Introduction
The initial stage of cardiac hypertrophy is an
adaptive response in volume related to hypertension, valvular
disease or neurohumoral stimuli. The decompensated stage of
pathological hypertrophy leads to contractile dysfunction and heart
failure (HF) (1,2). Mitochondria serve crucial biological
roles in excitation-contraction coupling in cardiac muscle,
cellular metabolism, HF progression and myocardial
ischemia/reperfusion (3-5).
It has been hypothesized that abnormalities in mitochondrial
function and endoplasmic reticulum (ER) may be related to
cardiomyopathy (6,7).
Increasing evidence suggests that mitochondria and
the ER are organelles that form an endomembrane network.
Mitochondria-associated membranes (MAM) appear to be the contact
point through which the ER directly communicates with mitochondria
(8-10).
The ER-mitochondria interface is enriched in various proteins, such
as the macromolecular complex composed of voltage-dependent anion
channels 1 (VDAC1), glucose regulated protein 75 (GRP75) and
inositol 1,4,5-triphosphate receptor (IP3R), which regulate
Ca2+ transfer from the ER to mitochondria (11,12).
The crosstalk between these organelle and MAM in cardiomyopathy
remains unknown and requires further investigation.
Polyamines, such as putrescine, spermidine and
spermine, are involved in cell proliferation and survival and in
numerous biological and pathological processes, including
apoptosis, regulation of Kir channels and oxidative stress
(13-15).
Previous studies have reported that elevated polyamine levels are
associated with cardiac hypertrophy (16,17).
Certain stimuli, such as growth factors and oncogenes (hormones,
Myc and Ras) can rapidly increase ornithine decarboxylase (ODC)
protein level, which is a key enzyme involved in cellular polyamine
biosynthesis (18,19). Difluoromethylornithine (DFMO) is an
ornithine analog that has previously been reported to deplete
cellular polyamines through inhibition of ODC and to promote
cardioprotection by attenuating apoptosis and the NO/cGMP-dependent
protein kinase-1 pathway (17,20,21).
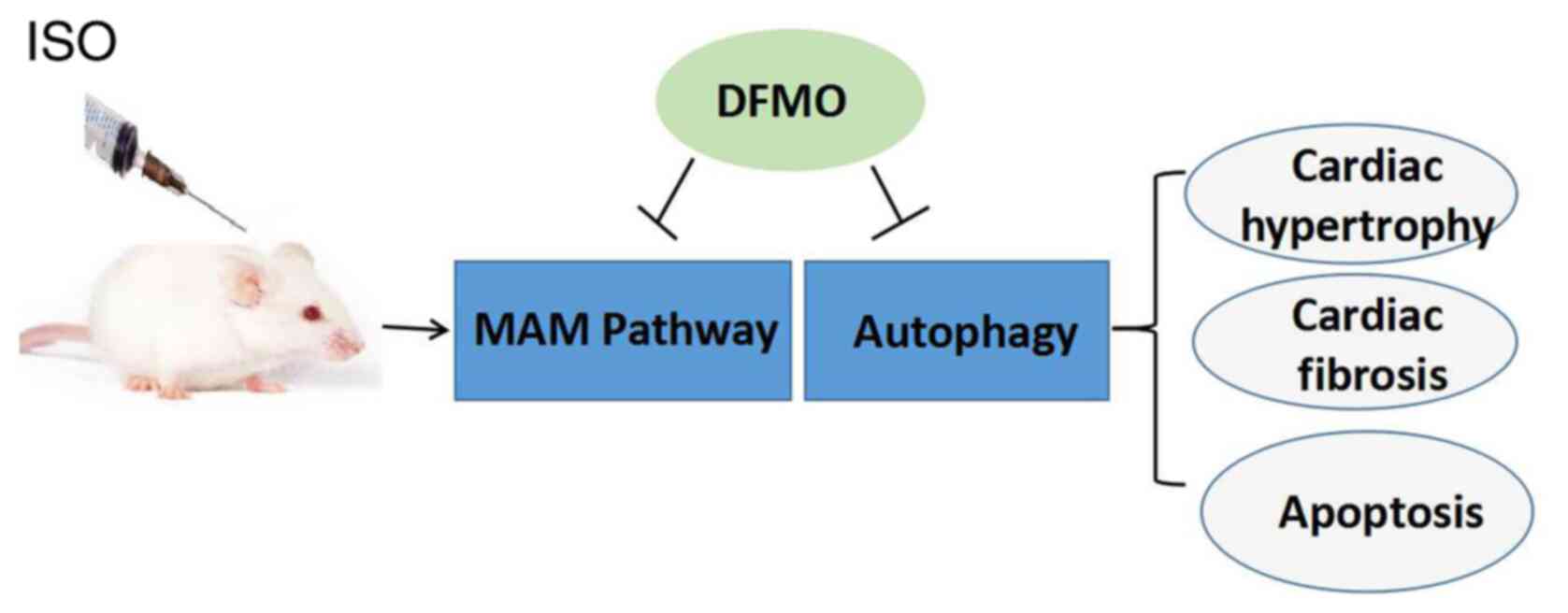
Subsequently, the present study investigated whether isoproterenol
(ISO)-induced cardiomyopathy could be associated with increased
apoptosis and MAM. In addition, this study examined if DFMO could
be considered as an effective pharmacological method to attenuate
ISO-induced cardiomyopathy and if this possible effect may be
mediated by MAM signaling pathway regulation. In the present study,
it was hypothesized that cardiac hypertrophy induced by ISO was
