1, an important EMT pathway
molecule (83,84). Therefore, salinomycin may be a novel
drug that could be used in drug combinations. However, weight loss
and nerve injury are side effects of salinomycin treatment and may
affect its application in a clinical setting (119).
Additionally, it has been demonstrated that
combination treatment reversed dox-induced EMT morphology, reversed
EMT marker protein (vimentin and E-cadherin) expression and
inhibited ARK5 expression (75).
This process was also demonstrated in breast cancer, gastric
cancer, non-small cell lung cancer, cholangiocarcinoma and hepatic
cancer (120).
ON123300
ON123300 is a novel second-generation oral drug and
CDK inhibitor, which exerts dual inhibitory effects on CDK4 and
ARK5(121). Compared with
first-generation drugs, the site-specific ON123300 reverses the
poor curative effects exerted by other drugs and the resultant poor
cancer prognosis, which was demonstrated in multiple myeloma
treatment (120). ON123300 was
also revealed to exert beneficial effects in breast cancer, glioma
and mantle cell lymphomas in vivo and in vitro
without obvious harm to normal tissue (122).
ARK5 is a molecule that may lead to first-generation
drug failure (20,28,122).
However, while ARK5 overexpression has been observed in primary
multiple myeloma cells, ON123300 caused cell cycle arrest and
apoptosis, serving as an ARK5 and CDK4 inhibitor (32,28).
MYC-CDK4 binding in multiple myeloma cells is a key interaction in
tumorgenesis and ON123300 also has an impact on the MYC pathway as
well as on the retinoblastoma/mTOR pathway (122). Furthermore, ARK5 knockdown
significantly increased first-generation drug sensitivity of
multiple myeloma (123,124).
7x
As 7x is a novel cyanopyridopyrimidine compound that
acts as a multi-kinase inhibitor, 7x targets CDK4/cyclin D1 and
ARK5 kinases (122). ARK5 is also
negatively regulated by 7x (124).
A previous study demonstrated that ARK5 targeting resulted in high
efficacy when cancer cell proliferation and metastasis were
inhibited with no significant signs of toxicity (122).
With the increasing number of compounds that have
been discovered as effective drugs targeting the ARK5 protein for
cancer treatment, the current study hypothesizes that ARK5 could be
considered a significant therapeutic strategy for cancer under 7x
treatment.
HTH-01-015
HTH-01-015 is a highly selective protein kinase
inhibitor, which mainly affects ARK5 by inhibiting the
phosphorylation of myosin phosphatase target subunit 1 (MYPT1), a
substrate of ARK5(125).
Experimentally, HTH-01-015 was revealed to slow mitosis by
inhibiting the phosphorylation of MYPT1 and preventing the entry of
cells into the M phase by regulating DNA replication in the S phase
(126). This indicated that
HTH-01-015 inhibited ARK5 and modulated cell migration and
adhesion. Additionally, HTH-01-015 was revealed to detect the
physiological function of ARK5 abnormalities, further elucidating
its role (126).
In summary, HTH-01-015 was demonstrated to regulate
the physiological functions of cells by acting on ARK5 and could
potentially serve as a novel drug to combat clinical drug
resistance.
5. Relationship between ARK5 and other
diseases
ARK5, diabetic nephropathy and renal
fibrosis
ARK5 is associated with Tau protein stability. This
may explain the potential link between ARK5 and a range of
diseases, including neurodegenerative diseases and diabetes
(55).
ARK5 serves an important role in human diabetic
nephropathy (DN) along with TGF-β1 (127,128). DN is often accompanied by a series
of renal diseases in which TGF-β1 mediates glomerular sclerosis and
tubular fibrosis (127).
In vitro, TGF-β1 was used to induce a model
of renal tubular fibrosis. The protein changes that occur during
the EMT process in epithelial cells include: E-cadherin
(epithelial) to N-cadherin (mesenchymal) transformation and
increased vimentin, α-smooth muscle actin, connective tissue growth
factor and Notch ligand Jagged-1(129). By silencing ARK5 and thereby
lowering TGF-β1 expression, fibrosis was reversed in renal tubular
cells. This also influenced downstream proteins of TGF-β1 by
preventing the stimulation of Jagged-1, verifying the link between
ARK5 and EMT (130,131).
ARK5 and striated muscle
generation
ARK5 protein was not previously identified in
murine skeletal muscle. However, in later studies, ARK5 was
demonstrated to be highly expressed in cardiac muscle and skeletal
muscle, and ARK5 mRNA was identified via reverse
transcription-quantitative PCR (130). Muscle contraction increased ARK5
phosphorylation (132). However,
this did not alter ARK5 activity, indicating that phosphorylation
of ARK5 at Ser400 cannot stimulate this kinase (132).
Uncordinated-82 (UNC-82) kinase serves a key role
in Caenorhabditis elegans, a nematode, and is an orthologue
of human ARK5 and SNF1/AMP kinase-regulated kinase (SNARK) that is
necessary for myosin filament reorganization during cellular
elongation (27). Research has
suggested that ARK5 may exert a similar function to UNC-82 in
striated muscle development (130). ARK5 knockout could lead to
alterations in contractile apparatus protein (the actin-myosin
cytoskeleton) phosphorylation and SNARK reduction may lead to
muscle mass disruption with increasing age. These results revealed
the conserved role of UNC-82/ARK5/SNARK in muscle generation across
diverse animal lineages (27).
