Introduction
Hyaline cartilage can be damaged by trauma and is
degraded in different forms of arthritis (1,2). Once
damaged, cartilage has a limited capacity to repair and does not
fully regenerate (3,4). In past decades, efforts have been made
to achieve functional repair of hyaline cartilage and to regenerate
hyaline cartilage-like tissue, but these clinical attempts have
consistently failed (5,6). This may be due to a lack of knowledge
regarding the formation and maintenance of distinct features of
hyaline cartilage and the components of its extracellular matrix.
Therefore, a biological understanding of chondrocytes derived from
human joints and extensive research may be necessary to achieve
these goals.
Cartilage is an unusual tissue in that chondrocytes
can be anabolic (synthesize matrix) or catabolic (degrade matrix)
(7). In adult cartilage, a balance
exists between the synthesis and degradation of the cartilaginous
extracellular matrix. However, inflammation can cause an imbalance
between expression of anabolic [aggrecan (ACAN) and collagen type
II (COL2)] and catabolic [matrix metalloproteinases (MMPs)] factors
in chondrocyte tissue (8-10).
A severe imbalance between anabolic and catabolic chondrocyte
factors leads to progressive degradation of the cartilage matrix
(11-13).
Specifically, pro-inflammatory stimuli in chondrocytes drive poor
regenerative capacity by inducing catabolic molecules (14).
Cartilage destruction is a key characteristic of
degenerative joint diseases, particularly osteoarthritis (OA); it
also features in chronic inflammatory joint diseases, such as
rheumatoid arthritis (RA) (2).
Tumor necrosis factor α (TNFα) is a pleotropic cytokine that plays
key regulatory roles in inflammation and the response to cellular
stressors such as oxidative stress (15,16).
It also serves a vital role in the pathological process of
inflammatory changes by promoting release of MMP and downregulation
of SRY-box transcription factor 9 (SOX9), COL2 and ACAN expression,
eventually leading to extracellular matrix destruction in hyaline
cartilage (17). Therefore, TNFα
has been highlighted as a pathological factor in both OA and RA.
Previous studies have reported that TNFα serves an important role
in hyaline cartilage destruction in both OA and RA pathophysiology
(18,19). Therefore, TNFα has proven to be a
potential effective therapeutic target for patients with RA
(15).
The present study defined specific surface markers
of primary chondrocytes derived from human OA hyaline cartilage and
investigated the role of TNFα in the chondrogenesis of hyaline
cartilage.
Materials and methods
Human OA and RA subjects
The present study was performed in accordance with
Hanyang University Hospital Institutional Review Board guidelines
and approved by the Ethics Committee of Hanyang University Hospital
(approval no. 2017-05-003) and Hanyang University Guri Hospital
(approval no. 2018-07-024). Written informed consent was obtained
from all subjects.
Between April 2018 and May 2019, 15 patients with OA
(9 females and 6 males; mean age, 70.5±8.7 years) and three with RA
(all females; mean age, 57.1±11.3 years) were enrolled and surgical
samples were obtained from total knee replacement at Hanyang
University Guri Hospital. Both OA and RA surgical knee samples were
fixed at room temperature (RT) for two weeks with 10% formalin,
decalcified with 10% formic acid and embedded routinely in a
paraffin block. The paraffin block was sectioned at a thickness of
3.5 µm and stained by hematoxylin and eosin, Safranin O and
Toluidine blue. The section slides were deparaffinized in the
tissue in 100% neoclear (cat. no. 109843; Merck) for 10 min and
rehydration in serial ethanol dilution (100, 90, 80, 70, and 50%
ethanol) for 2 min each step, and washed with tap water for 5 min.
For H&E staining, the slides were stained with 100% hematoxylin
for 3 min, washed with tap water for 10 min, 100% eosin stained for
3 min, washed with tap water, and mounted with a permanent mounting
medium (cat. no. H-5000; Vector Lab). For Safranin O staining, the
slides were stained with 0.1% fast green stained for 5 min, washed
with 1% acetic acid one tapping, washed with tap water for 1 min,
0.1% Safranin O for 6 min, washed with tap water for 1 min, and
mounted with a permanent mounting medium. For toluidine blue
staining, the slides were stained with 0.1% toluidine blue stained
for 2 min, washed with tap water for 10 min, and mounted with a
permanent mounting medium. All staining procedures performed at RT.
Stained slides were imaged by a Nikon eclipse Ti-U light microscope
(Nikon Corporation).
Between February 2017 and May 2019, synovial fluid
samples were collected from 34 patients with OA (eight men and 26
women; mean age, 53.7±16.1 years) and 25 with RA (all women; mean
age, 68.6±8.5 years) at Hanyang University Hospital for Rheumatic
Disease. For synovial fluid analysis, 10 ml fluid was incubated
with 1.5 mg hyaluronidase (cat. no. H3506; Sigma-Aldrich) for 15
min at 37˚C, followed by centrifugation at 1,400 x g for 15 min at
4˚C. After centrifugation, the fluids were immediately divided into
aliquots and stored at -80˚C for TNFα levels (cat. no. DTA00D;
R&D Systems, Inc.) measurement using ELISA according to
manufacturer's protocol.
Isolation of human OA
chondrocytes
Hyaline cartilage from OA knee joints was scraped
using a rongeur and collected in serum-free DMEM (cat. no.
L0103-500; Biowest) buffer containing 1 mg/ml Collagenase type 2
(cat. no. C6885; Sigma-Aldrich). The collected tissue samples were
incubated at 37˚C with agitation overnight. The next day, the
digested hyaline cartilage was filtered through a 70-µm strainer
(cat. no. 93070; SPL Life Sciences) and seeded ~60% cell density in
DMEM (cat. no. L0103-500; Biowest) supplemented with 10% FBS (cat.
no. 16000-044; Gibco; Thermo Fisher Scientific, Inc.), 1%
penicillin and streptomycin (cat. no. 15140122; Gibco; Thermo
Fisher Scientific, Inc.) at 37˚C and 5% CO2. Primary
chondrocytes at passage 2-5 were used in subsequent experiments.
Until the passage 2-5, chondrocytes were cultured at 60% density
before full cell density at 37˚C.
Reagents and biological agents
Recombinant human TNFα (cat. no. 300-01A; PeproTech,
Inc.), golimumab (Janssen Global Services, LLC; 1 µg/ml, 37˚C, 4
weeks) and BAY 11-7082 (cat. no. B5556; Sigma-Aldrich; 10 or 30 µM,
37˚C, 24 h) were obtained.
Flow cytometric analysis
The chondrocytes were fixed using a
fixation/permeabilization solution kit (cat. no. 554715; BD
Biosciences) and stained with CD34 (cat. no. 343607; BioLegend,
Inc.; 1:100), CD44 (cat. no. 338807; BioLegend, Inc.; 1:100), CD59
(cat. no. 304711; BioLegend, Inc.; 1:100), CD74 (cat. no. 326811;
BioLegend, Inc.; 1:100), CD90 (cat. no. 328109; BioLegend, Inc.;
1:100), CD105 (cat. no. 323205; BioLegend, Inc.; 1:100), CD146
(cat. no. 361015; BioLegend, Inc.; 1:100), CD164 (cat. no. 324805;
BioLegend, Inc.; 1:100), SOX9-Alexa-647 (cat. no. 565493; BD
Pharmingen; BD Biosciences; 1:200), ACAN-PE (cat. no. sc-33695;
Santa Cruz Biotechnology, Inc.; 1:100), IgG1-Alexa-647 (cat. no.
557732; BD Pharmingen; BD Biosciences; 1:100), IgG1-PE (cat. no.