associated with apoptosis and MAM and polyamine depletion by DFMO
exerts a protective effect on cardiac hypertrophy.
Materials and methods
Chemicals and reagents
ISO (cat. no. 51-30-9) and DFMO (cat. no.
70052-12-9) were both purchased from Sigma-Aldrich; Merck KGaA. The
TUNEL detection kit was purchased from Roche Diagnostics GmbH. The
primary antibodies against cleaved caspase-3/9, caspase-3/9, VDAC1,
GRP75, cyclophilin D (CypD), mitofusin 2 (Mfn2), autophagy related
5 (Atg5) and Beclin1 were obtained from Santa Cruz Biotechnology,
Inc. TRIzol reagent and a PrimeScript RT Reagent kit were from
Takara Biotechnology. Protein extraction kit and GAPDH antibody
(both Wuhan Boster Biological Technology, Ltd.) and Masson's
trichrome staining kit (Beijing Solarbio Science & Technology
Co., Ltd.) were also purchased.
Animals
Healthy male Wistar rats weighing 200-250 g were
used in the present study (Qiqihar Medicine University Animal
Center). Rats were housed on a 12 h dark-light cycle at 23±1˚C. The
study protocol was approved by the Institutional Animal Research
Committee (approval no. QMU-AECC-2019-53).
Rat cardiac hypertrophy model and drug
treatment
As described previously (15), rats were randomly divided into six
groups (n=10 per group) as follows: i) Control group, rats were
given subcutaneous injections of 0.9% saline; ii) ISO1d group, rats
were given ISO (5 mg/kg/day) for 1 day; iii) ISO3d group, rats were
given ISO (5 mg/kg/day) for 3 days; iv) ISO7d group, rats were
given ISO (5 mg/kg/day) for 7 days; v) ISO14d group, rats were
given ISO (5 mg/kg/day) for 14 days; and vi) DFMO treatment group,
rats were given ISO (5 mg/kg/day) for 14 days and 2% DFMO in their
water for 4 weeks. Following treatment, all rats were euthanized
using 3% pentobarbital sodium (80 mg/kg) and hearts were collected
for further experiments. The hearts were carefully isolated,
excised and weighed in cold buffer. The ventricles were separated
and weighed and the degree of ventricular hypertrophy was assessed
by evaluating the heart-to-body weight (HW/BW) ratio and left
ventricle-to-body weight (LVW/BW) ratio.
Quantification of fibrosis
Heart tissue samples were cut and fixed in 10%
formalin for 2-3 days and embedded in paraffin at room temperature.
To evaluate morphological changes, tissue samples were stained with
Masson's trichrome reagent at room temperature for 120-160 min. A
light microscope was used to visualize the cardiac sections at x200
magnification. The degree of collagen deposition was analyzed using
Image-Pro Plus v6.0 (Media Cybernetics, Inc.). A total of ≥100
myocardial cells in each group was analyzed.
