Introduction
The typical clinical manifestation of psoriasis
include widely distributed scaly erythema or plaques that can
affect the skin, joints and fingernails (1). Skin, as the first barrier against
infection and injury, contains a large number of immune cells from
the epidermis and dermis, including keratinocytes (KCs), Langerhans
cells, dermal dendritic cells, macrophages and T lymphocyte
(2). Psoriasis is characterized by
the excessive proliferation and aberrant differentiation of KCs,
but is fully reversible with appropriate therapy (3). Previous studies have reported that
psoriasis can be complicated with diabetes, vascular disease,
arthritis, Crohn's disease, lymphoma and depression (4,5). The
World Health Organization reported in 2016 that the global
prevalence of psoriasis ranged from 0.09-11.43%, and it can occur
at any age (6). Psoriasis has
therefore emerged as a public health concern.
MicroRNAs (miRNAs/miRs) are a class of small,
endogenous, single-stranded, non-coding RNAs that serve a key role
in proliferation, apoptosis, differentiation, invasion and
metabolism (7). miRNAs bind to the
3' untranslated regions (UTR) of specific target mRNAs via
complementary base pairing, leading to mRNA degradation and the
inhibition of translation (8).
miRNAs can directly or indirectly interact with key genes of the
inflammatory pathway to regulate inflammatory responses, and serve
an important role in classic inflammatory pathways (9). In recent years, the specific roles of
miRNAs in psoriasis have become a hot topic of research. For
example, Chang et al (10)
revealed that miR-126 served an important role in promoting the
proliferation and migration of KCs during skin wound healing, but
the mechanism of action of miR-489-3p was not reported. The present
study performed bioinformatics predictions that identified the
ability of miR-489-3p to directly bind to the 3'UTR of TLR4 and
downregulate its expression. The activation of TLR4 triggers its
downstream effector NF-κB, which translocates to the nucleus
(11) and enhances the expression
of IL-1β, IL-6, TNF-α and other pro-inflammatory cytokines
(12,13). Therefore, preventing inflammatory
responses in psoriasis could alleviate disease progression.
KCs can sense injury-related molecular model
molecules and/or pathogenesis-related molecular model molecules and
activate inflammatory bodies, which induces the inflammatory
response in psoriasis (14). The
activation of NF-κB in psoriatic KCs, leading to the production of
a variety of immune-related proteins, and lead to further
inflammatory response (15). In the
present study, human KCs (HaCaT cell line) were used for studying
psoriasis in vitro, and to evaluate the molecular mechanism
of miR-489-3p in inhibiting the inflammatory response of
psoriasis.
Materials and methods
Research subjects and sample
collection
The specimens were collected from 15 patients with
psoriasis between March 2018 and March 2019 in The Third People's
Hospital of Hangzhou, including 8 men and 7 women, with an average
age of 33.5 years (age range, 24-68 years). The course of disease
ranged from 14 months to 12 years (mean, 6.5 years). All patients
with psoriasis were diagnosed clinically and pathologically, and
all plaques were of progressive plaque type. The Psoriasis Area and
Severity Index (PASI) score ranged from 10-20(16). The inclusion criteria were as
follows: Patients with psoriasis had typical erythematous; scaling
plaques confirmed by histopathology; systemic therapy including
immunosuppressants, biological agents, glucocorticoids and retinoic
acids were not used within the first 3 months, and external drug
therapy and phototherapy ceased for 1 month prior to the study; no
accompanying autoimmune disease, neoplastic disease or inflammatory
disease. The exclusion criteria were as follows: Patients with
chronic plaque psoriasis in the involuting and stable stage;
pregnancy, lactation and menstruation in female patients. The
healthy controls showed no history of psoriasis or other skin
defects, and no autoimmune or systemic disease, as well as were
comparable to the sex and age of the patients.
All participants provided written informed consent
for their participation. The study protocol was designed and
implemented in accordance with the principles of the Helsinki
Declaration and was approved by The Third People's Hospital of
Hangzhou Ethics Review Committee.
Cell culture
Human KCs (HaCaT cell line) were purchased from
Guangzhou Jennio Biological Technology (Guangzhou, China). Cells
were cultured in DMEM (Gibco; Thermo Fisher Scientific Inc.)
containing 1% non-essential amino acids and 10% FBS (Thermo Fisher
Scientific, Inc.) at 37˚C with 5% CO2. Medium was
replaced every 2 days and cells were passaged when 70-80%
confluent.
Cell transfections
Cells were digested with trypsin and then seeded in
6-well cell culture plates at 5x105/well. When the cells
reached 60% confluence, they were transfected with 100 nM
miR-489-3p mimic, 100 nM miR-489-3p mimic negative control using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's procedure for 24
h at 37˚C with 5% CO2. miR-489-3p mimic, miR-489-3p
mimic negative control (mimic-con), miR-489-3p inhibitor and
miR-489-3p inhibitor negative control (inhibitor-con) were
purchased from Shanghai GenePharma Co., Ltd. (miR-489-3p
mimics-con, 5'-UUCUCCGAACGUGUCACGUTT-3'; miR-489-3p mimic
5'-GUGACAUCACAUAUACGGCAGC-3'; miR-489-3p inhibitor-con,
5'-CAGUACUUUUGUGUAGUACAA-3'; and miR-489-3p inhibitor
5'-GCUGCCGUAUAUGUGAUGUCAC-3'). Transfection efficiency was verified
24 h later via reverse transcription-quantitative (RT-q)PCR.
TLR4 silencing
Cells (1x106) were transfected with 1.0
µg TLR4 small interfering (si)
RNA5'-CCGGCCGCTGGTGTATCTTTGAATACTCGAGTATTCAAAGATACA
CCAGCGGTTTTTG-3') or scrambled siRNA (si-con,
5'-CCGGCTCCGGGTGTATCGTTTAATACTCGAGTCTAT AGAAATACACCAGGGCTTTTTG-3')
as a negative control (cat. no. sc-40260; Santa Cruz Biotechnology,
Inc.) using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's procedure
for 24 h at 37˚C with 5% CO2.
Post-transfection, cells were cultured in complete
media for 48 h. TLR4-siRNAs were purchased from Shanghai GenePharma
Co., Ltd. Transfection efficiency was verified 48 h later via
RT-qPCR.
miRNA target prediction and dual
luciferase reporter assays
TargetScan v.7.2 (http://www.targetscan.org/mamm_31/) and miRDB
(http://mirdb.org/) miRNA target prediction database
were used to identify the target genes of has-miR-489-3p. TLR4
3'UTR wild-type (WT) and TLR4 3'UTR mutant (MUT) plasmids were
cloned into pGL3 luciferase reporter vectors (Promega Corporation).
HaCaT cells were seeded into 6-well plates at a density of
5xl05/well. Cells were co-transfected with TLR4
luciferase reporter constructs (1 µg), miR-489-3p mimics (100 nM)
or miR-489-3p mimic-con using Lipofectamine 2000 at 37˚C with 5%
CO2 for 24 h. Luciferase activity was assessed using a
Dual-Luciferase Reporter Assay system (Promega Corporation) after
24 h post-transfection. Relative luciferase activities were
calculated by firefly luciferase activities/Renilla
luciferase activities.
Cell proliferation assays
Cell proliferation was determined using Cell
Counting Kit-8 (CCK-8) assays (MedChemExpress) according to the
manufacturer's protocol Cells were seeded at the density of
4x104 cells/well into 96-well plates and incubated for
0-3 days, and then CCK-8 (10 µl) were added at different time
points (0, 24, 48 and 72 h) at 37˚C and further cultured for 2 h.