400112; BioLegend, Inc.; 1:100), IgG1-APC (cat. no. 400122;
BioLegend, Inc.; 1:100), IgG1-FITC (cat. no. 400109; BioLegend,
Inc.; 1:100) or IgG2a-APC (cat. no. 400221; BioLegend, Inc.; 1:100)
for 30 min at 4˚C. The dilution of the antibody for FACS was ranged
from 1:100-1:200. After staining, cells were washed with Perm/Wash
Buffer (cat. no. 554715; BD Biosciences) and analyzed by flow
cytometry (FACS Canto II; BD Biosciences). Data were analyzed using
FlowJo version 10.7 software (FlowJo LLC).
Proliferation assay
The water-soluble tetrazolium salt (WST) assay was
performed with EZ-CYTOX (cat. no. EZ-1000; Dogen Bio Co., Ltd.)
according to the manufacturer's instructions. Primary chondrocytes
were plated into 96-well plate (1x103 cells/well) and
treated with 10 or 25 ng/ml TNFα in 37˚C CO2 incubator
for 1-6 days. WST solution was added to the cells, which were
incubated for 1 h. The absorbance at 450 nm was measured with a
microplate reader (Thermo Fisher Scientific, Inc.).
Human MMP antibody array
Chondrocytes were stimulated with 10 ng/ml human
TNFα at RT for 1 day and collected for human MMP analysis. The
stimulated cells were lysed in 1X RIPA buffer including phosphatase
and proteinase inhibitors and assessed according to the
manufacturer's protocol (cat. no. ab134004; Abcam). To analyze the
array data, comparison of signaling intensities for individual
spots was detected using the UVItech system (Cleaver Scientific
Ltd.) and analyzed with ImageJ 1.52a version software (National
Institutes of Health).
Reverse transcription-quantitative
(RT-q)PCR
RT-qPCR was performed as previously described
(20). Briefly, total RNA of the
TNF-treated cells extracted with TRIzol® reagent (cat.
no. 15596018; Invitrogen; Thermo Fisher Scientific, Inc.) was used
to generate complementary DNA (42˚C for 1 h and then 70˚C for 10
min) using a RevertAid First Strand cDNA Synthesis kit (cat. no.
K1622; Thermo Fisher Scientific, Inc.). qPCR was performed on a
CFX96 Real-time PCR detection system (cat. no. 1855201; Bio-Rad
Laboratories, Inc.) following the manufacturer's procedure and the
following thermal cycling: Initial denaturation for 3 min at 95˚C;
two step-cycling: Denaturation for 10 sec at 95˚C, combined
annealing/extension for 30 sec at 60˚C for 35 cycle. The expression
of each target gene was normalized to GAPDH. Normalized
expression values were averaged and the relative levels of gene
expression were quantified by using the comparative CT method
(21). Primers used for PCR were as
follows: MMP1 forward, 5'-AGAGCAGATGTGGACCATGC-3' and
reverse, 5'-TTGTCCCGATGATCTCCCCT-3'; MMP3 forward,
5'-TCTATGGACCTCCCCCTGAC-3' and reverse, 5'-GATTTGCGCCAAAAGTGCCT-3';
MMP13 forward, 5'-GCCATTACCAGTCTCCGAGG-3' and reverse,
5'-TACGGTTGGGAAGTTCTGGC-3'; SOX9 forward,
5'-CTGAACGAGAGCGAGAAGCG-3' and reverse, 5'-CCCGTTCTTCACCGACTTCC-3';
ACAN forward, 5'-TGGGAACCAGCCTATACCCCAG-3' and reverse,
5'-CAGTTGCAGAAGGGCCTTCTGTAC-3'; COL2 forward,
5'-GCCGGATCTGTGTCTGTGAC-3' and reverse, 5'-TGTCCCTTTGGTCCTGGTTG-3';
and GAPDH forward, 5'-CAAGATCATCAGCAATGCC-3' and reverse,
5'-CTGTGGTCATGAGTCCTTCC-3'.
Immunoblotting
Immunoblot analysis was performed as previously
described (22). The treated cells
were lysed in RIPA buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl,
0.1% SDS, 0.6% Na-deoxycholate, 1% Triton X-100) supplemented with
protease (cat. no. 535140, Calbiochem) and phosphatase (cat. no.
5870, Cell signaling) inhibitor cocktails. Lysed samples were
incubated on ice for 1 h followed by a centrifugation at 12,000 x g
for 30 min at 4˚C. The protein of whole lysates were determined
using a Bradford protein assay (cat. no. 5000006; Bio-Rad
Laboratories, Inc.). Protein (30~50 µg) were separated by SDS-PAGE
and electrophoretically transferred onto nitrocellulose membranes
(cat. no. 10600002, Cytiva) in a transbuffer. Membranes were
blocked with 5% non-fat milk in Tris-buffered saline (TBS) with
0.1% Tween-20 and incubated with specific primary antibodies,
followed by incubation with horseradish peroxidase-conjugated
secondary antibodies. The dilution of the primary antibody for
Immunoblotting was ranged from 1:500 to 1:1,000. Diluted primary
antibodies were incubated at 4˚C overnight and secondary antibodies
incubation diluted 1:1,000 at RT for 1 h. Membranes were visualized
with Pierce ECL (cat. no. 34580; Thermo Fisher Scientific, Inc.)
and the visualized images were collected by chemiluminescence
imaging system (Alliance Q9 advanced, Uvitech System). The primary
antibodies for TNF receptor 1 (cat. no. sc-8436; 1:1,000) and NF-κB
p65 (cat. no. sc-372; 1:1,000) were from Santa Cruz Biotechnology,
Inc. Phosphorylated (p-)NF-κB p65 (cat. no. 3033; 1:1,000), p-ERK
(cat. no. 9101s; 1:1,000), total-ERK (cat. no. 9102s; 1:1,000),
p-p38 (cat. no. 9215s; 1:500), total-p38 (cat. no. 9212; 1:1,000),
and β-actin (cat. no. 4970; 1:5,000) antibodies were from Cell
Signaling Technology, Inc. MMP-1 (cat. no. MAB901; 1:500), MMP-3
(cat. no. MAB513; 1:500), and MMP-13 (cat. no. MAB511; 1:500)
antibodies were from R&D Systems, Inc. Goat anti-rabbit (cat.
no. 111-035-003; 1:2,000) and anti-mouse (cat. no. 115-035-003;
1:2,000) secondary antibodies were from Jackson ImmunoResearch
Laboratories, Inc.
Trichloroacetic acid (TCA) assay
TCA precipitation was performed as previously
described (23). In brief,
chondrocytes were seeded at 80-90% confluence. The next day, growth
medium was replaced with serum-free DMEM including 10 ng/ml TNFα or
distilled water for 1 day. The cell supernatant obtained by
centrifugation at 1,224 x g at 4˚C for 10 min was collected for TCA
precipitation, subjected to 12.5% SDS-PAGE and then stained with
Coomassie Brilliant Blue R-250 solution (cat. no. C2006; Biosesang)
as a loading control. After destaining (acetic acid: Methanol:
Water; 7.5:5:87.5) overnight, the gel was washed five times with
distilled water for 10 min and followed by immunoblotting
procedures.
Immunofluorescence
The TNFα-stimulated chondrocytes were washed twice
with 1X PBS and fixed with 10% formalin at RT for 15 min, followed
by permeabilization with 1X PBS containing 0.1% Triton X-100 and 1%
BSA (cat. no. BSA-BSH-1XG; Rocky Mountain Biologicals, Inc.) at RT
for 1 h, incubation with a primary antibody at 4˚C overnight,
washing with 1X PBS and incubation with Cy3-conjugated anti-rabbit
antibody (cat. no. 111-165-144; Jackson Immunoresearch) or Alexa
488-conjugated anti-mouse antibody (cat. no. A-11001; Invitrogen)
for 1 h. All primary and secondary antibodies were used at 1:100
dilution. The stained cells were washed with distilled water and
mounted with DAPI (cat. no. H1200; Vector Laboratories, Inc.;
Maravai Life Sciences). In order to visualize stained cells,
immunofluorescence images were collected with a confocal microscope
(TCS SP5; Leica Microsystems GmbH). Images were captured using LAS
version 4.2.1 software (Leica Microsystems GmbH).