Additionally, ARK5 was demonstrated to be a direct
target of large musculoaponeurotic fibrosarcoma proteins and its
activation was mediated by MAF-recognition element sequences
(133). Activation of ARK5 in
muscle allows it to interact with several types of myosin
phosphatases (134). Furthermore,
the ARK5-MYPT1-serine/threonine-protein phosphatase 1B complex
promoted the interaction between ARK5 and 1433 protein, which
inhibited phosphatase activity (134).
Ultimately, ARK5 controls cell adhesion and as a
regulator of myosin phosphatase compounds; it prevents the
phosphorylation of AMPK targets by LKB1 and controls phosphatase
compounds to influence the phosphorylation of its targets (135).
Furthermore, experiments have demonstrated that
ARK5 is involved in the negative feedback regulation of insulin
signaling transduction and inhibits insulin-mediated glucose uptake
in skeletal muscle (136).
ARK5 and neurodegenerative
diseases
The effects of ARK5 in neurological diseases
primarily manifest in three aspects: Promoting the polarization and
migration of neurons, affecting the expression and accumulation of
Tau protein and affecting neuron apoptosis through CASP6(137).
Neuronal axons need energy to grow and branch, and
this energy comes from the large consumption of ATP in the
mitochondria (138). As a
mediating factor, ARK5 regulates the growth of axons and cortical
neuron branches. The LKB1-ARK5 pathway controls the fixation of
mitochondria in axons and ARK5 mediates axon growth and growth of
cortical neuron branches (138).
Overexpression of ARK5 can increase the axon branch, and knockout
can cause growth stagnation (138).
Numerous neurodegenerative proteinopathies share a
common pathogenesis: Abnormal accumulation of disease-related
proteins (139). ARK5 was revealed
to regulate Tau by stabilizing the protein via specific Ser356
phosphorylation and its inhibition inhibited Tau expression in
Drosophila neurodegeneration (139). The results suggested that reducing
Tau expression is potentially an effective strategy to alleviate
Tau-related neurodegenerative changes. Furthermore, ARK5 may serve
as a new entry point for the treatment of Tau-associated diseases
(139).
CASP6 was demonstrated to be an important molecule
in neurodegenerative diseases, particularly in Alzheimer's disease
(AD) and Huntington's disease (137). Research has revealed that avoiding
destruction of CASP6 protected mice from neural diseases, such as
AD (140). This protection is the
result of ARK5 phosphorylating CASP6 at Ser257, which caused CASP6
inhibition and prevented neural cell death (86). The phosphorylation achieved by AKR5
is indirect and is mediated via the downregulation of p53
expression (106) due to the
direct relationship between p53 and CASP6(141). Furthermore, ARK5-mediated CASP6
phosphorylation inhibited its activation, mediating CASP6 activity
(86). This phosphorylation site is
specific for CASP6 (86,142).
The aforementioned study offered a potential site
for future drug discovery by targeting this unique site to
gradually inhibit neural death. Since ARK5 is a negative regulator
of CASP6, a potential strategy could involve activating ARK5
expression or interfering with CASP6 phosphorylation at this
specific site.
6. Conclusion and future perspectives
The current review summarized all known functions
of ARK5 and potential drugs from a limited study size. Being a
member of the AMPK family, ARK5 serves a metabolic role through its
expression: Promoting the survival of cancer cells in harsh
microenvironments. Additionally, ARK5 serves an important role in
cell adhesion and metastasis (84).
Various processes are involved in cancer cell drug
resistance and further research into this resistance mechanism is
required to fully elucidate it. As an emerging molecule, ARK5
exerts multiple functions in certain diseases, particularly in
malignant tumors. Since AKR5 can be used to evaluate the malignancy
of tumors, metastasis and drug restistancy, it is a potential
therapeutic target for cancer and drug resistance. Targeting ARK5
in combination with other drugs could potentially improve drug
resistance and inhibit tumor metastasis.
The current review hypothesized that in the future,
drugs targeting ARK5 will have impactful and specific effects that
will improve clinical drug resistance. However, further research
into the role of ARK5 in normal cells may be beneficial as it could
reduce the targeting side effects of ARK5 therapy. The present
review also hypothesized that the relationship between ARK5 and TAU
stability is conducive to further elucidating the molecular
mechanism of neurodegenerative diseases in future studies.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (no. 81560397). But it was neither enrolled in
the design of the study, collection, analysis, and interpretation
of the data, writing of the report, nor in the decision to submit
the article for publication. Every author had full access to all
data of the study, and the corresponding author had final
responsibility for the decision to submit the article for
publication.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
GM and QJ contributed to the concept and design of
this manuscript. BZ contributed to data reviewing and part of the
analysis GM, QJ and BZ wrote the manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Esumi H, Izuishi K, Kato K, Hashimoto K,
Kurashima Y, Kishimoto A, Ogura T and Ozawa T: Hypoxia and nitric
oxide treatment confer tolerance to glucose starvation in a
5'-AMP-activated protein kinase-dependent manner. J Biol Chem.