The optical densities were measured at 450 nm using an automatic
microplate reader (INFINITE M200; Tecan Group, Ltd.).
Colony formation assays
Cells were seeded in 12-well plates (~300
cells/well) 24 h post-transfection. Cells were cultured at 37˚C
with 5% CO2 for 14 days, and the medium was replaced
every 3 days. Cells were stained with crystal violet (2%) at 37˚C
for 30 min and observed and photographed under a light microscope
(Olympus Soft Imaging Solutions GmbH) and colonies ≥50 cells were
counted with Image J software v.1.52 (National Institutes of
Health). Experiments were performed on a minimum of three
independent occasions.
Cell cycle assays
Cell cycle analysis was performed via flow
cytometry. Cells (2x106 cells/ml) in 6-well plates were
trypsinized and fixed in pre-chilled 75% ethanol at 4˚C overnight.
Cells were harvested by centrifugation at 800 x g at 4˚C for 10
min. The supernatants were discarded and then resuspended in PBS
with 50 µg/ml propidium iodide (PI), 100 µg/ml RNase A and 0.2%
Triton X-100 and incubated in the dark for 30 min at room
temperature. PI, RNase A and Triton X-100 were all purchased from
Sigma-Aldrich; Merck KGaA. Cells were captured on a flow cytometer
(FACSCalibur; BD Biosciences), and cell cycle data was analyzed
using CellQuest software v.2.0 (BD Biosciences) and ModFit LT v.2.0
(Verity Software House, Inc.).
RT-qPCR
Total RNA was extracted using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), and RNA
concentrations and purity were measured via UV spectrophotometry
(Nanodrop Technologies). A total of 1 µg total RNA was reverse
transcribed using PrimeScript RT reagent kit (Takara Bio, Inc.).
The conditions for RT were as follows: 37˚C for 10 min, followed by
85˚C for 5 sec and followed by holding at 4˚C. SYBR Premix Ex Taq
II (Tli RNaseH Plus) (Takara Bio, Inc.) was used to detect
miR-489-3p and TLR mRNA expression levels using the ABI PRISM 7000
instrument (Applied Biosystems; Thermo Fisher Scientific Inc.). The
housekeeping gene GAPDH was used as the control of TLR4 and the
small nuclear RNA U6 was used as the control of miR-489-3p. The
thermocycling conditions used were as follows: Denaturation at 95˚C
for 30 sec, followed by 40 cycles at 95˚C for 5 sec and 60˚C for 30
sec. The following primers were used: miR-489-3p, forward,
5'-GCGCGGTGACATCACATATAC-3' and reverse,
5'-AGTGCAGGGTCCGAGGTATT-3'; U6 forward,
5'-GCTTCGGCAGCACATATACTAAAAT-3'and reverse,
5'-CGCTTCACGAATTTGCGTGTCAT-3'; TLR4 forward,
5'-TTTGGACAGTTTCCCACATTGA-3' and reverse,
5'-AAGCATTCCCACCTTTGTTGG-3'; and GAPDH forward,
5'-ACAACTTTGGTATCGTGGAAGG-3' and reverse,
5'-GCCATCACGCCACAGTTTC-3'. Melting curves were used to monitor
non-specific amplifications. Relative Ct values were quantitated
using the 2-∆ΔCq method (17) were ΔΔCq=(Cq target gene-Cq control
gene) experimental group-(Cq target gene-Cq control gene) control
group.
Western blot analysis
Cell proteins were extracted in RIPA buffer
(Sigma-Aldrich; Merck KGaA). Nuclear proteins were extracted using
CelLytic™ NuCLEAR™ reagent (Sigma-Aldrich; Merck KGaA). Proteins
were quantified using a BCA assay, and equal concentrations (50
µg/lane) were resolved on 12.5% polyacrylamide gels at 130 V.
Proteins were transferred to PVDF membranes and blocked in 2.5%
skimmed milk at 37˚C for 1 h. Membranes were probed with primary
antibodies overnight at 4˚C, including anti-rabbiti-TLR4 (1:1,000;
cat no. 13867; Abcam), anti-rabbit-NF-κB p65 (1:1,000; cat no.
3034; Cell Signaling Technologies, Inc.), phospho-NF-κB p65
(1:1,000; cat no. 3033; Cell Signaling Technologies, Inc.),
anti-rabbit-Lamin B (1:2,000; cat no. 194109; Abcam) and
anti-rabbit-β-actin (1:2,000; cat no. 115777; Abcam). Membranes
were washed in TBS containing 0.1% Tween 20 and labeled with Goat
Anti-Rabbit IgG H&L (HRP) antibodies (1:3,000; cat no. 7090;
Abcam) for 1 h at room temperature. Membranes were then washed and
proteins were visualized using the ECL detection system (Amersham;
Cytiva) on a ChemiDoc Bio-Rad system (Bio-Rad Laboratories, Inc.).
Band intensities were semi-quantified using ImageJ software v.1.52v
(National Institutes of Health).
ELISA
HaCaT cells (1x106) were seeded into
6-well plates to 80-85% confluence. The cell culture supernatant
were collected by centrifugation at 2,000 x g for 10 min at 4˚C in
order to remove debris. ELISA assays were performed according to
the manufacturer's instructions to detect the secretion of TNF-α
(cat no. ab181421; Abcam), IFN-γ (cat. no. ab46025; Abcam), IL-22
(cat. no. ab216170; Abcam), IL-4 (cat no. ab215089; Abcam) and
IL-1β (cat no. ab214025; Abcam). Gradient dilution of the standard
was used to draw a standard curve. The OD value was evaluated at
450 nm using an automatic microplate reader (INFINITE M200; Tecan
Group, Ltd.). Three replicates were performed.
Statistical analysis
Data were analyzed using SPSS 20.0 (IBM Corp.).
Unpaired t-tests were used to compare between two groups, and a
one-way ANOVA was used to compare multiple groups, followed by
Dunnett's post hoc analysis. Pearson's correlation coefficient
analysis was used to determine the correlation between two
variables. All experiments were repeated three times. P<0.05 was
considered to indicate a statistically significant difference. Data
are presented as the mean ± SD.