Promoter assay
NF-κB p65 wild-type and two p65 S536A (substitution
of alanine for serine) or S536E (substitution of glutamate for
serine 536) mutant promoters in pGL3-Basic were a gift from Dr
Heekyoung Chung (Hanyang University, Seoul, South Korea) (24). 293T cells were a generous gift from
Dr Heekyoung Chung (Hanyang University, Seoul, Republic of Korea)
and seeded into 60-mm culture plates (2x105 cells per
well) and co-transfected with p65 wild-type, p65 S536A or p65 S536E
(1 µg/well), and Renilla (0.25 µg/well) plasmids as control
for 48 h using Lipofectamine® 3000 (cat. no. L3000-015;
Invitrogen; Thermo Fisher Scientific, Inc.). The transfected cells
were reseeded on a 12-well culture plate (5x104 cells
per well) for treatment at 37˚C for 24 h with distilled water or 10
or 25 ng/ml TNFα and then analyzed with Dual-Luciferase Reporter
Assay system (cat. no. E1500; Promega Corporation). The procedure
was performed according to the manufacturer's instructions and
activity was measured with a luminometer (Titertek-Berthold). The
measured values were analyzed by comparison with Renilla
luciferase. This ratio was then normalized to the averaged ratio of
vehicle.
Chondrogenic differentiation of
chondrocytes with pellet culture
The ‘pellet culture’ method was performed as
previously described (25).
Briefly, primary chondrocytes (2x105) were collected
following centrifugation at 25˚C for 3 min at 441 x g, replaced and
cultured in chondrogenic medium with distilled water or TNFα for 4
weeks. The medium was changed every 3 days. Chondrogenic medium was
composed of serum-free DMEM/F12 (cat. no. 11320033; Gibco; Thermo
Fisher Scientific, Inc.) with 10% Insulin-Transferrin-Selenium
premix tissue culture supplement (cat. no. I3146; Sigma-Aldrich;
Merck KGaA), 10 µM dexamethasone (cat. no. D-2915; Sigma-Aldrich;
Merck KGaA), 1 µM ascorbate-2-phosphate (cat. no. 49752;
Sigma-Aldrich; Merck KGaA), 1% sodium pyruvate (cat. no. 11360070;
Gibco; Thermo Fisher Scientific, Inc.) and 10 ng/ml TGF-β1 (cat.
no. 0218209-1; PeproTech, Inc.). 10% Formalin-fixed pellets at RT
for 24 h were washed with 1X PBS and transferred to 30% sucrose
solution overnight. Pellets were soaked in OCT compound (cat. no.
4583; Sakura Finetek USA) and sectioned on a cryotome (cat. no.
CM1850; Leica Microsystems GmbH) to create 15-µm sections on
gelatin-coated slides. Then, the slides were stained with Safranin
O (cat. no. 1446640250; ACROS Organics) and Toluidine blue (cat.
no. T3260-5g; Sigma-Aldrich; Merck KGaA). Each staining was washed
with water for 1 min and then stained with 0.1% dyeing solution for
1 min at RT. Stained slides were imaged under a Nikon eclipse Ti-U
light microscope (Nikon Corporation).
Statistical analysis
Data were analyzed with GraphPad Prism 6 software
(GraphPad Software, Inc.). A two-tailed Student t-test was used to
compare data between two unpaired groups. One-way ANOVA with
Tukey's post hoc test was used to compare data between more than
two groups. All data are expressed as the mean ± SD (n≥3).
P<0.05 was considered to indicate a statistically significant
difference.
Results
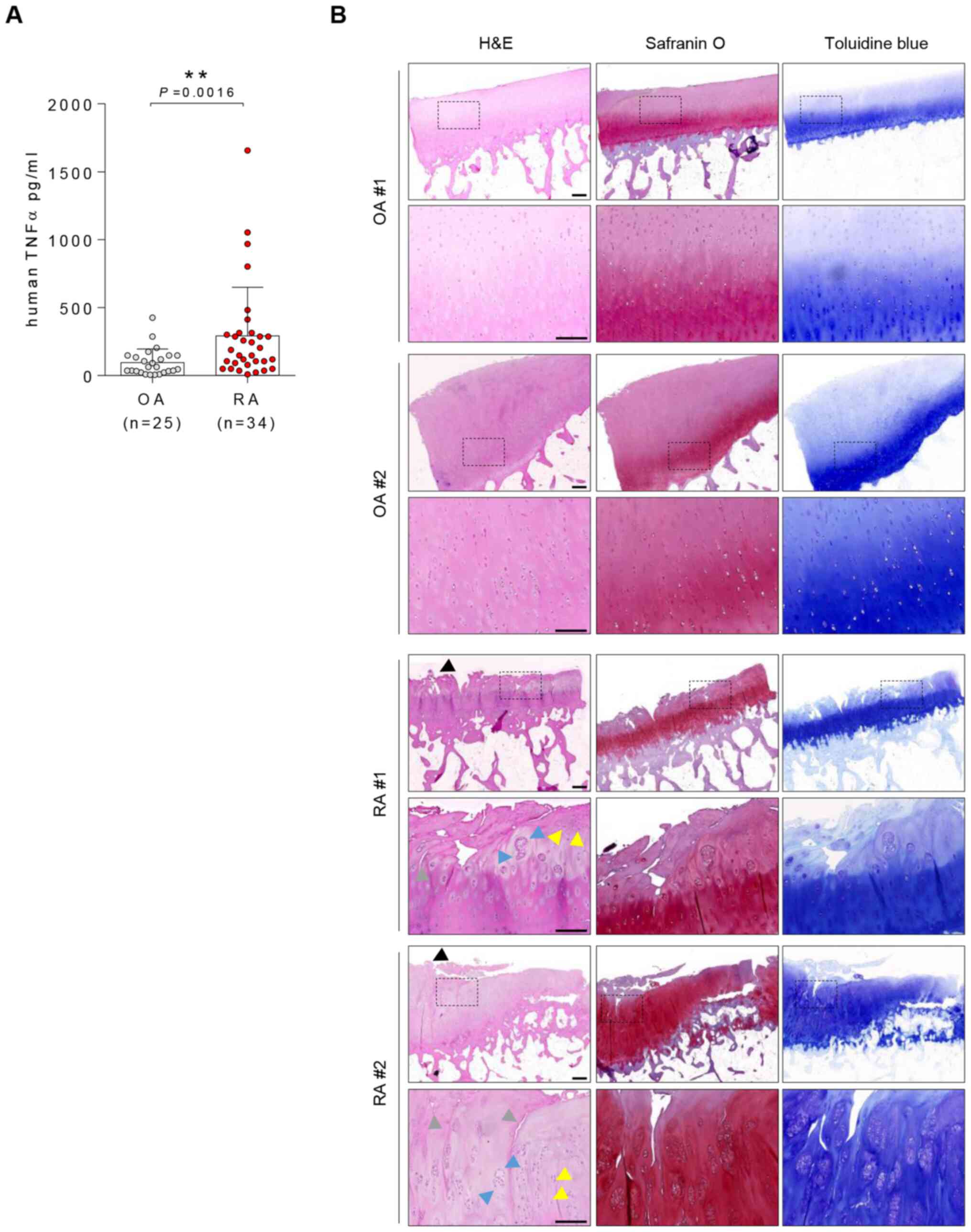
High TNFα levels in synovial fluid and
destruction of hyaline cartilage are observed in RA
TNFα levels in synovial fluid were significantly
higher in patients with RA than OA (mean, 168.7 vs. 64.28 pg/ml;
Fig. 1A). Although superficial
fibrillation and loss of proteoglycan detection with safranin O
staining were observed in OA hyaline cartilage, patients with RA
exhibited more severe hyaline cartilage damage than patients with
OA. Overt fibrillation (black arrows), chondrocyte clustering (blue
arrows), changes in chondrocyte morphology/distribution (yellow
arrows) and matrix destruction (grey arrows) were observed in
patients with RA (Fig. 1B).