277:32791–32798. 2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ivanov S, Liao SY, Ivanova A,
Danilkovitch-Miagkova A, Tarasova N, Weirich G, Merrill MJ,
Proescholdt MA, Oldfield EH, Lee J, et al: Expression of
hypoxia-inducible cell-surface transmembrane carbonic anhydrases in
human cancer. Am J Pathol. 158:905–919. 2001.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kaluz S, Kaluzova M, Chrastina A, Olive
PL, Pastoreková S, Pastorek J, Lerman MI and Stanbridge EJ: Lowered
oxygen tension induces expression of the hypoxia marker MN/carbonic
anhydrase IX in the absence of hypoxia-inducible factor 1 alpha
stabilization: A role for phosphatidylinositol 3'-kinase. Cancer
Res. 62:4469–4477. 2002.PubMed/NCBI
|
|
4
|
Suzuki A, Kusakai G, Kishimoto A, Lu J,
Ogura T, Lavin MF and Esumi H: Identification of a novel protein
kinase mediating Akt survival signaling to the ATM protein. J Biol
Chem. 278:48–53. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li B, Tsao SW, Li YY, Wang X, Ling MT,
Wong YC, He QY and Cheung AL: Id-1 promotes tumorigenicity and
metastasis of human esophageal cancer cells through activation of
PI3K/AKT signaling pathway. Int J Cancer. 125:2576–2585.
2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ohta T, Isobe M, Takahashi T,
Saitoh-Sekiguchi M, Motoyama T and Kurachi H: The Akt and ERK
activation by platinum-based chemotherapy in ovarian cancer is
associated with favorable patient outcome. Anticancer Res.
29:4639–4647. 2009.PubMed/NCBI
|
|
7
|
Renton A, Llanos S and Lu X: Hypoxia
induces p53 through a pathway distinct from most DNA-damaging and
stress-inducing agents. Carcinogenesis. 24:1177–1182.
2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Suzuki A, Iida S, Kato-Uranishi M, Tajima
E, Zhan F, Hanamura I, Huang Y, Ogura T, Takahashi S and Ueda R:
ARK5 is transcriptionally regulated by the Large-MAF family and
mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a
new molecular determinant of malignant multiple myeloma. Oncogene.
24:6936–6944. 2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Simon PO Jr, McDunn JE, Kashiwagi H, Chang
K, Goedegebuure PS, Hotchkiss RS and Hawkins WG: Targeting AKT with
the proapoptotic peptide, TAT-CTMP: A novel strategy for the
treatment of human pancreatic adenocarcinoma. Int J Cancer.
125:942–951. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kato K, Ogura T, Kishimoto A, Minegishi Y,
Nakajima N, Miyazaki M and Esumi H: Critical roles of AMP-activated
protein kinase in constitutive tolerance of cancer cells to
nutrient deprivation and tumor formation. Oncogene. 21:6082–6090.
2002.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Izuishi K, Kato K, Ogura T, Kinoshita T
and Esumi H: Remarkable tolerance of tumor cells to nutrient
deprivation: Possible new biochemical target for cancer therapy.
Cancer Res. 60:6201–6207. 2000.PubMed/NCBI
|
|
12
|
Suzuki A, Kusakai G, Kishimoto A, Lu J,
Ogura T and Esumi H: ARK5 suppresses the cell death induced by
nutrient starvation and death receptors via inhibition of caspase 8
activation, but not by chemotherapeutic agents or UV irradiation.
Oncogene. 22:6177–6182. 2003.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Suzuki A, Kusakai G, Shimojo Y, Chen J,
Ogura T, Kobayashi M and Esumi H: Involvement of transforming
growth factor-beta 1 signaling in hypoxia-induced tolerance to
glucose starvation. J Biol Chem. 280:31557–31563. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kuehl WM and Bergsagel PL: Molecular
pathogenesis of multiple myeloma and its premalignant precursor. J
Clin Invest. 122:3456–3463. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Benboubker L, Dimopoulos MA, Dispenzieri
A, Catalano J, Belch AR, Cavo M, Pinto A, Weisel K, Ludwig H,
Bahlis N, et al: Lenalidomide and dexamethasone in
transplant-ineligible patients with myeloma. N Engl J Med.
371:906–917. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rajkumar SV, Blood E, Vesole D, Fonseca R
and Greipp PR: Eastern Cooperative Oncology Group. Phase III
clinical trial of thalidomide plus dexamethasone compared with
dexamethasone alone in newly diagnosed multiple myeloma: A clinical
trial coordinated by the Eastern Cooperative Oncology Group. J Clin
Oncol. 24:431–436. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Richardson PG, Weller E, Lonial S,
Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM,
Schlossman RL, et al: Lenalidomide, bortezomib, and dexamethasone
combination therapy in patients with newly diagnosed multiple
myeloma. Blood. 116:679–686. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
San Miguel JF, Schlag R, Khuageva NK,
Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT,
Palumbo A, Samoilova OS, et al: Bortezomib plus melphalan and
prednisone for initial treatment of multiple myeloma. N Engl J Med.
359:906–917. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kumar SK, Dispenzieri A, Lacy MQ, Gertz
MA, Buadi FK, Pandey S, Kapoor P, Dingli D, Hayman SR, Leung N, et
al: Continued improvement in survival in multiple myeloma: Changes
in early mortality and outcomes in older patients. Leukemia.
28:1122–1128. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kawauchi S, Takahashi S, Nakajima O, Ogino
H, Morita M, Nishizawa M, Nishizawa M, Yasuda K and Yamamoto M:
Regulation of lens fiber cell differentiation by transcription
factor c-Maf. J Biol Chem. 274:19254–19260. 1999.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kim JI, Li T, Ho IC, Grusby MJ and
Glimcher LH: Requirement for the c-Maf transcription factor in
crystallin gene regulation and lens development. Proc Natl Acad Sci
USA. 96:3781–3785. 1999.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ring BZ, Cordes SP, Overbeek PA and Barsh
GS: Regulation of mouse lens fiber cell development and
differentiation by the Maf gene. Development. 127:307–317.