Results
miR-489-3p targets the TLR4/NF-κB axis
in psoriasis
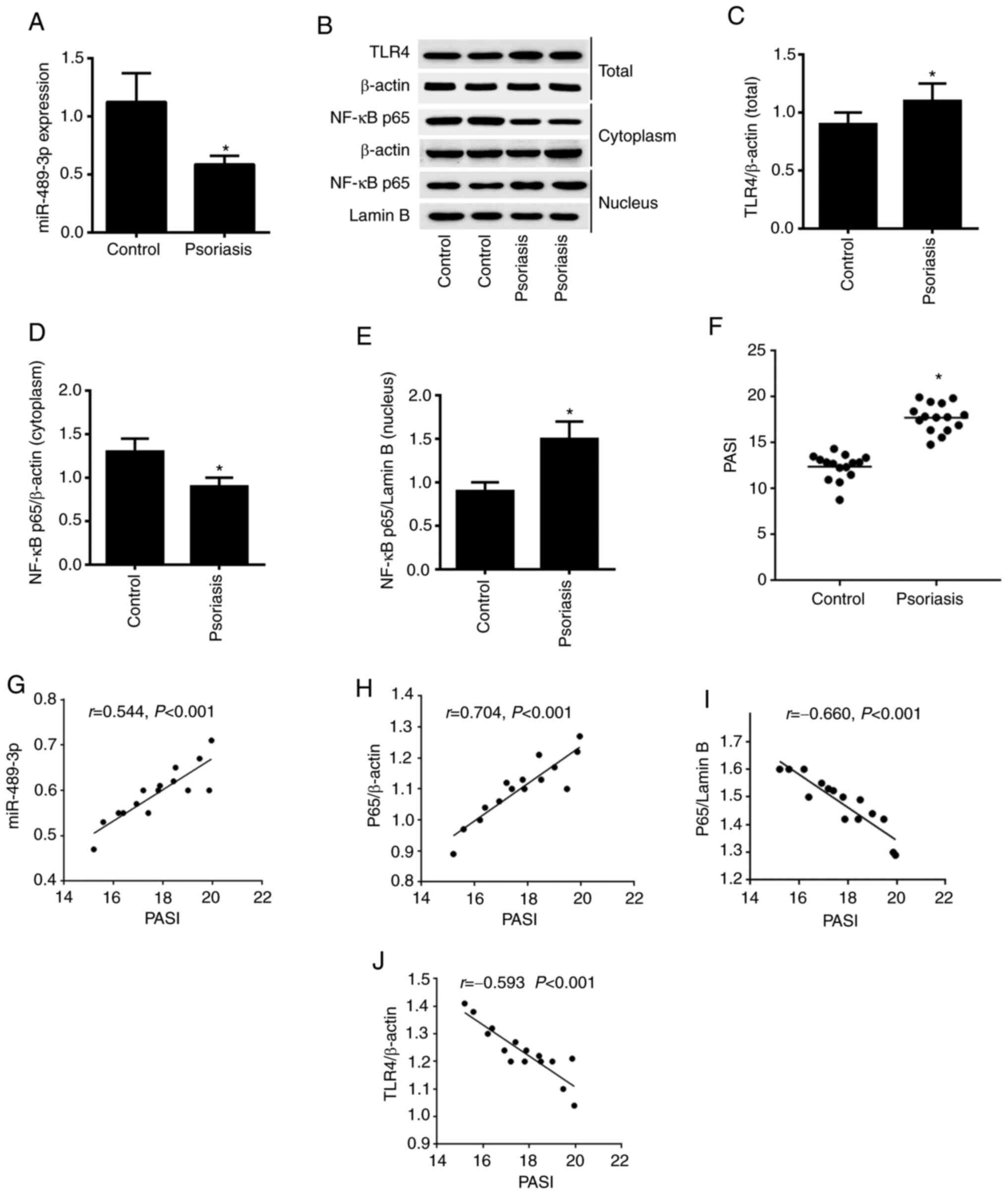
The present study first assessed the expression
level of miR-489-3p via RT-qPCR analysis. Downregulation of
miR-489-3p expression was observed in psoriasis vs. healthy tissue
(Fig. 1A). In addition,
upregulation of TLR4 and nuclear NF-κB expression levels,
downregulation of cytoplasmic NF-κB expression levels were detected
using western blot analysis (Fig.
1B-E). A comparison of the PASI between the patient and health
control samples revealed that the PASI was significantly higher in
patients with psoriasis (P<0.05; Fig. 1F). The correlation between PASI and
miR-489-3p, NF-κB p65 and TLR4 expression levels was determined
using Pearson's correlation coefficient analysis. The results
demonstrated that PASI was positively correlated with miR-489-3p
expression and cytoplasmic NF-κB p65 expression. Moreover, PASI was
negatively correlated with NF-κB p65/Lamin B and TLR4 expression
(P<0.001; Fig. 1G-J). These
results suggested that miR-489-3p expression may promote enhanced
inflammation in psoriasis by suppressing NF-κB/TLR4 expression.
miR-489-3p inhibits TLR4/NF-κB
signaling
To confirm the regulation of TLR4 by miR-489-3p,
TargetScan and miRDBA tools were used to predict potential target
genes. From this analysis, it was found that miR-489-3p could bind
to the 3'UTR of TLR4 (Fig. 2A).
Dual luciferase assays in HaCaT cells transfected with TLR4-WT
luciferase reporter identified a significant loss of TLR4 activity
in cells co-expressing miR-489-3p mimic (P<0.05). However, no
such effect was observed for the TLR4-MUT reporter (P>0.05) in
miR-489-3p overexpressing cells (Fig.
2B). RT-qPCR analysis was conducted to verify the transfection
efficiency, and the results demonstrated that the expression levels
of miR-489-3p were significantly upregulated in HaCaT cells
transfected with miR-489-3p mimic, compared with cells transfected
with miR-489-3p mimic-con. Furthermore, the expression levels of
miR-489-3p were significantly downregulated in HaCaT cells
transfected with miR-489-3p inhibitor, compared with cells
transfected with miR-489-3p inhibitor-con (P<0.05; Fig. 2C). Western blot analysis confirmed
that the overexpression of miR-489-3p inhibited TLR4 and nuclear
NF-κB p65 expression and upregulated cytoplasmic NF-κB p65; when
miR-489-3p expression was inhibited, the TLR4 and nuclear NF-κB p65
expression were upregulated and the cytoplasmic NF-κB p65 was
downregulated (P<0.05; Fig.
2D-G). The results suggested that miR-489-3p upregulation
inhibits the inflammatory response of skin cells via TLR4/NF-κB
signaling.