TNFα has no significant effects on
proliferation and extracellular molecule expression in OA
chondrocytes
The present study aimed to detect known chondrocyte
surface markers and investigate the effects of TNFα on them
(22,23). CD44, CD59 and CD90 were highly
expressed in chondrocytes, whereas CD34, CD74 and CD146 were not;
in addition, there was partial positive expression of CD105 and
CD164. (Fig. 2A). Following TNFα
treatment, there were no significant changes in the rate of cell
proliferation (Fig. 2B) or
chondrocyte expression of SOX9 and ACAN (Fig. 2C).
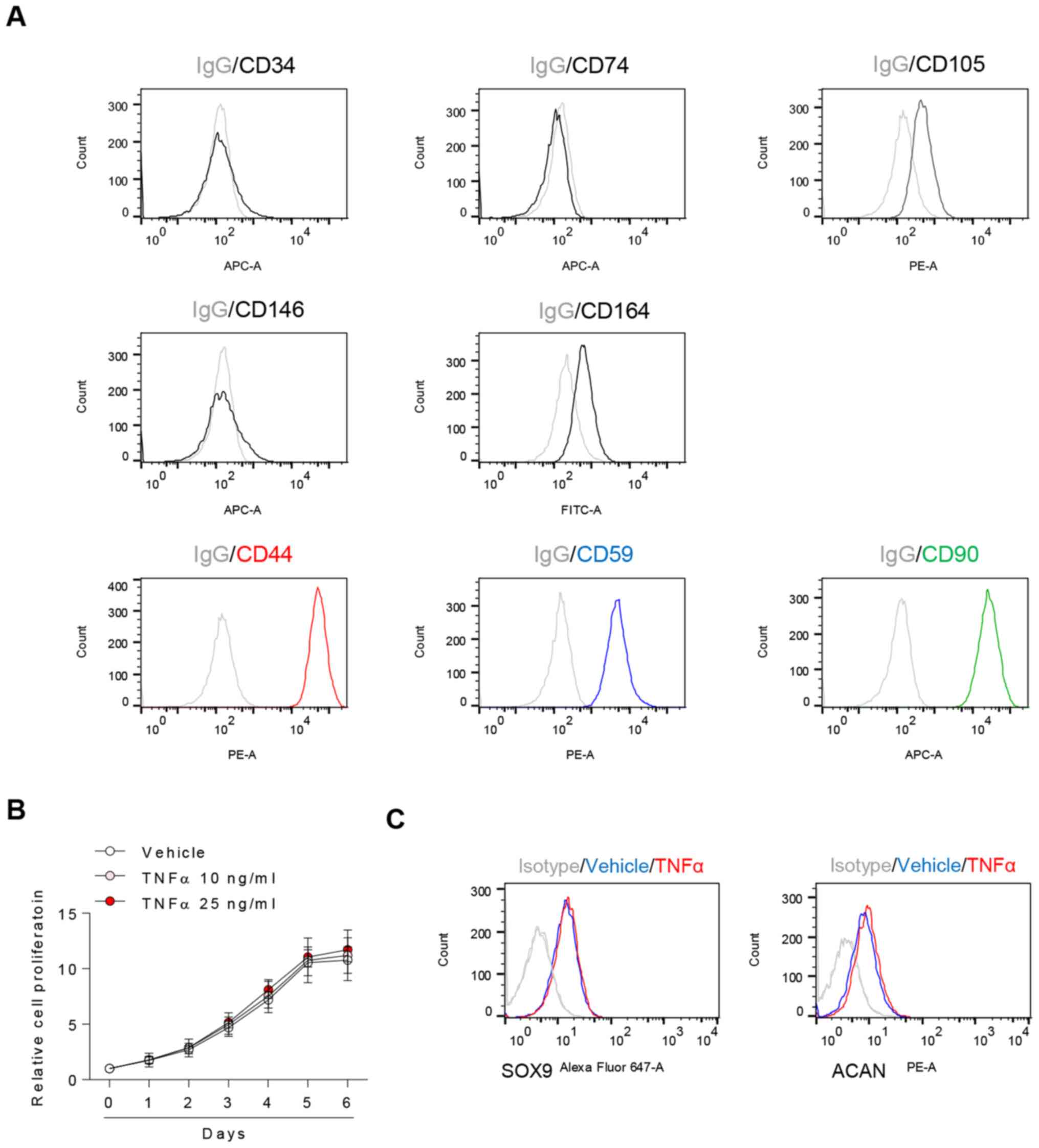 | Figure 2TNFα does not significantly affect
proliferation and extracellular molecule expression in OA
chondrocytes. (A) CD34, CD44, CD59, CD74, CD90, CD105, CD146 and
CD164 surface markers of chondrocytes were evaluated by flow
cytometry. (B) Chondrocytes were exposed to TNFα and assessed by
water-soluble tetrazolium salt assay. n=5, one-way ANOVA with
Tukey's post hoc test. (C) Chondrocytes were exposed to 25 ng/ml
TNFα for 24 h and assessed by flow cytometry. IgG was used as a
control. OA, osteoarthritis; TNFα, tumor necrosis factor α; ACAN,
aggrecan; SOX9, SRY-box transcription factor 9. |
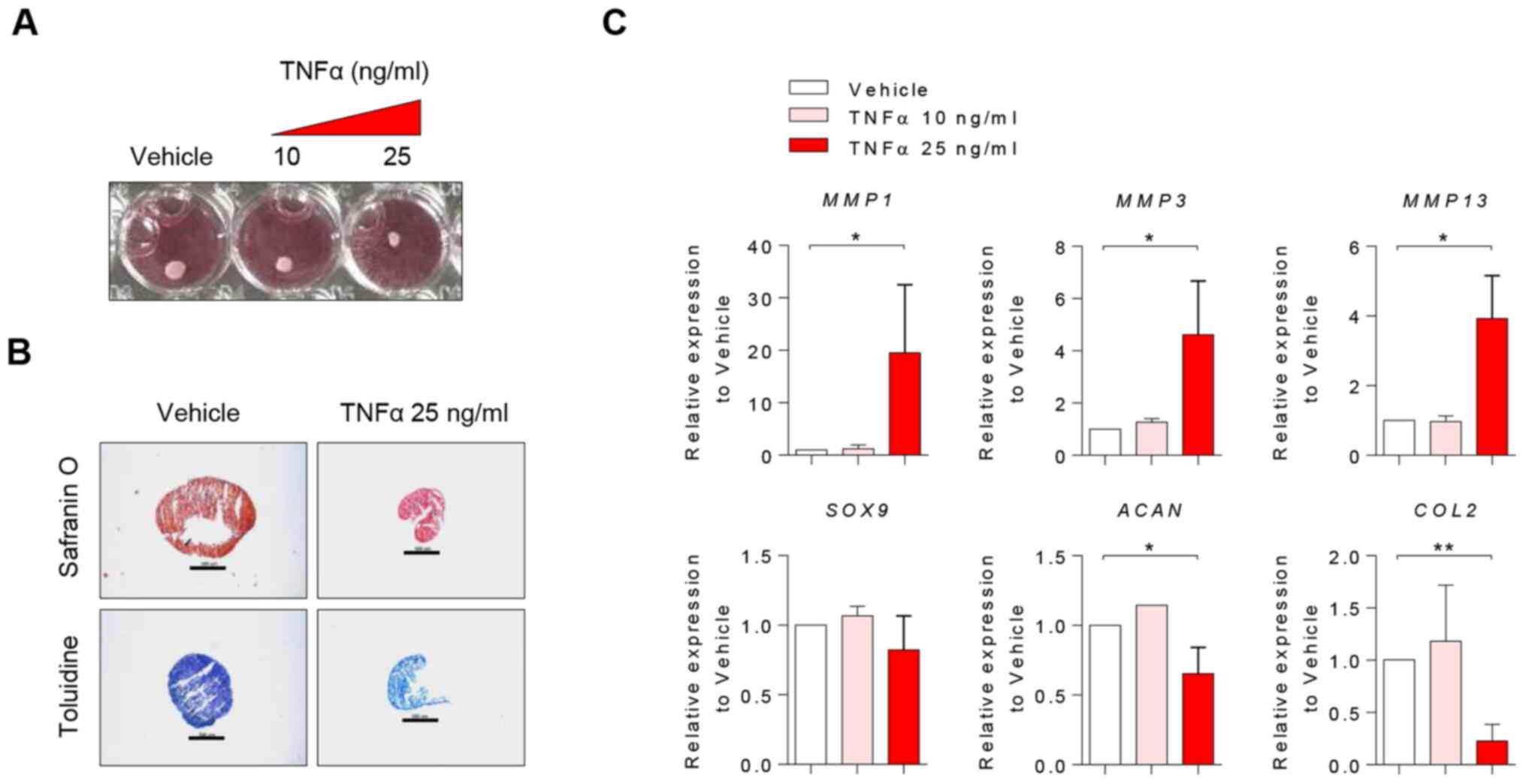
TNFα induces MMP expression in human
OA chondrocytes
TNFα-treated chondrocytes were used as a model for
inflammatory arthritis. In order to determine whether TNFα may
affect the destructive hyaline cartilage of patients with
inflammatory arthritis, chondrocytes were stimulated with TNFα. To
confirm dose effects of TNFα in chondrocytes, we treated with 1, 5,
10, 25, 50 ng/ml TNFα dose and analyzed MMP expression levels using
RT-qPCR and immunoblotting. We confirmed an increase in MMP1, 3, 13
expression in dose-dependently TNFα treatment (Fig. S1). Strong increases in the protein
expression levels of MMP1, 3 and 13, and a decrease in TIMP
metallopeptidase inhibitor (TIMP)1 and TIMP2 expression levels were
observed (Figs. 3A, S2 and S3). Changes in the expression levels of
MMPs were validated by RT-qPCR (Fig.
3B) and immunoblotting (Fig.
3C). TNFα stimulation of chondrocytes elevated MMP1, 3, and 13
protein expression levels in the cytoplasm and led to extracellular
secretion of these proteins (Fig.
3C, D and F). Moreover, TNFα induced NF-κB
phosphorylation and nuclear translocation in chondrocytes, and
significantly augmented p65 promoter activity, but not that of