2000.PubMed/NCBI
|
|
24
|
Zhang Z, Tong J, Tang X, Juan J, Cao B,
Hurren R, Chen G, Taylor P, Xu X, Shi CX, et al: The ubiquitin
ligase HERC4 mediates c-Maf ubiquitination and delays the growth of
multiple myeloma xenografts in nude mice. Blood. 127:1676–1686.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang S, Juan J, Zhang Z, Du Y, Xu Y, Tong
J, Cao B, Moran MF, Zeng Y and Mao X: Inhibition of the
deubiquitinase USP5 leads to c-Maf protein degradation and myeloma
cell apoptosis. Cell Death Dis. 8(e3058)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Qiang YW, Ye S, Chen Y, Buros AF, Edmonson
R, van Rhee F, Barlogie B, Epstein J, Morgan GJ and Davies FE: MAF
protein mediates innate resistance to proteasome inhibition therapy
in multiple myeloma. Blood. 128:2919–2930. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Schiller NR, Duchesneau CD, Lane LS, Reedy
AR, Manzon ER and Hoppe PE: The Role of the UNC-82 protein kinase
in organizing myosin filaments in striated muscle of caenorhabditis
elegans. Genetics. 205:1195–1213. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Perumal D, Kuo PY, Leshchenko VV, Jiang Z,
Divakar SK, Cho HJ, Chari A, Brody J, Reddy MV, Zhang W, et al:
Dual targeting of CDK4 and ARK5 using a novel kinase inhibitor
ON123300 exerts potent anticancer activity against multiple
myeloma. Cancer Res. 76:1225–1236. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liu L, Ulbrich J, Müller J, Wüstefeld T,
Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, et
al: Deregulated MYC expression induces dependence upon AMPK-related
kinase 5. Nature. 483:608–612. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hoppe S, Bierhoff H, Cado I, Weber A,
Tiebe M, Grummt I and Voit R: AMP-activated protein kinase adapts
rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA.
106:17781–17786. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kusakai G, Suzuki A, Ogura T, Miyamoto S,
Ochiai A, Kaminishi M and Esumi H: ARK5 expression in colorectal
cancer and its implications for tumor progression. Am J Pathol.
164:987–995. 2004.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li X, Zhang XA, Li X, Xie W and Huang S:
MYC-mediated synthetic lethality for treating tumors. Curr Cancer
Drug Targets. 15:99–115. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Colon Cancer Treatment (PDQ®):
Patient Version. PDQ Cancer Information Summaries. Bethesda, MD,
2002.
|
|
34
|
Peng JK, Shen SQ, Wang J, Jiang HW and
Wang YQ: Ηypoxia-inducible factor 1-α promotes colon cell
proliferation and migration by upregulating AMPK-related protein
kinase 5 under hypoxic conditions. Oncol Lett. 15:3639–3645.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang JS, Jing CQ, Shan KS, Chen YZ, Guo
XB, Cao ZX, Mu LJ, Peng LP, Zhou ML and Li LP: Semaphorin 4D and
hypoxia-inducible factor-1alpha overexpression is related to
prognosis in colorectal carcinoma. World J Gastroenterol.
21:2191–2198. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sirri E, Castro FA, Kieschke J, Jansen L,
Emrich K, Gondos A, Holleczek B, Katalinic A, Urbschat I, Vohmann C
and Brenner H: Recent trends in survival of patients with
pancreatic cancer in Germany and the United States. Pancreas.
45:908–914. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rajamani D and Bhasin MK: Identification
of key regulators of pancreatic cancer progression through
multidimensional systems-level analysis. Genome Med.
8(38)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Berlin JD, Catalano P, Thomas JP, Kugler
JW, Haller DG and Benson AB III: Phase III study of gemcitabine in
combination with fluorouracil versus gemcitabine alone in patients
with advanced pancreatic carcinoma: Eastern Cooperative Oncology
Group Trial E2297. J Clin Oncol. 20:3270–3275. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Oettle H, Richards D, Ramanathan RK, van
Laethem JL, Peeters M, Fuchs M, Zimmermann A, John W, Von Hoff D,
Arning M and Kindler HL: A phase III trial of pemetrexed plus
gemcitabine versus gemcitabine in patients with unresectable or
metastatic pancreatic cancer. Ann Oncol. 16:1639–1645.
2005.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wang Y, Kuramitsu Y, Tokuda K, Baron B,
Kitagawa T, Akada J, Maehara S, Maehara Y and Nakamura K:
Gemcitabine induces poly (ADP-ribose) polymerase-1 (PARP-1)
degradation through autophagy in pancreatic cancer. PLoS One.
9(e109076)2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gilkes DM, Semenza GL and Wirtz D: Hypoxia
and the extracellular matrix: Drivers of tumour metastasis. Nat Rev
Cancer. 14:430–439. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wu J, Yang B, Zhang Y, Feng X, He B, Xie
H, Zhou L, Wu J and Zheng S: miR-424-5p represses the metastasis
and invasion of intrahepatic cholangiocarcinoma by targeting ARK5.
Int J Biol Sci. 15:1591–1599. 2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang X, Song Z, Chen F, Yang X, Wu B, Xie
S, Zheng X, Cai Y, Chen W and Zhong Z: AMPK-related kinase 5 (ARK5)
enhances gemcitabine resistance in pancreatic carcinoma by inducing
epithelial-mesenchymal transition. Am J Transl Res. 10:4095–4106.
2018.PubMed/NCBI
|
|
48
|
Jemal A, Murray T, Samuels A, Ghafoor A,
Ward E and Thun MJ: Cancer statistics, 2003. CA Cancer J Clin.