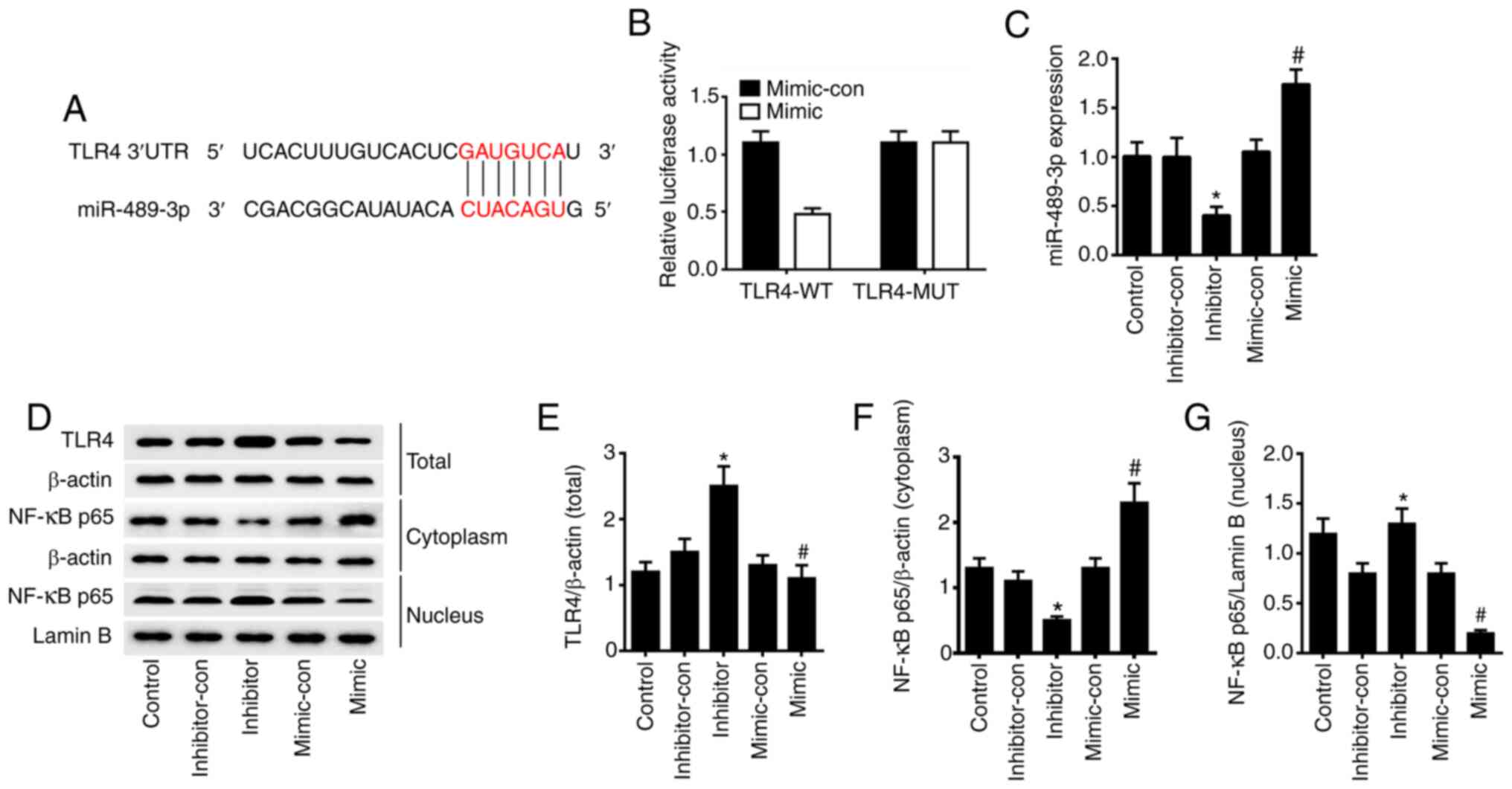 | Figure 2miR-489-3p inhibits TLR4/NF-κB
signaling. (A) TLR4 was the target gene of miR-489-3p, as predicted
via bioinformatics analysis. (B) HaCaT cells were transfected with
luciferase reporter plasmids containing WT or MUT TLR4 3'UTR,
together with miR-489-3p mimic or miR-489-3p mimic-con. (C)
miR-489-3p expression in HaCaT cells transfected with different
plasmids, as detected via reverse transcription-quantitative PCR.
(D) Western blot analysis of the protein expression levels of (E)
TLR4, (F) cytoplasmic NF-κB p65 and (G) nuclear NF-κB p65 in HaCaT
cells transfected with different plasmids. *P<0.05
vs. inhibitor-con; #P<0.05 vs. mimic-con. WT,
wild-type; MUT, mutant; UTR, untranslated region; con, negative
control; miR, microRNA; TLR4, Toll-like receptor 4. |
miR-489-3p inhibits KC
hyperproliferation and inflammation
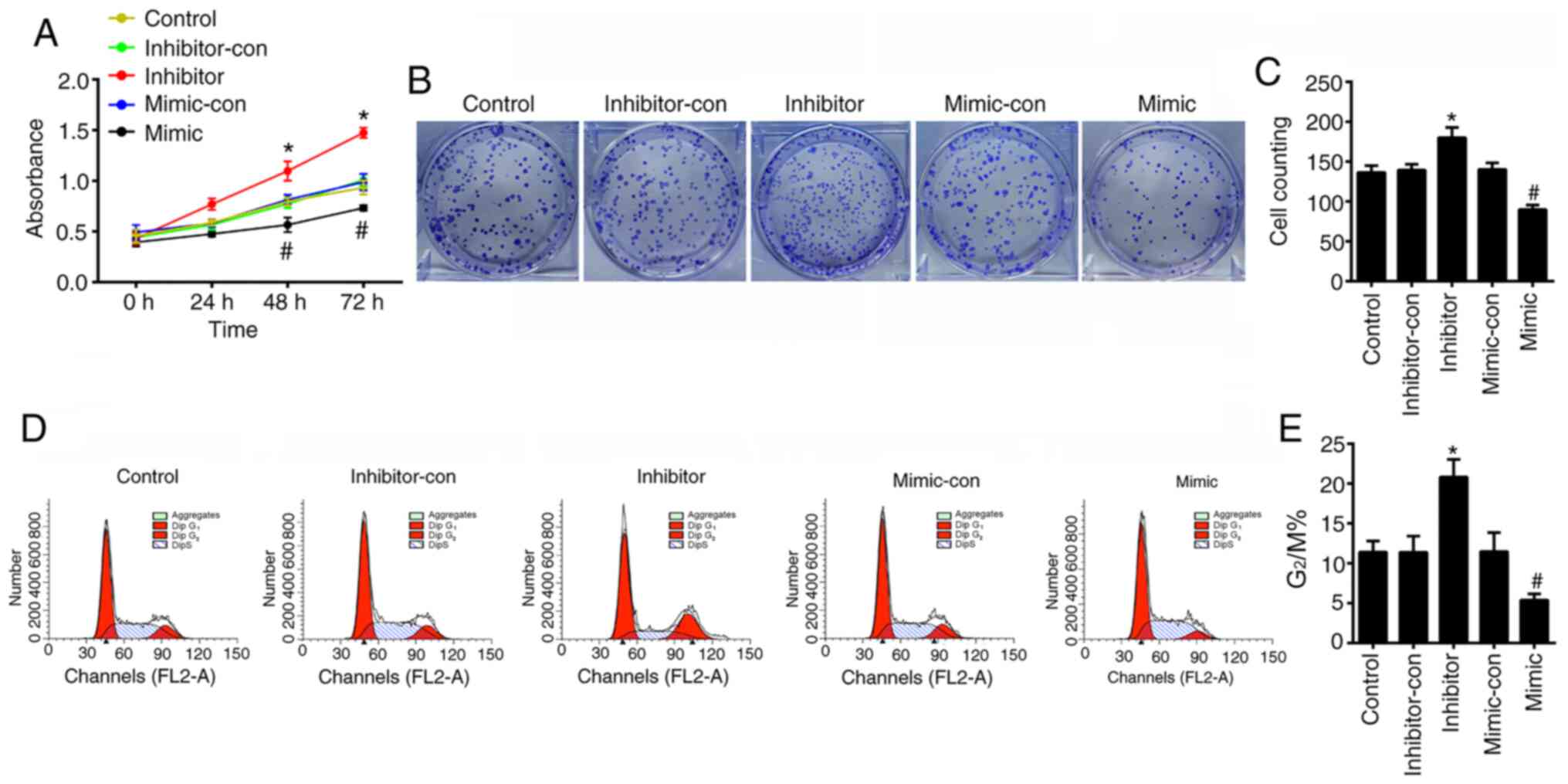
To verify the effects of miR-489-3p on the
proliferation of KCs in vitro, the viability of HaCaT cells
was assessed using CCK-8, cell cycle and colony formation assays to
determine cell proliferation. The CCK-8 assay revealed that the
viability of HaCaT cells transfected with miR-489-3p mimic was
significantly decreased (P<0.05), whilst cells transfected with
miR-489-3p inhibitors showed higher levels of viability in a
time-dependent manner (P<0.05; Fig.
3A). Colony formation was impaired in HaCaT cells transfected
with miR-489-3p mimic (P<0.05), whilst the colony forming
ability of HaCaT cells transfected with miR-489-3p inhibitor was
increased (P<0.05; Fig. 3B and
C). Flow cytometry assays
identified that miR-489-3p overexpression inhibited G2/M
cell cycle progression (P<0.05; Fig.