mutants (Fig. 3E and F). Therefore, it was concluded that TNFα
promoted the expression of MMP1, 3 and 13 in human chondrocytes,
which was accompanied by activation of NF-κB signaling.
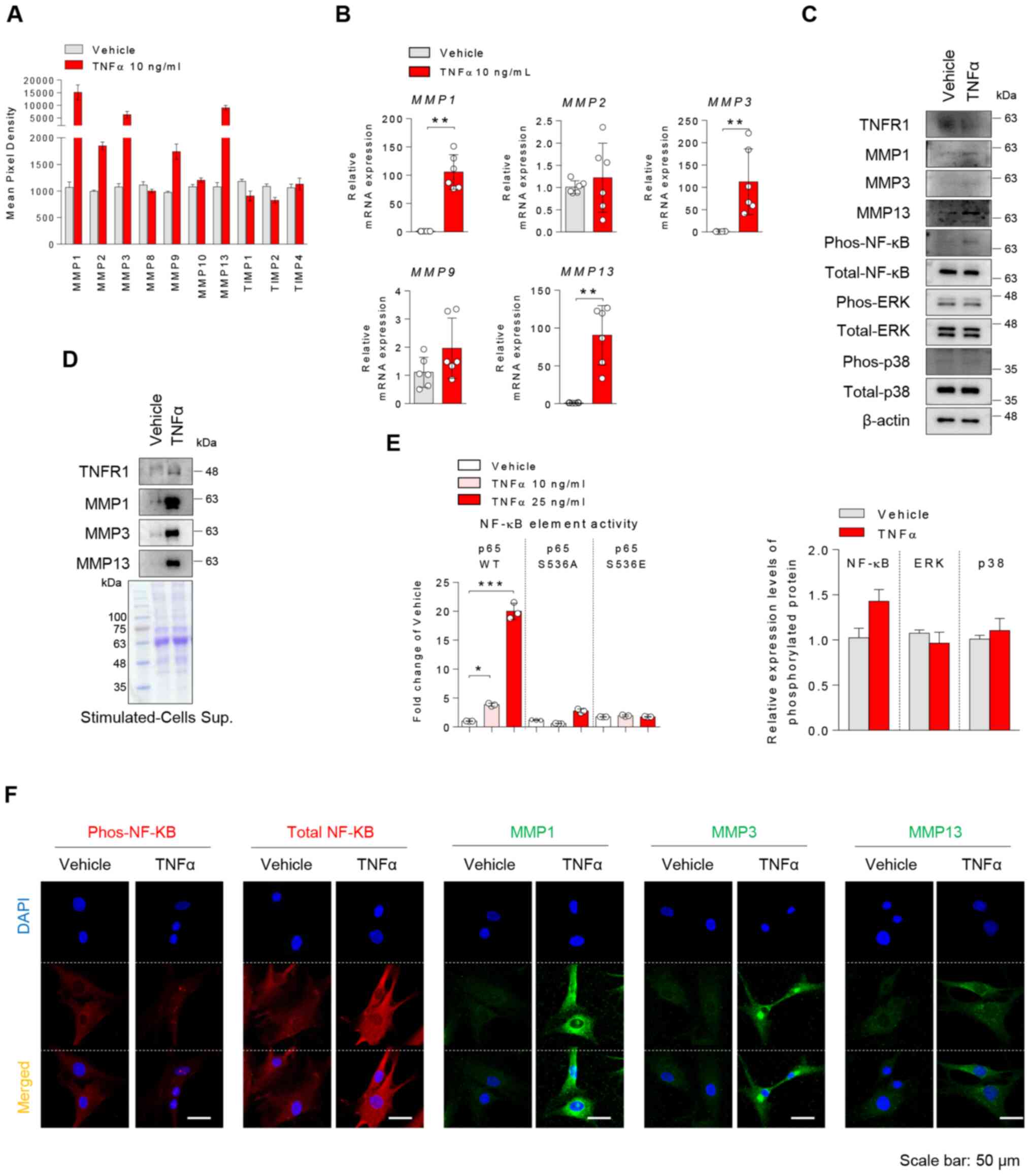 | Figure 3TNFα induces the expression of MMPs
in OA chondrocytes via NF-κB activation. (A) Chondrocytes were
treated with vehicle or 10 ng/ml TNFα for 24 h. MMP protein
expression levels in stimulated cell lysates were semi-quantified
via a human MMP antibody array (n=2). (B) MMP mRNA
expression in stimulated cells was determined by reverse
transcription-quantitative PCR. (C) TNFα-treated chondrocytes were
assessed by immunoblotting. Phosphorylation was semi-quantified
with ImageJ and calculated relative to each total protein. (D)
Proteins secreted in the cell supernatant by TNFα stimulation were
precipitated with trichloroacetic acid and detected by
immunoblotting. Coomassie blue staining was used as a loading
control. (E) 293T cells were transfected with p65 wild type or
double mutants (S536S or S536E), followed by treatment with
vehicle, 10 or 25 ng/ml TNFα for 24 h; promoter activity analysis
was subsequently performed. n=3, one-way ANOVA with Tukey's post
hoc test. (F) Protein expression levels of p-NF-κB, total NF-κB and
MMP1, 3 and 13 in TNFα-stimulated cells were analyzed by
immunofluorescence. Representative data are shown. Scale bar, 50
µm. *P<0.05, **P<0.01;
***P<0.001 (mean ± SD; n=6). OA, osteoarthritis;
phos, phosphorylated; TNFα, tumor necrosis factor α; MMP, matrix
metalloproteinase; NF-κB, nuclear factor κB. |
TNFα decreases the regenerative
capacity of OA chondrocytes during differentiation
In order to elucidate the effect of TNFα on
regenerative capacity in chondrocytes, chondrogenic differentiation
of chondrocytes was induced in the presence or absence of TNFα
treatment. Chondrocytes treated with TNFα exhibited lower
differentiation capacity; they showed weaker intensity of staining
and smaller pellet size compared with controls (Fig. 4A and B). Furthermore, upregulation of
MMP1, MMP3 and MMP13, and downregulation of
ACAN and COL2 was observed (Fig. 4C), which suggested that TNFα
stimulation induced a metabolic shift from anabolism to catabolism
during chondrogenic differentiation. Thus, it was concluded that
TNFα decreased the regenerative capacity of chondrocytes during
differentiation.