53:5–26. 2003.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Schiller JH, Harrington D, Belani CP,
Langer C, Sandler A, Krook J, Zhu J and Johnson DH: Eastern
Cooperative Oncology Group. Comparison of four chemotherapy
regimens for advanced non-small-cell lung cancer. N Engl J Med.
346:92–98. 2002.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Li Y, Qi K, Zu L, Wang M, Wang Y and Zhou
Q: Anti-apoptotic brain and reproductive organ-expressed proteins
enhance cisplatin resistance in lung cancer cells via the protein
kinase B signaling pathway. Thorac Cancer. 7:190–198.
2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhang HY, Li JH, Li G and Wang SR:
Activation of ARK5/miR-1181/HOXA10 axis promotes
epithelial-mesenchymal transition in ovarian cancer. Oncol Rep.
34:1193–1202. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yu HG, Wei W, Xia LH, Han WL, Zhao P, Wu
SJ, Li WD and Chen W: FBW7 upregulation enhances cisplatin
cytotoxicity in non-small cell lung cancer cells. Asian Pac J
Cancer Prev. 14:6321–6326. 2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chang XZ, Yu J, Liu HY, Dong RH and Cao
XC: ARK5 is associated with the invasive and metastatic potential
of human breast cancer cells. J Cancer Res Clin Oncol. 138:247–254.
2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Cui J, Yu Y, Lu GF, Liu C, Liu X, Xu YX
and Zheng PY: Overexpression of ARK5 is associated with poor
prognosis in hepatocellular carcinoma. Tumour Biol. 34:1913–1918.
2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sun X, Gao L, Chien HY, Li WC and Zhao J:
The regulation and function of the NUAK family. J Mol Endocrinol.
51:R15–R22. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Suzuki A, Ogura T and Esumi H: NDR2 acts
as the upstream kinase of ARK5 during insulin-like growth factor-1
signaling. J Biol Chem. 281:13915–13921. 2006.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Lizcano JM, Göransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG and
Alessi DR: LKB1 is a master kinase that activates 13 kinases of the
AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833–843.
2004.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Davis LE, Jeng S, Svalina MN, Huang E,
Pittsenbarger J, Cantor EL, Berlow N, Seguin B, Mansoor A, McWeeney
SK and Keller C: Integration of genomic, transcriptomic and
functional profiles of aggressive osteosarcomas across multiple
species. Oncotarget. 8:76241–76256. 2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Ryu S and Tjian R: Purification of
transcription cofactor complex CRSP. Proc Natl Acad Sci USA.
96:7137–7142. 1999.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Itoh N, Semba S, Ito M, Takeda H, Kawata S
and Yamakawa M: Phosphorylation of Akt/PKB is required for
suppression of cancer cell apoptosis and tumor progression in human
colorectal carcinoma. Cancer. 94:3127–3134. 2002.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signalling pathway in human malignancy. Cell Signal.
14:381–395. 2002.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ruggeri BA, Huang L, Wood M, Cheng JQ and
Testa JR: Amplification and overexpression of the AKT2 oncogene in
a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog.
21:81–86. 1998.PubMed/NCBI
|
|
64
|
Higuchi M, Masuyama N, Fukui Y, Suzuki A
and Gotoh Y: Akt mediates Rac/Cdc42-regulated cell motility in
growth factor-stimulated cells and in invasive PTEN knockout cells.
Curr Biol. 11:1958–1962. 2001.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Lawlor MA and Alessi DR: PKB/Akt: A key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
66
|
Taatjes DJ, Naar AM, Andel F III, Nogales
E and Tjian R: Structure, function, and activator-induced
conformations of the CRSP coactivator. Science. 295:1058–1062.
2002.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Kusakai G, Suzuki A, Ogura T, Kaminishi M
and Esumi H: Strong association of ARK5 with tumor invasion and
metastasis. J Exp Clin Cancer Res. 23:263–268. 2004.PubMed/NCBI
|
|
68
|
Cox AD and Der CJ: The dark side of Ras:
Regulation of apoptosis. Oncogene. 22:8999–9006. 2003.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Suzuki A, Lu J, Kusakai G, Kishimoto A,
Ogura T and Esumi H: ARK5 is a tumor invasion-associated factor
downstream of Akt signaling. Mol Cell Biol. 24:3526–3535.
2004.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Xie M, Wu X, Zhang J, Zhang J and Li X:
Ski regulates Smads and TAZ signaling to suppress lung cancer
progression. Mol Carcinog. 56:2178–2189. 2017.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Sato H, Takino T, Okada Y, Cao J,
Shinagawa A, Yamamoto E and Seiki M: A matrix metalloproteinase
expressed on the surface of invasive tumour cells. Nature.
370:61–65. 1994.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Kaufhold S and Bonavida B: Central role of
Snail1 in the regulation of EMT and resistance in cancer: A target
for therapeutic intervention. J Exp Clin Cancer Res.
33(62)2014.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Li M, Zheng C, Xu H, He W, Ruan Y, Ma J,
Zheng J, Ye C and Li W: Inhibition of AMPK-related kinase 5 (ARK5)
enhances cisplatin cytotoxicity in non-small cell lung cancer cells
through regulation of epithelial-mesenchymal transition. Am J
Transl Res. 9:1708–1719. 2017.PubMed/NCBI
|
|
76
|
Liu Y, Du F, Zhao Q, Jin J, Ma X and Li H:
Acquisition of 5-fluorouracil resistance induces
epithelial-mesenchymal transitions through the Hedgehog signaling
pathway in HCT-8 colon cancer cells. Oncol Lett. 9:2675–2679.