3D and E), which was enhanced
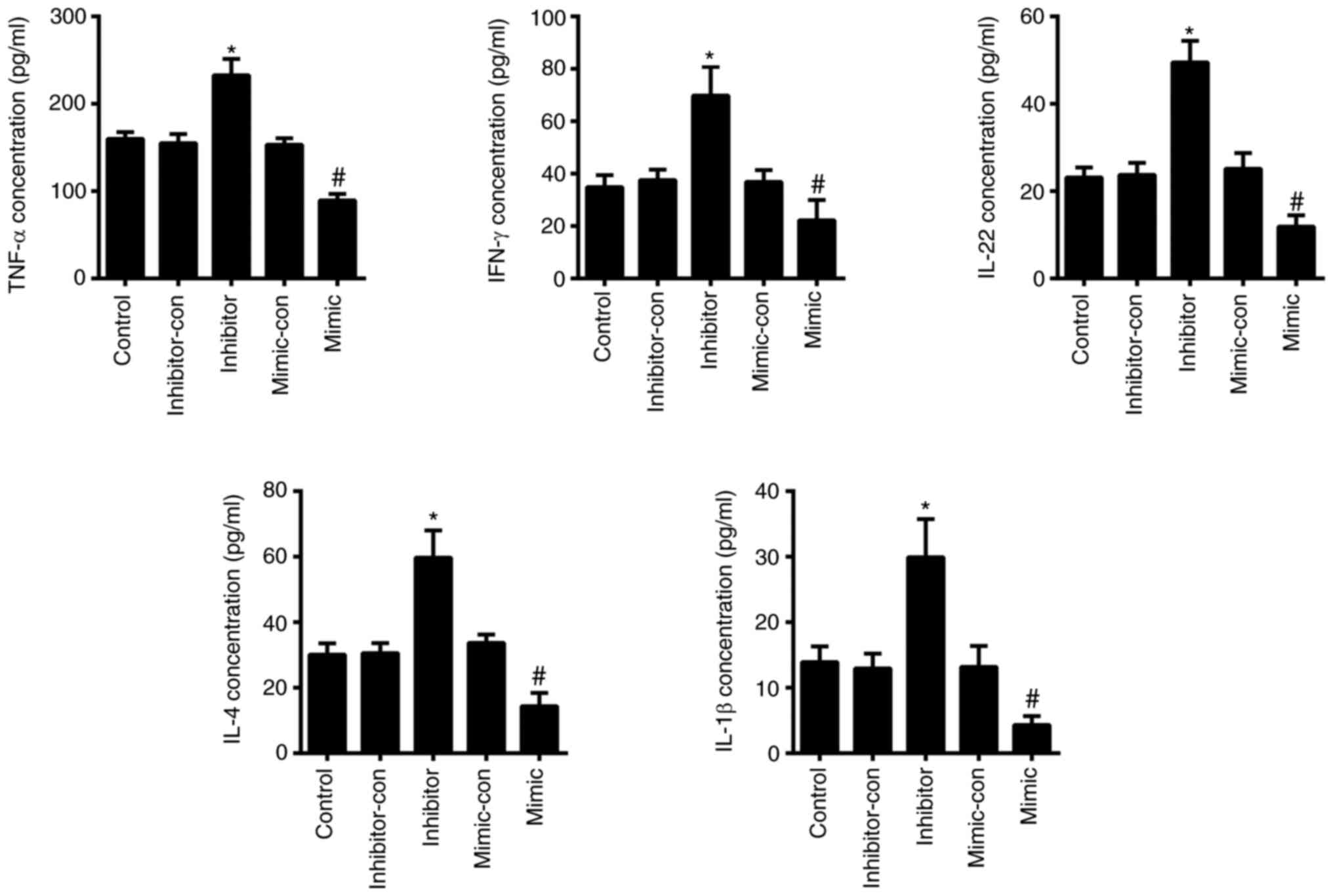
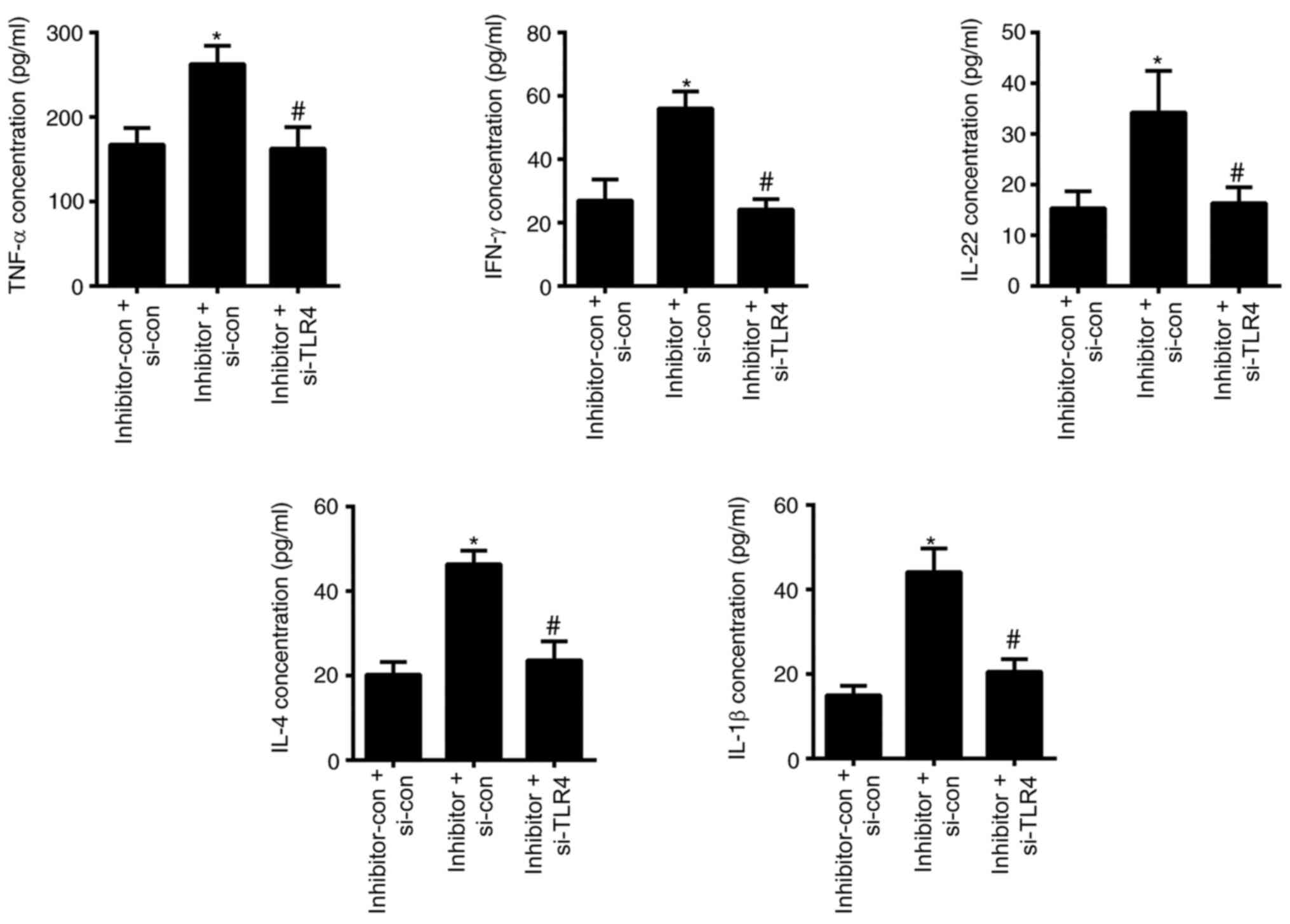
by the transfection of miR-489-3p inhibitor (P<0.05; Fig. 3D and E). The ELISA results demonstrated that the
levels of TNF-α, IFN-γ, IL-22, IL-4 and IL-1β were significantly
increased in cells transfected with miR-489-3p inhibitor
(P<0.05; Fig. 4), but were
declined in cells transfected with miR-489-3p mimic (P<0.05;
Fig. 4). These data indicated that
miR-489-3p inhibited the proliferation of KCs and the secretion of
inflammatory cytokines.