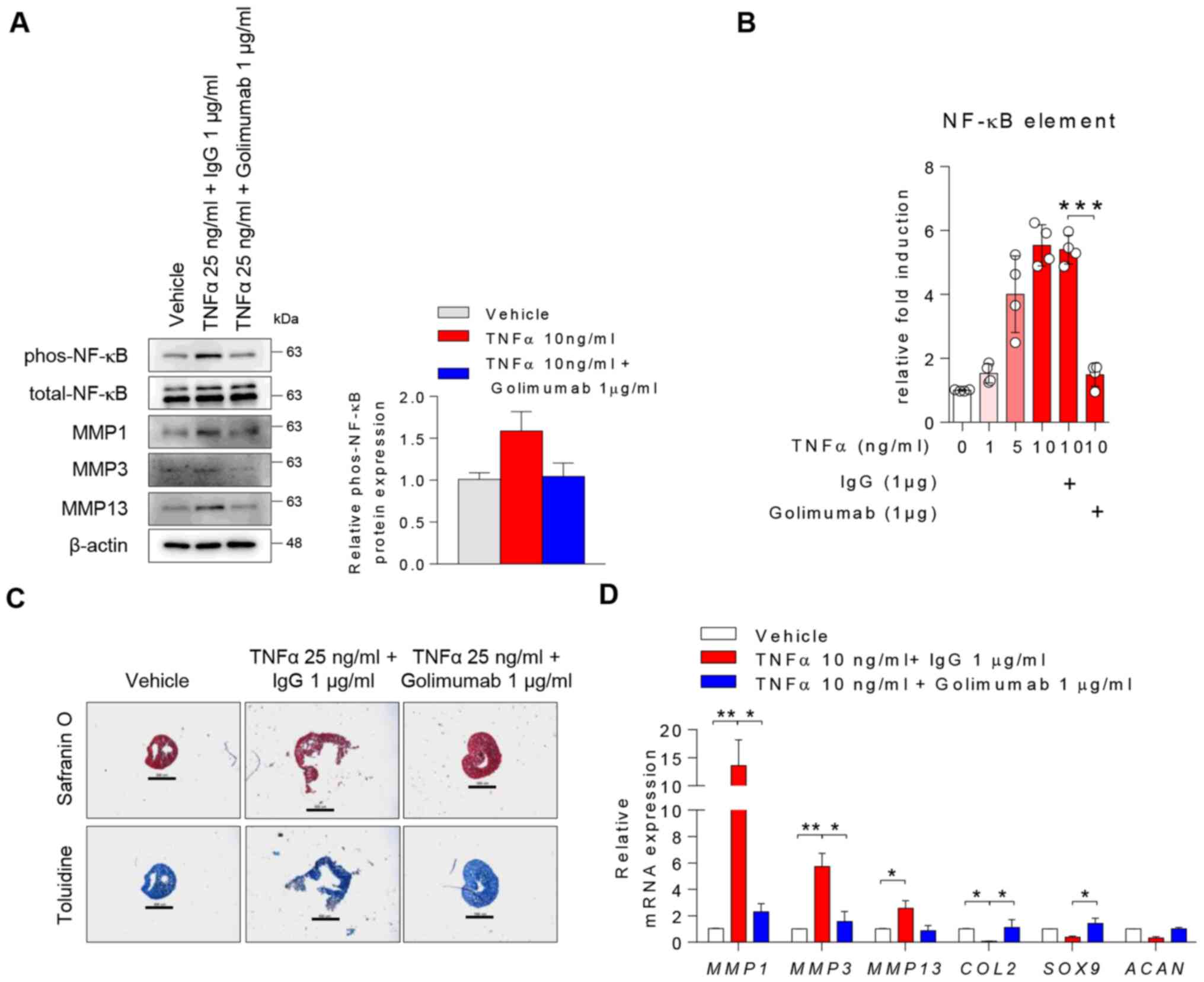
Blocking TNFα attenuates TNFα-driven
cartilage matrix degradation in OA chondrocytes
To determine whether the modulation of TNFα affected
catabolic and anabolic gene expression levels, human chondrocytes
were treated with 1 µg/ml golimumab (a TNFα blocker) in the
presence of TNFα. TNFα stimulation upregulated p-NF-κB, MMP1, MMP3
and MMP13 in chondrocytes, whereas treatment with golimumab impeded
these changes mediated by TNFα (Fig.
5A). In the line with Fig. 5A,
changes in TNF-mediated p-NF-κB were quantified (Fig. 5A, right panel). TNFα activated the
WT NF-κB promoter in a dose-dependent manner but blocking TNFα
diminished this activation (Fig.
5B). In addition, treatment with 10 or 30 µM BAY, an NF-κB
inhibitor, suppressed TNFα-mediated induction of MMP1,
MMP3 and MMP13 mRNA expression in chondrocytes
(Fig. S4). During chondrogenic
differentiation, treatment with the TNFα blocker interfered with
the destructive effect of TNFα on physical changes of
chondrogenesis (Fig. 5C) and its
mRNA expressions (Fig. 5D).
Therefore, blocking TNFα may attenuate TNFα-driven cartilage matrix
degradation in inflammatory arthritis.
Discussion
The present study revealed that TNFα levels were
increased in synovial fluid and the destruction of hyaline
cartilage was greater in RA compared with in OA. To demonstrate the
association between TNFα and hyaline cartilage destruction in OA,
chondrocytes were stimulated with TNFα; the results indicated that
TNFα induced a metabolic shift in chondrocytes via NF-κB signaling.
Based on these findings, a model for the pathogenesis of hyaline
cartilage degradation in inflammatory arthritis was proposed. TNFα,
an inflammatory cytokine that is increased by excessive
inflammation, may modulate anabolic and catabolic factors, thereby
revealing potential therapeutic targets for inflammatory arthritis
progression.