2015.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Mallini P, Lennard T, Kirby J and Meeson
A: Epithelial-to-mesenchymal transition: What is the impact on
breast cancer stem cells and drug resistance. Cancer Treat Rev.
40:341–348. 2014.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Xu T, Zhang J, Chen W, Pan S, Zhi X, Wen
L, Zhou Y, Chen BW, Qiu J, Zhang Y, et al: ARK5 promotes
doxorubicin resistance in hepatocellular carcinoma via
epithelial-mesenchymal transition. Cancer Lett. 377:140–148.
2016.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Chen D, Liu G, Xu N, You X, Zhou H, Zhao X
and Liu Q: Knockdown of ARK5 expression suppresses invasion and
metastasis of gastric cancer. Cell Physiol Biochem. 42:1025–1036.
2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Liotta LA: Tumor invasion and
metastases-role of the extracellular matrix: Rhoads memorial award
lecture. Cancer Res. 46:1–7. 1986.PubMed/NCBI
|
|
81
|
Chen XF, Zhang HJ, Wang HB, Zhu J, Zhou
WY, Zhang H, Zhao MC, Su JM, Gao W, Zhang L, et al: Transforming
growth factor-β1 induces epithelial-to-mesenchymal transition in
human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling
pathways. Molr Biol Reps. 39:3549–3556. 2012.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Park NR, Cha JH, Jang JW, Bae SH, Jang B,
Kim JH, Hur W, Choi JY and Yoon SK: Synergistic effects of CD44 and
TGF-β1 through AKT/GSK-3β/β-catenin signaling during
epithelial-mesenchymal transition in liver cancer cells. Biochem
Biophys Res Commun. 477:568–574. 2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Shiraki K, Tsuji N, Shioda T, Isselbacher
KJ and Takahashi H: Expression of Fas ligand in liver metastases of
human colonic adenocarcinomas. Proc Natl Acad Sci USA.
94:6420–6425. 1997.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Yoong KF, Afford SC, Randhawa S, Hubscher
SG and Adams DH: Fas/Fas ligand interaction in human colorectal
hepatic metastases: A mechanism of hepatocyte destruction to
facilitate local tumor invasion. Am J Pathol. 154:693–703.
1999.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Sánchez-Tilló E, Fanlo L, Siles L,
Montes-Moreno S, Moros A, Chiva-Blanch G, Estruch R, Martinez A,
Colomer D, Győrffy B, et al: The EMT activator ZEB1 promotes tumor
growth and determines differential response to chemotherapy in
mantle cell lymphoma. Cell Death Differ. 21:247–257.
2014.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Suzuki A, Kusakai G, Kishimoto A, Shimojo
Y, Miyamoto S, Ogura T, Ochiai A and Esumi H: Regulation of
caspase-6 and FLIP by the AMPK family member ARK5. Oncogene.
23:7067–7075. 2004.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Restifo NP: Not so Fas: Re-evaluating the
mechanisms of immune privilege and tumor escape. Nat Med.
6:493–495. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
88
|
Suzuki A, Kusakai G, Kishimoto A,
Minegichi Y, Ogura T and Esumi H: Induction of cell-cell detachment
during glucose starvation through F-actin conversion by SNARK, the
fourth member of the AMP-activated protein kinase catalytic subunit
family. Biochem Biophys Res Commun. 311:156–161. 2003.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Conacci-Sorrell M, McFerrin L and Eisenman
RN: An overview of MYC and its interactome. Cold Spring Harb
Perspect Med. 4(a014357)2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Evan GI, Christophorou M, Lawlor EA,
Ringshausen I, Prescott J, Dansen T, Dansen T, Finch A, Martins C
and Murphy D: Oncogene-dependent tumor suppression: Using the dark
side of the force for cancer therapy. Cold Spring Harb Symp Quant
Biol. 70:263–273. 2005.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Murphy DJ, Junttila MR, Pouyet L, Karnezis
A, Shchors K, Bui DA, Brown-Swigart L, Johnson L and Evan GI:
Distinct thresholds govern Myc's biological output in vivo. Cancer
Cell. 14:447–457. 2008.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Ross FA, MacKintosh C and Hardie DG:
AMP-activated protein kinase: A cellular energy sensor that comes
in 12 flavours. Febs J. 283:2987–3001. 2016.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Woods A, Dickerson K, Heath R, Hong SP,
Momcilovic M, Johnstone SR, Carlson M and Carling D:
Ca2+/calmodulin-dependent protein kinase kinase-beta
acts upstream of AMP-activated protein kinase in mammalian cells.
Cell Metab. 2:21–33. 2005.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Ciccarese F, Zulato E and Indraccolo S:
LKB1/AMPK pathway and drug response in cancer: A therapeutic
perspective. Oxid Med Cell Longev. 2019(8730816)2019.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Kawakami Y, Nishimoto H, Kitaura J,
Maeda-Yamamoto M, Kato RM, Littman DR, Leitges M, Rawlings DJ and
Kawakami T: Protein kinase C betaII regulates Akt phosphorylation
on Ser-473 in a cell type- and stimulus-specific fashion. J Biol
Chem. 279:47720–4725. 2004.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Partovian C and Simons M: Regulation of
protein kinase B/Akt activity and Ser473 phosphorylation by protein
kinase Calpha in endothelial cells. Cell Signal. 16:951–957.