miR-489-3p targets TLR4 to inhibit KC
hyperproliferation and inflammation
To confirm that miR-489-3p inhibits the TLR-mediated
inflammatory response, HaCaT cells were co-transfected with
TLR4-siRNA and miR-489-3p inhibitor plasmids, and cell
proliferation and inflammatory cytokine secretion were assessed. A
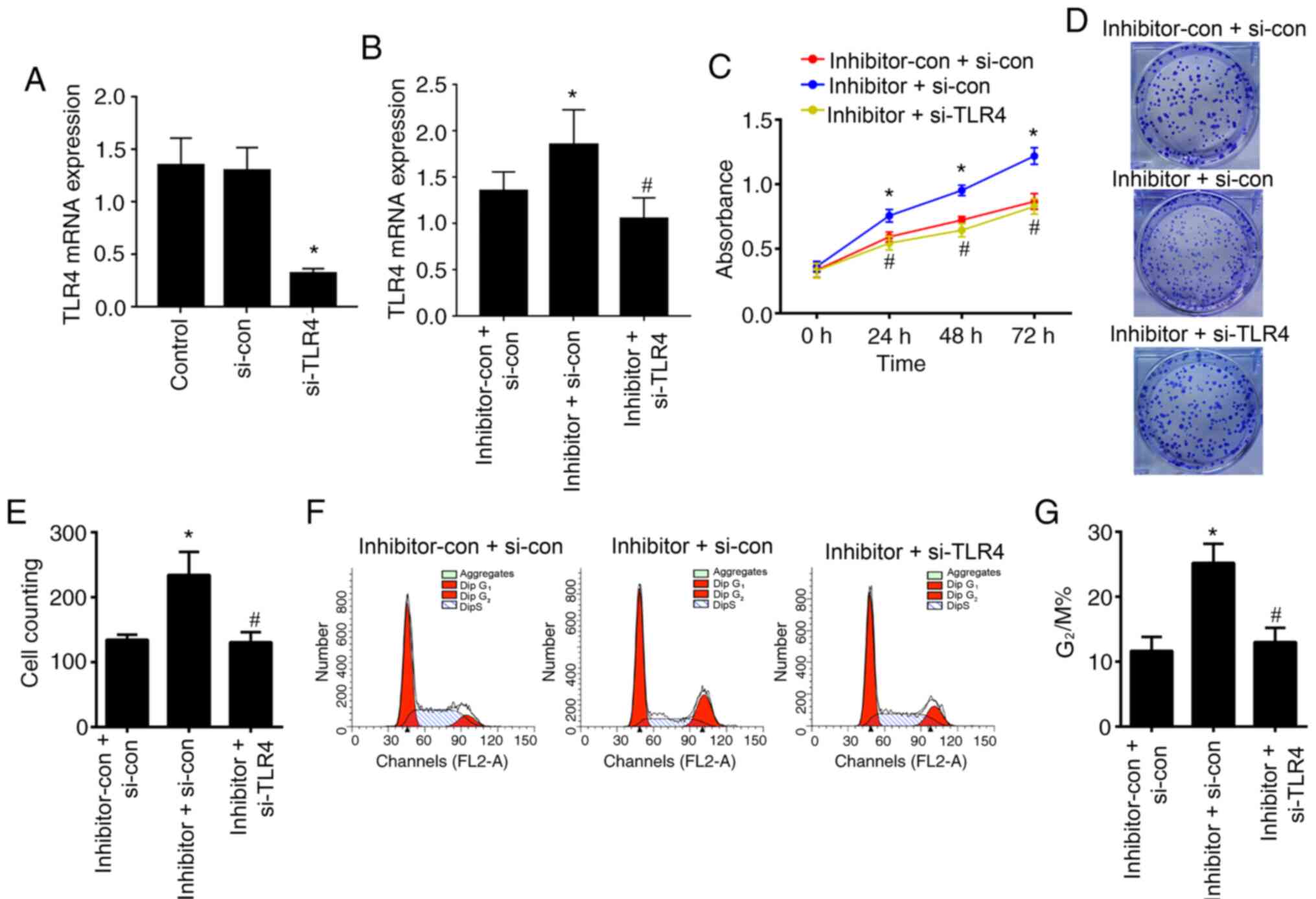
RT-qPCR assay was conducted to verify the transfection efficiency,
and the results indicated that the expression levels of TLR4 were
significantly downregulated in HaCaT cells transfected with TLR4
siRNA compared with those transfected with TLR4 si-con (P<0.05;
Fig. 5A). Moreover, the expression
levels of TLR4 were significantly upregulated in HaCaT cells
transfected with miR-489-3p inhibitor + TLR4 si-con compared with
transfected with TLR4 si-con + inhibitor-con (P<0.05; Fig. 5B), while the expression levels of
TLR4 were significantly downregulated in HaCaT cells transfected
with miR-489-3p inhibitor + TLR4 si-RNA compared with cells
transfected with TLR4 si-con + miR-489-3p inhibitor (P<0.05;
Fig. 5B).
The ability of cells to survival and form colonies
were significantly upregulated in the cells transfected with
miR-489-3p-inhibitor + si-TLR4-con compared with the cells
transfected with miR-489-3p-inhibitor-con + i-TLR4-con and
significantly downregulated in the cells transfected with inhibitor
+ si-TLR4 compared with the cells transfected with
miR-489-3p-inhibitor + si-TLR4-con (P<0.05; Fig. 5C-E). Consistent with these findings,
G2/M progression was also significantly increased in the
miR-489-3p-inhibitor+ si-TLR4-con group compared with the
miR-489-3p-inhibitor-con + si-TLR4-con group; and decreased in the
miR-489-3p-inhibitor + si-TLR4 group compared with the
miR-489-3p-inhibitor + si-TLR4-con group (P<0.05; Fig. 5F and G). The secretion levels of the
inflammatory cytokines TNF-α, IFN-γ, IL-22, IL-4 and IL-1β in cells
transfected with miR-489-3p-inhibitor + si-TLR4-con were
significantly higher compared with those transfected with
inhibitor-con + TLR4 si-con and the secretion levels were
significantly lower in the cells transfected with
miR-489-3p-inhibitor + si-TLR4 compared with the cells transfected
with miR-489-3p-inhibitor + si-TLR4-con (P<0.05; Fig. 6). These results suggested that
miR-489-3p regulated KC proliferation by inhibiting TLR4 and the
inflammatory response.