The precise mechanism by which TNFα suppresses the
expression of matrix proteoglycans, such as ACAN and COL2, requires
further study. It was hypothesized that MMP 1, 3 and 13 are
indicators of TNFα-driven matrix proteoglycan degradation. Although
24-h TNFα exposure did not affect SOX9 and ACAN expression in
chondrocytes, exposure to TNF during chondrogenic differentiation
resulted in decreased mRNA expression levels of anabolic mediators,
such as ACAN and COL2. Exposure of chondrocytes to
TNFα did not affect cell proliferation, but high dose-exposure may
induce cell death and senescence (26,27).
Moreover, loss of matrix proteoglycans was revealed to be
TNFα-dependent because TNFα blockade inhibited destruction of
chondrocytes by TNFα. Therefore, anti-TNFα therapy may alleviate
the destruction of matrix proteoglycans and hyaline cartilage in
inflammatory arthritis.
When chondrocytes become hypertrophic, they express
unique genes, such as COL10 and MMP13; these genes
indicate calcification or matrix mineralization (28,29).
In line with these results, the present study showed that TNFα
treatment upregulated COL10 mRNA expression (data not
shown). Collectively, these data suggested that TNFα stimulation of
chondrocytes regulated MMP13 and COL10 expression, leading to
chondrocyte hypertrophy, calcification or matrix
mineralization.
TNFα plays a critical role in bone destruction in
several types of inflammatory joint disease (30,31).
Particularly in RA, focal bone loss occurs due to excessive bone
resorption by osteoclasts. Moreover, bone formation by osteoblasts
is impaired at the bone erosion site. Thus, anti-TNF therapies
could delay the progression of bone destruction without bone
erosion in patients with RA, leading to significant clinical
improvement. TNFα in inflammatory arthritis has a destructive
function in all of the components that comprise joints, namely
osteoclasts, osteoblasts, and chondrocytes (32-34).
Blocking studies with an anti-TNFα agent in an
arthritis model have shown that TNFα may drive cartilage and bone
destruction (35,36). Treatment with TNFα inhibitors
decreased inflammation, as well as bone and cartilage damage, in
patients with RA (30,31). Although osteoclastogenesis is a more
dominant mechanism in the bone erosion and cartilage destruction of
inflammatory arthritis (8,34), there are fewer reports on the
pathological mechanisms by which TNFα and its inhibitors influence
physiological changes of hyaline cartilage in human knee
joints.
OA is a degenerative joint disease caused by the
degradation of hyaline cartilage. Cartilage loss in OA is
associated with aging and mechanical stress, as well as
inflammation. The primary cause of OA is aging and mechanical
stress, which suppress the regenerative capacity of chondrocytes
involved in extracellular matrix components comprising hyaline
cartilage. Moreover, total knee replacement for OA is a significant
health burden, and there are no therapeutic drugs available yet for
the regeneration of hyaline cartilage (37,38).
Thus, there is a significant unmet need for effective medical
therapies for OA. The present study demonstrated that TNFα
contributed to OA pathogenesis via NF-κB mechanisms affecting
metabolic imbalance in chondrocytes, leading to structural joint
damage (39). Similarly, it was
recently demonstrated that patients with OA exhibited a notable
response to anti-TNF therapy (40),
indicating a pivotal role for TNFα in cartilage catabolism. Thus,
TNF inhibitors may be a therapeutic option for patients with OA
with severe inflammation or TNFα-driven cartilage destruction.
In conclusion, the present study showed that TNFα
was associated with progressive destruction of hyaline cartilage in
OA. Mechanistically, chondrocyte exposure to TNFα increased
MMP1, MMP3 and MMP13 gene expression, causing
degradation of ACAN and COL2 structural proteins in hyaline
cartilage. During chondrogenic differentiation, TNFα stimulation
decreased the expression of COL2 and ACAN, but not
SOX9, thereby decreasing relative chondrogenic size; these
effects were reversed following treatment with anti-TNF agents.
Therefore, treatment with anti-TNF therapy may relieve chondrocyte
destruction in inflammatory arthritis.
Supplementary Material
TNFα dose-dependently activates MMP1,
3 and 13 expression in chondrocytes. Chondrocytes were treated with
TNFα for 24 h and analyzed by (A) RT-qPCR and (B) immunoblotting.
The results of qPCR did not reach statistical significance. phos,
phosphorylated; MMP, matrix metalloproteinase; TNFα, tumor necrosis
factor α; RT-qPCR, reverse transcription quantitative PCR.
TNFα upregulates MMP1, 3 and 13, and
downregulates TIMP1 and TIMP2 expression in chondrocytes. Raw
images for human MMP array. Significant differences are marked in
red compared with the vehicle. TIMP, TIMP metallopeptidase
inhibitor; MMP, matrix metalloproteinase; TNFα, tumor necrosis
factor α.
TNFα inhibits TIMP1 and TIMP2
expression in chondrocytes. Chondrocytes were treated with TNFα for
24 h and subjected to immunofluorescence to detect TIMP1 and TIMP2
expression. Representative images are shown. Scale bar, 50 μm.
TIMP, TIMP metallopeptidase inhibitor; TNFα, tumor necrosis factor
α.
NF-κB inhibitor suppresses
TNF-activated MMP expression in chondrocytes. Chondrocytes were
stimulated with vehicle or TNFα in the presence of high- or
low-dose BAY (NF-κB inhibitor) for 24 h and mRNA expression was
analyzed by RT-qPCR. The results of RT-qPCR did not reach
statistical significance. MMP, matrix metalloproteinase; TNFα,
tumor necrosis factor α; RT-qPCR, reverse
transcription-quantitative PCR.
Acknowledgements
The authors would like to thank Dr Dae Hyun Yoo, Dr
Sang-Cheol Bae and Dr Jae-Bum Jun of the Hanyang University
Hospital for Rheumatic Disease for helping to collect synovial
fluid from patients. Immunofluorescence images were analyzed by
confocal microscopy (Leica Microsystems GmbH) at Hanyang LINC
Analytical Equipment Center (Seoul, Korea).
Funding
Funding: The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) and funded by the Ministry of Science, ICT, and Future (grant
nos. NRF-2016R1A2B4008606 and 2019R1A2C2004214). It was also
supported by a Korea Health Technology R&D grant through the
Korea Health Industry Development Institute, which is funded by the
Ministry of Health and Welfare, Republic of Korea (grant no.
HI17C0888).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP, HP, YLL and SW performed all experiments. SJ,
BN, JHY, and YGK designed the experiments. JP, HP, and SJ analyzed
experimental data. JHY provided human knee joint samples. YGK and
BN provided synovial fluid of patients with OA and RA. SJ and THK
wrote the manuscript. JP, HP, and SJ confirm the authenticity of
all the raw data. THK conceptualized and supervised the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Studies involving human materials were performed in
compliance with the Helsinki Declaration and were approved by the
Ethics Committee of Hanyang University Hospital (approval no.
IRB-2017-05-003) and Hanyang University Guri Hospital (approval no.
2018-07-024). All subjects provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goldring MB: Articular cartilage
degradation in osteoarthritis. HSS J. 8:7–9. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Goldring MB and Marcu KB: Cartilage
homeostasis in health and rheumatic diseases. Arthritis Res Ther.