2004.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Delbridge AR and Strasser A: The BCL-2
protein family, BH3-mimetics and cancer therapy. Cell Death Differ.
22:1071–1080. 2015.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Muthalagu N, Junttila MR, Wiese KE, Wolf
E, Morton J, Bauer B, Evan GI, Eilers M and Murphy DJ: BIM is the
primary mediator of MYC-induced apoptosis in multiple solid
tissues. Cell Rep. 8:1347–1353. 2014.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Cermelli S, Jang IS, Bernard B and
Grandori C: Synthetic lethal screens as a means to understand and
treat MYC-driven cancers. Cold Spring Harb Perspect Med.
4(a014209)2014.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Carling D: AMPK signalling in health and
disease. Curr Opin Cell Biol. 45:31–37. 2017.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Jones RG, Plas DR, Kubek S, Buzzai M, Mu
J, Xu Y, Birnbaum MJ and Thompson CB: AMP-activated protein kinase
induces a p53-dependent metabolic checkpoint. Mol Cell. 18:283–293.
2005.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Hou X, Liu JE, Liu W, Liu CY, Liu ZY and
Sun ZY: A new role of NUAK1: Directly phosphorylating p53 and
regulating cell proliferation. Oncogene. 30:2933–2942.
2011.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Baker DJ, Jeganathan KB, Cameron JD,
Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC,
Roche P and van Deursen JM: BubR1 insufficiency causes early onset
of aging-associated phenotypes and infertility in mice. Nat Genet.
36:744–749. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
108
|
Chesnokova V, Zonis S, Kovacs K,
Ben-Shlomo A, Wawrowsky K, Bannykh S and Melmed S: p21(Cip1)
restrains pituitary tumor growth. Proc Natl Acad Sci USA.
105:17498–17503. 2004.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Takahashi A, Ohtani N, Yamakoshi K, Iida
S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H and Hara E:
Mitogenic signalling and the p16INK4a-Rb pathway cooperate to
enforce irreversible cellular senescence. Nat Cell Biol.
8:1291–1297. 2006.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Zhang D, Shimizu T, Araki N, Hirota T,
Yoshie M, Ogawa K, Nakagata N, Takeya M and Saya H: Aurora A
overexpression induces cellular senescence in mammary gland
hyperplastic tumors developed in p53-deficient mice. Oncogene.
27:4305–4314. 2008.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Holland B, Wong J, Li M and Rasheed S:
Identification of human microRNA-like sequences embedded within the
protein-encoding genes of the human immunodeficiency virus. PLoS
One. 8(e58586)2013.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Obayashi M, Yoshida M, Tsunematsu T, Ogawa
I, Sasahira T, Kuniyasu H, Imoto I, Abiko Y, Xu D, Fukunaga S, et
al: microRNA-203 suppresses invasion and epithelial-mesenchymal
transition induction via targeting NUAK1 in head and neck cancer.
Oncotarget. 7:8223–8239. 2016.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Shenouda SK and Alahari SK: MicroRNA
function in cancer: Oncogene or a tumor suppressor? Cancer
Metastasis Rev. 28:369–378. 2009.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Yu Y, Wang Y, Xiao X, Chang W, Hu L, Yao
W, Qian Z and Wu W: MiR-204 inhibits hepatocellular cancer drug
resistance and metastasis through targeting NUAK1. Biochem Cell
Biol. 97:563–570. 2019.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Xiong X, Sun D, Chai H, Shan W, Yu Y, Pu L
and Cheng F: MiR-145 functions as a tumor suppressor targeting
NUAK1 in human intrahepatic cholangiocarcinoma. Biochem Biophys Res
Commun. 465:262–269. 2015.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Bell RE, Khaled M, Netanely D, Schubert S,
Golan T, Buxbaum A, Janas MM, Postolsky B, Goldberg MS, Shamir R
and Levy C: Transcription factor/microRNA axis blocks melanoma
invasion program by miR-211 targeting NUAK1. J Invest Dermatol.
134:441–451. 2014.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Huang X, Lv W, Zhang JH and Lu DL: miR96
functions as a tumor suppressor gene by targeting NUAK1 in
pancreatic cancer. Int J Mol Med. 34:1599–1605. 2014.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Monteverde T, Tait-Mulder J, Hedley A,
Knight JR, Sansom OJ and Murphy DJ: Calcium signalling links MYC to
NUAK1. Oncogene. 37:982–992. 2018.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ojo OO, Bhadauria S and Rath SK:
Dose-dependent adverse effects of salinomycin on male reproductive
organs and fertility in mice. PLoS One. 8(e69086)2013.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Zhou Y, Liang C, Xue F, Chen W, Zhi X,
Feng X, Bai X and Liang T: Salinomycin decreases doxorubicin
resistance in hepatocellular carcinoma cells by inhibiting the
β-catenin/TCF complex association via FOXO3a activation.
Oncotarget. 6:10350–10365. 2015.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Yu Z, Cheng H, Zhu H, Cao M, Lu C, Bao S,
Pan Y and Li Y: Salinomycin enhances doxorubicin sensitivity
through reversing the epithelial-mesenchymal transition of
cholangiocarcinoma cells by regulating ARK5. Braz J Med Biol Res.
50(e6147)2017.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Reddy MV, Akula B, Cosenza SC,
Athuluridivakar S, Mallireddigari MR, Pallela VR, Billa VK,
Subbaiah DR, Bharathi EV, Vasquez-Del Carpio R, et al: Discovery of
8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile
(7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and
AMPK-related kinase 5 (ARK5). J Med Chem. 57:578–599.