Discussion
The pathogenesis of psoriasis is both complex and
poorly characterized, involving multiple factors such as immunity,
inflammation, cell proliferation and apoptosis, as well as a common
immune-mediated pathway to cause abnormal proliferation,
differentiation, apoptosis and local inflammation of KCs (3). Various cells (including dendritic
cells, T lymphocytes, vascular endothelial cells and KCs),
cytokines, chemokines and intracellular signal transduction are
involved in this pathological process (18,19).
Abnormal KC hyperproliferation, accompanied by hyperkeratosis,
disfunction and inflammatory cell infiltration, are the main
pathological features of psoriasis (20), and KCs serve an important role in
the development of psoriasis. KCs form the epidermis of the skin,
and maintain its mechanical barrier function by promoting wound
healing. KCs, which have undergone a complete differentiation
cycle, overlap and arrange with adjacent cells in the stratum
corneum, forming a strong skin barrier (21). As key players in innate immune
defenses, KCs express a variety of pattern recognition receptors,
including TLRs 1-6 and 9, and retinoic acid-inducible gene I-like
receptors (22), that form the
initial responses to internal and external environmental stimuli.
KCs undergo terminal differentiation via the spinous layer and
granular layer of the epidermis, and begin to secrete keratin (K) 1
and K10 from the spinous layer, resulting in a loss of the nucleus
in the stratum corneum (23).
The excessive proliferation of KCs is an important
histopathological cause of psoriasis (19). During the terminal differentiation
process, the nucleus is retained and the differentiation cycle is
incomplete, which is accompanied by lipid secretion and reduced
horny transparent particles, thus disrupting the normal skin
barrier (24). In recent years, the
regulation of KCs via psoriatic-associated miRNAs has been an area
of intense research interest. Tsuru et al (25) reported that miR-424 was poorly
expressed in psoriasis and influenced cell proliferation by
regulating the expression levels of mitogen-activated protein
kinase 1 and cyclin E1 in KCs. Moreover, Xu et al (26) revealed that miR-125b was lowly
expressed in patients with psoriasis and that its overexpression
inhibited KC proliferation and differentiation. Chang et al
(10) also observed that miR-126
served an important role in the proliferation and migration of KCs
during wound healing. However, to the best of our knowledge, the
relationship between miR-489-3p and psoriasis has not been
previously reported, and studies on miR-489-3p have focused on its
role in cancer development. The overexpression of miR-489-3p may
can delay the development of various diseases. For example, the
overexpression of miR-489-3p in bladder cancer cells can inhibit
tumor cell proliferation and invasion (27), whilst in osteosarcoma, miR-489-3p
inhibits tumor cell metastasis via the paired box 3/MET pathway
(28). Furthermore, Jiang et
al (29) reported that
miR-489-3p inhibited neuronal growth via its regulation of PI3K/AKT
signaling during spinal cord injury. The present study demonstrated
that miR-489-3p expression was downregulated in psoriasis, and that
its overexpression in human immortalized KCs in vitro
inhibited cell proliferation and colony forming ability. This
indicated that the overexpression of miR-489-3p in psoriasis
inhibited the proliferation of KCs.
Bioinformatics predictions identified that TLR4 was
a target of miR-489-3p, the negative regulation of which was
confirmed using a luciferase assay. To further corroborate the
present findings, the expression levels of TLR4 and NF-κB were
detected in KCs transfected with miR-489-3p-mimic or
miR-489-3p-inhibitor plasmids. To confirm these findings, TLR
expression was silenced using siRNA technology. The results
demonstrated that miR-489-3p negatively regulated TLR4 and NF-κB
expression in KCs.
TLRs serve a crucial role in protecting the host
from a variety of exogenous and endogenous pathogens (30). Moreover, TLRs are key mediators of
innate and adaptive immunity, and are activated upon stimulation by
pro-inflammatory factors to activate the inflammatory response
(31). NF-κB is a multi-directional
nuclear transcription factor that is ubiquitous in the cytoplasm
(32,33). In response to TLR activation, NF-κB
translocates to the nucleus and induces the expression of an array
of genes that mediate innate and acquired immune regulation, cell
adhesion, inflammatory response and anti-apoptotic stimuli
(34,35). NF-κB activates TNF-α, IFN-γ, IL-22,
IL-4, IL-1β and other cytokines, all of which serve key roles in
the inflammatory response (36).
Thus, inflammatory cytokines can be targeted therapeutically for
the treatment of psoriasis (37).
The relationship between TNF-α and miRNAs in psoriasis is also
well-characterized. For instance, Pivarcsi et al (37) reported that miR-146a and miR-125b
regulated the secretion of TNF-α. After treatment with TNF-α
targeting compounds, miR-128a was highly expressed, while
miR-142-3p and miR-181a were downregulated (37). The present study demonstrated that
miR-489-3p inhibited the secretion of TNF-α, IFN-γ and IL-22 in
KCs. It has been shown that TLR4/NF-κB negatively regulated the
inflammatory responses of miRNAs. For example, Loubaki et al
(38) reported that miR-146a
regulated immune responses via TLR4 signaling in sepsis, while Liu
et al (39) revealed that
miR-129-5p targeted high mobility group box 1 to inhibit autoimmune
encephalomyelitis-related epilepsy via the TLR4/NF-κB axis.
Psoriasis is caused by the involvement of the skin
in an autoinflammatory reaction caused by the abnormal interaction
between epidermal KCs and immune cells (40). The present study simply used HaCaT
cells as the cellular model, and this was a single in vitro
study that did not consider the complex immune mechanism of
psoriasis. In future studies, animal models will be established to
further examine the role of miR-489-3p in the treatment of
psoriasis.
In summary, the present study demonstrated that
miR-489-3p negatively regulated the expression of TLR4 at the
post-transcriptional level and inhibited the proliferation of KCs
and prevented the secretion of inflammatory cytokines via
inhibition of the TLR4/NF-κB pathway in psoriasis. These results
highlight miR-489-3p as a promising therapeutic target for
psoriasis.
Acknowledgements
Not applicable.
Funding
Funding: This research was financially supported by a grant from
Zhejiang Traditional Chinese Medicine Administration (grant no.
2020ZA090).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY designed the study and performed the experiments.
PW collected data and interpreted the results. FZ contributed to
the preparation and revision of the manuscript for important
intellectual content. YY and FZ evaluated the authenticity of the
original data. All authors agreed to be accountable for the content
of the work, and read and approved the final manuscript.
Ethics approval and consent to
participate
The study complied with ethics committee regulations
of The Third People's Hospital of Hangzhou and was performed with
informed consent of the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Langley RG, Krueger GG and Griffiths CE:
Psoriasis: Epidemiology, clinical features, and quality of life.
Ann Rheum Dis. 64 (Suppl 2):ii18–ii25. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhang K and Shen ZA: Research advances on
immunological properties and gene regulation of skin keratinocytes.
Zhonghua Shao Shang Za Zhi. 37:89–92. 2021.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
3
|
Lowes MA, Bowcock AM and Krueger JG:
Pathogenesis and therapy of psoriasis. Nature. 445:866–873.
2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Qureshi AA, Choi HK, Setty AR and Curhan
GC: Psoriasis and the risk of diabetes and hypertension: A
prospective study of US female nurses. Arch Dermatol. 145:379–382.
2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Guttman-Yassky E, Nograles KE and Krueger
JG: Contrasting pathogenesis of atopic dermatitis and
psoriasis-part I: Clinical and pathologic concepts. J Allergy Clin
Immunol. 127:1110–1118. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rizwan M and Khan A: Association of
psoriasis and serum uric acid levels: A case control study.