11(224)2009.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Langer R and Vacanti JP: Tissue
engineering. Science. 260:920–926. 1993.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mao AS and Mooney DJ: Regenerative
medicine: Current therapies and future directions. Proc Natl Acad
Sci USA. 112:14452–14459. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jiang S, Guo W, Tian G, Luo X, Peng L, Liu
S, Sui X, Guo Q and Li X: Clinical application status of articular
cartilage regeneration techniques: Tissue-engineered cartilage
brings new hope. Stem Cells Int. 2020(5690252)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Malda J, Groll J and van Weeren PR:
Rethinking articular cartilage regeneration based on a 250-year-old
statement. Nat Rev Rheumatol. 15:571–572. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mueller MB and Tuan RS: Anabolic/Catabolic
balance in pathogenesis of osteoarthritis: Identifying molecular
targets. PM R. 3 (Suppl 1):S3–S11. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Little CB, Flannery CR, Hughes CE, Mort
JS, Roughley PJ, Dent C and Caterson B: Aggrecanase versus matrix
metalloproteinases in the catabolism of the interglobular domain of
aggrecan in vitro. Biochem J. 344:61–68. 1999.PubMed/NCBI
|
|
9
|
Matyas JR, Ehlers PF, Huang D and Adams
ME: The early molecular natural history of experimental
osteoarthritis. I. Progressive discoordinate expression of aggrecan
and type II procollagen messenger RNA in the articular cartilage of
adult animals. Arthritis Rheum. 42:993–1002. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Park EH, Kim JS, Lee JS, Lee YJ, Song YW
and Lee EY: Compound K inhibits interleukin-1β-induced expression
of inflammatory mediators and matrix metalloproteinases by
inhibiting mitogen-activated protein kinase activation in
chondrocytes. J Rheumatic Dis. 25(188)2018.
|
|
11
|
Billinghurst RC, Dahlberg L, Ionescu M,
Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O,
Tschesche H, et al: Enhanced cleavage of type II collagen by
collagenases in osteoarthritic articular cartilage. J Clin Invest.
99:1534–1545. 1997.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Shlopov BV, Gumanovskaya ML and Hasty KA:
Autocrine regulation of collagenase 3 (matrix metalloproteinase 13)
during osteoarthritis. Arthritis Rheum. 43:195–205. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tetlow LC, Adlam DJ and Woolley DE: Matrix
metalloproteinase and proinflammatory cytokine production by
chondrocytes of human osteoarthritic cartilage: Associations with
degenerative changes. Arthritis Rheum. 44:585–594. 2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aigner T, Fundel K, Saas J, Gebhard PM,
Haag J, Weiss T, Zien A, Obermayr F, Zimmer R and Bartnik E:
Large-scale gene expression profiling reveals major pathogenetic
pathways of cartilage degeneration in osteoarthritis. Arthritis
Rheum. 54:3533–3544. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tracey KJ and Cerami A: Tumor necrosis
factor: A pleiotropic cytokine and therapeutic target. Annu Rev
Med. 45:491–503. 1994.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu G: Molecular mechanism and TNF
signaling and beyond. Cell Res. 15:24–27. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kammermann JR, Kincaid SA, Rumph PF, Baird
DK and Visco DM: Tumor necrosis factor-alpha (TNF-alpha) in canine
osteoarthritis: Immunolocalization of TNF-alpha, stromelysin and
TNF receptors in canine osteoarthritic cartilage. Osteoarthritis
Cartilage. 4:23–34. 1996.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Arend WP and Dayer JM: Inhibition of the
production and effects of interleukin-1 and tumor necrosis factor
alpha in rheumatoid arthritis. Arthritis Rheum. 38:151–160.
1995.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kapoor M, Martel-Pelletier J, Lajeunesse
D, Pelletier JP and Fahmi H: Role of proinflammatory cytokines in
the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 7:33–42.
2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jo S, Yoon S, Lee SY, Kim SY, Park H, Han
J, Choi SH, Han JS, Yang JH and Kim TH: DKK1 induced by 1,25D3 is
required for the mineralization of osteoblasts. Cells.
9(236)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jo S, Wang SE, Lee YL, Kang S, Lee B, Han
J, Sung IH, Park YS, Bae SC and Kim TH: IL-17A induces osteoblast
differentiation by activating JAK2/STAT3 in ankylosing spondylitis.
Arthritis Res Ther. 20(115)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Koontz L: TCA precipitation. Methods
Enzymol. 541:3–10. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kim H, Chung H, Kim HJ, Lee JY, Oh MY, Kim
Y and Kong G: Id-1 regulates Bcl-2 and Bax expression through p53
and NF-kappaB in MCF-7 breast cancer cells. Breast Cancer Res
Treat. 112:287–296. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lee JK, Jo S, Lee YL, Park H, Song JS,
Sung IH and Kim TH: Anterior cruciate ligament remnant cells have
different potentials for cell differentiation based on their
location. Sci Rep. 10(3097)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li X, Du W, Ma FX, Feng X, Bayard F and
Han ZC: High concentrations of TNF-α induce cell death during
interactions between human umbilical cord mesenchymal stem cells
and peripheral blood mononuclear cells. PLoS One.
10(e0128647)2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li P, Gan Y, Xu Y, Song L, Wang L, Ouyang
B, Zhang C and Zhou Q: The inflammatory cytokine TNF-α promotes the
premature senescence of rat nucleus pulposus cells via the PI3K/Akt
signaling pathway. Sci Rep. 7(42938)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kishimoto H, Akagi M, Zushi S, Teramura T,
Onodera Y, Sawamura T and Hamanishi C: Induction of hypertrophic
chondrocyte-like phenotypes by oxidized LDL in cultured bovine
articular chondrocytes through increase in oxidative stress.
Osteoarthritis Cartilage. 18:1284–1290. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Borzi RM, Olivotto E, Pagani S, Vitellozzi
R, Neri S, Battistelli M, Falcieri E, Facchini A, Flamigni F, Penzo
M, et al: Matrix metalloproteinase 13 loss associated with impaired
extracellular matrix remodeling disrupts chondrocyte
differentiation by concerted effects on multiple regulatory
factors. Arthritis Rheum. 62:2370–2381. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jung SM, Kim KW, Yang CW, Park SH and Ju
JH: Cytokine-mediated bone destruction in rheumatoid arthritis. J
Immunol Res. 2014(263625)2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Karmakar S, Kay J and Gravallese EM: Bone
damage in rheumatoid arthritis: Mechanistic insights and approaches
to prevention. Rheum Dis Clin North Am. 36:385–404. 2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Azuma Y, Kaji K, Katogi R, Takeshita S and
Kudo A: Tumor necrosis factor-alpha induces differentiation of and
bone resorption by osteoclasts. J Biol Chem. 275:4858–4864.
2000.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gilbert L, He X, Farmer P, Boden S,
Kozlowski M, Rubin J and Nanes MS: Inhibition of osteoblast
differentiation by tumor necrosis factor-alpha. Endocrinology.
141:3956–3964. 2000.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lofvall H, Newbould H, Karsdal MA,
Dziegiel MH, Richter J, Henriksen K and Thudium CS: Osteoclasts
degrade bone and cartilage knee joint compartments through
different resorption processes. Arthritis Res Ther.
20(67)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
van Schouwenburg PA, Rispens T and Wolbink
GJ: Immunogenicity of anti-TNF biologic therapies for rheumatoid
arthritis. Nat Rev Rheumatol. 9:164–172. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Scott DL and Kingsley GH: Tumor necrosis
factor inhibitors for rheumatoid arthritis. N Engl J Med.
355:704–712. 2006.
|
|
37
|
Bedair H, Cha TD and Hansen VJ: Economic
benefit to society at large of total knee arthroplasty in younger
patients: A Markov analysis. J Bone Joint Surg Am. 96:119–126.
2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Global Burden of Disease Study 2013
Collaborators. Global, regional, and national incidence,
prevalence, and years lived with disability for 301 acute and
chronic diseases and injuries in 188 countries, 1990-2013: A
systematic analysis for the Global Burden of Disease Study 2013.
Lancet. 386:743–800. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Chow YY and Chin KY: The role of
inflammation in the pathogenesis of osteoarthritis. Mediators
Inflamm. 2020(8293921)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chisari E, Yaghmour KM and Khan WS: The
effects of TNF-alpha inhibition on cartilage: A systematic review
of preclinical studies. Osteoarthritis Cartilage. 28:708–718.
2020.PubMed/NCBI View Article : Google Scholar
|