2014.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Huang X, Di Liberto M, Jayabalan D, Liang
J, Ely S, Bretz J, Shaffer AL III, Louie T, Chen I, Randolph S, et
al: Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition
sensitizes myeloma cells to cytotoxic killing through cell
cycle-coupled loss of IRF4. Blood. 120:1095–1106. 2012.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Niesvizky R, Badros AZ, Costa LJ, Ely SA,
Singhal SB, Stadtmauer EA, Haideri NA, Yacoub A, Hess G, Lentzsch
S, et al: Phase 1/2 study of cyclin-dependent kinase (CDK)4/6
inhibitor palbociclib (PD-0332991) with bortezomib and
dexamethasone in relapsed/refractory multiple myeloma. Leuk
Lymphoma. 56:3320–3328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Banerjee S, Buhrlage SJ, Huang HT, Deng X,
Zhou W, Wang J, Traynor R, Prescott AR, Alessi DR and Gray NS:
Characterization of WZ4003 and HTH-01-015 as selective inhibitors
of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem J.
457:215–225. 2014.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Banerjee S, Zagorska A, Deak M, Campbell
DG, Prescott AR and Alessi DR: Interplay between Polo kinase,
LKB1-activated NUAK1 kinase, PP1βMYPT1 phosphatase complex and the
SCFβTrCP E3 ubiquitin ligase. Biochem J. 461:233–245.
2014.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Reeves WB and Andreoli TE: Transforming
growth factor beta contributes to progressive diabetic nephropathy.
Proc Natl Acad Sci USA. 97:7667–7669. 2000.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Zhang X, Lv H, Zhou Q, Elkholi R, Chipuk
JE, Reddy MV, Reddy EP and Gallo JM: Preclinical pharmacological
evaluation of a novel multiple kinase inhibitor, ON123300, in brain
tumor models. Mol Cancer Ther. 13:1105–1116. 2014.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Brennan EP, Morine MJ, Walsh DW, Roxburgh
SA, Lindenmeyer MT, Brazil DP, Gaora PÓ, Roche HM, Sadlier DM,
Cohen CD, et al: Next-generation sequencing identifies
TGF-β1-associated gene expression profiles in renal epithelial
cells reiterated in human diabetic nephropathy. Biochim Biophys
Acta. 1822:589–599. 2012.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Russo LM, del Re E, Brown D and Lin HY:
Evidence for a role of transforming growth factor (TGF)-beta1 in
the induction of postglomerular albuminuria in diabetic
nephropathy: Amelioration by soluble TGF-beta type II receptor.
Diabetes. 56:380–388. 2007.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Sakamoto K, Goransson O, Hardie DG and
Alessi DR: Activity of LKB1 and AMPK-related kinases in skeletal
muscle: Effects of contraction, phenformin, and AICAR. Am J Physiol
Endocrinol Metab. 287:E310–E317. 2004.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Fisher JS, Ju JS, Oppelt PJ, Smith JL,
Suzuki A and Esumi H: Muscle contractions, AICAR, and insulin cause
phosphorylation of an AMPK-related kinase. Am J Physiol Endocrinol
Metab. 289:E986–E992. 2005.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Hoppe PE, Chau J, Flanagan KA, Reedy AR
and Schriefer LA: Caenorhabditis elegans unc-82 encodes a
serine/threonine kinase important for myosin filament organization
in muscle during growth. Genetics. 184:79–90. 2010.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Zagorska A, Deak M, Campbell DG, Banerjee
S, Hirano M, Aizawa S, Prescott AR and Alessi DR: New roles for the
LKB1-NUAK pathway in controlling myosin phosphatase complexes and
cell adhesion. Sci Signal. 3(ra25)2010.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Inazuka F, Sugiyama N, Tomita M, Abe T,
Shioi G and Esumi H: Muscle-specific knock-out of NUAK family
SNF1-like kinase 1 (NUAK1) prevents high fat diet-induced glucose
intolerance. J Biol Chem. 287:16379–16389. 2012.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Yap JKY, Pickard BS, Chan EWL and Gan SY:
The role of neuronal NLRP1 inflammasome in Alzheimer's disease:
Bringing neurons into the neuroinflammation game. Mol Neurobiol.
56:7741–7753. 2019.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Courchet V, Roberts AJ, Meyer-Dilhet G,
Del Carmine P, Lewis TL Jr, Polleux F and Courchet J:
Haploinsufficiency of autism spectrum disorder candidate gene NUAK1
impairs cortical development and behavior in mice. Nat Commun.
9(4289)2018.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Soto C and Pritzkow S: Protein misfolding,
aggregation, and conformational strains in neurodegenerative
diseases. Nat Neurosci. 21:1332–1340. 2018.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Cao Q, Wang XJ, Liu CW, Liu DF, Li LF, Gao
YQ and Su XD: Inhibitory mechanism of caspase-6 phosphorylation
revealed by crystal structures, molecular dynamics simulations, and
biochemical assays. J Biol Chem. 287:15371–15379. 2012.PubMed/NCBI View Article : Google Scholar
|
|
141
|
MacLachlan TK and El-Deiry WS: Apoptotic
threshold is lowered by p53 transactivation of caspase-6. Proc Natl
Acad Sci USA. 99:9492–9497. 2002.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Kurokawa M and Kornbluth S: Caspases and
kinases in a death grip. Cell. 138:838–854. 2009.PubMed/NCBI View Article : Google Scholar
|