Pakistan Armed Forces Med J. 69:408–412. 2019.
|
|
7
|
Pradyuth S, Rapalli VK, Gorantla S,
Waghule T, Dubey SK and Singhvi G: Insightful exploring of
microRNAs in psoriasis and its targeted topical delivery. Dermatol
Ther. 33(e14221)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rebane A and Akdis CA: MicroRNAs:
Essential players in the regulation of inflammation. J Allergy Clin
Immunol. 132:15–26. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chang L, Liang J, Xia X and Chen X:
miRNA-126 enhances viability, colony formation, and migration of
keratinocytes HaCaT cells by regulating PI3 K/AKT signaling
pathway. Cell Biol Int. 43:182–191. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Covert MW, Leung TH, Gaston JE and
Baltimore D: Achieving stability of lipopolysaccharide-induced
NF-kappaB activation. Science. 309:1854–1857. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wei W, Dejie L, Xiaojing S, Tiancheng W,
Yongguo C, Zhengtao Y and Naisheng Z: Magnolol inhibits the
inflammatory response in mouse mammary epithelial cells and a mouse
mastitis model. Inflammation. 38:16–26. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lowes MA, Suárez-Fariñas M and Krueger JG:
Immunology of psoriasis. Annu Rev Immunol. 32:227–255.
2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang C, Xiao C, Dang E, Cao J, Zhu Z, Fu
M, Yao X, Liu Y, Jin B, Wang G and Li W: CD100-Plexin-B2 promotes
the inflammation in psoriasis by activating NF-κB and the
inflammasome in keratinocytes. J Invest Dermatol. 138:375–383.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wu AG, Conway J, Barazani L, Roy B, Cline
A and Pereira F: Is clear always clear? Comparison of psoriasis
area and severity index (PASI) and the Physician's global
assessment (PGA) in psoriasis clearance. Dermatol Ther (Heidelb).
10:1155–1163. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cai Y, Fleming C and Yan J: New insights
of T cells in the pathogenesis of psoriasis. Cell Mol Immunol.
9:302–309. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Griffiths CE and Barker JN: Pathogenesis
and clinical features of psoriasis. Lancet. 370:263–271.
2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Han G, Williams CA, Salter K, Garl PJ, Li
AG and Wang XJ: A role for TGFbeta signaling in the pathogenesis of
psoriasis. J Invest Dermatol. 130:371–377. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tattersall D, Scott CA, Gray C, Zicha D
and Kelsell DP: EKV mutant connexin 31 associated cell death is
mediated by ER stress. Hum Mol Genet. 18:4734–4745. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kalali BN, Köllisch G, Mages J, Müller T,
Bauer S, Wagner H, Ring J, Lang R, Mempel M and Ollert M:
Double-stranded RNA induces an antiviral defense status in
epidermal keratinocytes through TLR3-, PKR-, and
MDA5/RIG-I-mediated differential signaling. J Immunol.
181:2694–2704. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aldehlawi H, Usman S, Lalli A, Ahmad F,
Williams G, Teh MT and Waseem A: Serum lipids, retinoic acid and
phenol red differentially regulate expression of keratins K1, K10
and K2 in cultured keratinocytes. Sci Rep. 10(4829)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ni X and Lai Y: Keratinocyte: A trigger or
an executor of psoriasis? J Leukoc Biol. 108:485–491.
2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tsuru Y, Jinnin M, Ichihara A, Fujisawa A,
Moriya C, Sakai K, Fukushima S and Ihn H: miR-424 levels in hair
shaft are increased in psoriatic patients. J Dermatol. 41:382–385.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Xu N, Brodin P, Wei T, Meisgen F, Eidsmo
L, Nagy N, Kemeny L, Ståhle M, Sonkoly E and Pivarcsi A: MiR-125b,
a microRNA downregulated in psoriasis, modulates keratinocyte
proliferation by targeting FGFR2. J Invest Dermatol. 131:1521–1529.
2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li J, Qu W, Jiang Y, Sun Y, Cheng Y, Zou T
and Du S: miR-489 suppresses proliferation and invasion of human
bladder cancer cells. Oncol Res. 24:391–398. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu Q, Yang G and Qian Y: Loss of
MicroRNA-489-3p promotes osteosarcoma metastasis by activating
PAX3-MET pathway. Mol Carcinog. 56:1312–1321. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Jiang R, Zhang C, Gu R and Wu H:
MicroRNA-489-3p inhibits neurite growth by regulating PI3K/AKT
pathway in spinal cord injury. Pharmazie. 72:272–278.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Banerjee S, Thompson WE and Chowdhury I:
Emerging roles of microRNAs in the regulation of Toll-like receptor
(TLR)-signaling. Front Biosci (Landmark Ed). 26:771–796.
2021.PubMed/NCBI
|
|
31
|
Yamamoto M, Sato S, Hemmi H, Uematsu S,
Hoshino K, Kaisho T, Takeuchi O, Takeda K and Akira S: TRAM is
specifically involved in the Toll-like receptor 4-mediated
MyD88-independent signaling pathway. Nat Immunol. 4:1144–1150.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Faure E, Equils O, Sieling PA, Thomas L,
Zhang FX, Kirschning CJ, Polentarutti N, Muzio M and Arditi M:
Bacterial lipopolysaccharide activates NF-kappaB through Toll-like
receptor 4 (TLR-4) in cultured human dermal endothelial cells.
Differential expression of TLR-4 and TLR-2 in endothelial cells. J
Biol Chem. 275:11058–11063. 2000.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Muzio M, Natoli G, Saccani S, Levrero M
and Mantovani A: The human Toll signaling pathway: Divergence of
nuclear factor kappaB and JNK/SAPK activation upstream of tumor
necrosis factor receptor-associated factor 6 (TRAF6). J Exp Med.
187:2097–2101. 1998.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hayden MS and Ghosh S: . NF-κB, the first
quarter-century: Remarkable progress and outstanding questions.
Genes Dev. 26:203–234. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Vallabhapurapu S and Karin M: Regulation
and function of NF-kappaB transcription factors in the immune
system. Ann Rev Immunol. 27:693–733. 2009.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sun W, Gao Y, Yu X, Yuan Y, Yi J, Zhang Z,
Cheng Y, Li Y, Peng X and Cha X: ‘Psoriasis 1’ reduces
psoriasis-like skin inflammation by inhibiting the VDR-mediated
nuclear NF-κB and STAT signaling pathways. Mol Med Rep.
18:2733–2743. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Pivarcsi A, Meisgen F, Xu N, Ståhle M and
Sonkoly E: Changes in the level of serum microRNAs in patients with
psoriasis after antitumour necrosis factor-α therapy. Br J
Dermatol. 169:563–570. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Loubaki L, Chabot D, Paré I, Drouin M and
Bazin R: MiR-146a potentially promotes IVIg-mediated inhibition of
TLR4 signaling in LPS-activated human monocytes. Immunol Lett.
185:64–73. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Liu AH, Wu YT and Wang YP: MicroRNA-129-5p
inhibits the development of autoimmune encephalomyelitis-related
epilepsy by targeting HMGB1 through the TLR4/NF-κB signaling
pathway. Brain Res Bull. 132:139–149. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Nicolas JF: Psoriasis: How the epithelium
influences the immune response: Keratinocytes, dendritic cells and
T lymphocytes. Bull Acad Natl Med. 198:17–30. 2014.PubMed/NCBI(In French).
|