Reverse transcription quantitative
(RT-q)PCR
As previously described (22,23),
total RNA was isolated using TRIzol according to the manufacturer's
instruction (Invitrogen; Thermo Fisher Scientific, Inc.) from left
ventricular tissues at room temperature. A cDNA synthesis kit
(Takara Biotechnology Inc.) and oligo(dT) primers were subsequently
used to synthesize the first strand cDNA. RT-qPCR was performed
using PowerUp SYBR-Green Master Mix and the results were analyzed
using the StepOne™ (Thermo Fisher Scientific, Inc.) system with
GAPDH as an internal control. The primers for ANP (a marker gene of
cardiac hypertrophy) were as follows: forward
5'-GGGAAGTCAACCCGTCTCA-3' and reverse 5'-GGGCTC CAATCCTGTCAAT-3'.
The primers for GAPDH were as follows: Forward,
5'-GAGACAGCCGCATCTTCTTG-3' and reverse, 5'-ATACGGCCAAATCCGTTCAC-3'.
The thermocycling conditions were as follows: Initial denaturation
at 95˚C for 30 sec; 40 cycles of annealing at 95˚C for 5 sec and
extension at 60˚C for 30 sec. The relative expression levels were
normalized to endogenous control and were expressed as
2-ΔΔCq (20).
Western blotting
Harvested left ventricle tissues were frozen in
liquid nitrogen and stored at -80˚C until prepared as described
previously (19). Heart tissues
were homogenized in RIPA lysis buffer containing protease and
phosphate inhibitors on ice (cat. no. P0013B; Beyotime Institute of
Biotechnology) and centrifuged at 12,000 x g for 20 min at 4˚C. The
protein concentration was determined using a BCA protein assay.
Proteins samples were separated by electrophoresis in 10% SDS-PAGE
gels (20 µg protein/lane) and transferred onto PVDF membranes.
After blocking with 5% skimmed milk in Tris-buffered saline for 1.5
h at 25˚C, membranes were incubated with primary antibodies against
GRP75 (1:1,000; cat. no. ab171089), VDAC1 (1:500; cat. no.
ab14734), CypD (1:500; cat. no. ab231155), Mfn2 (1:500; cat. no.
11925S), GAPDH (1:1,000; cat. no. ab8245), cleaved caspase-3
(1:1,000; cat. no. 9661S), cleaved caspase-9 (1:1,000; cat. no.
20750S), Atg5 (1:500; cat. no. 2630S) and Beclin1 (1:500; cat. no.
3738S) overnight at 4˚C. The membrane was washed three times in
TBST and incubated with horseradish peroxidase-conjugated secondary
antibody (1:5,000; cat. no. BA1058; Wuhan Boster Biological
Technology, Ltd.) in TBST for 1 h at 25˚C. The bands were detected
using ECL chemiluminescence kit (cat. no. P0018M; Beyotime
Institute of Biotechnology). Membranes were stripped with stripping
buffer (cat. no. P0025; Beyotime Institute of Biotechnology) and
re-probed with different antibodies. Relative expression levels
were normalized to endogenous control GAPDH using ImageJ software
(version 1.4.3.67; National Institutes of Health).
TUNEL staining
Cell apoptosis was assessed using TUNEL assay
according to the manufacturers' instructions. The sections of heart
tissue were cut into 6 µm slices and then dewaxed. After xylene
dewaxing and ethanol dehydration, the cardiac tissue sections were
incubated with 20 µg/ml proteinase K for 20 min and
fluorescein-labelled dUTP for 1 h at 37˚C. Fluorescence microscopy
(x200 magnification; Olympus Corporation) was used to analyze ≥100
myocardial cells in each group. Image-Pro Plus 6.0 (Media
Cybernetics, Inc.) was used to calculate the results.
Immunohistochemical staining
Cardiomyocyte apoptosis was evaluated using
caspase-3 and caspase-9 immunohistochemistry. Heart sections were
cut into 4 µm slices according to the protocol using the citric
acid method (Citrate Antigen Retrieval Solution; cat. no. P0081;
Beyotime Institute of Biotechnology). Sections were blocked with 8%
goat serum for 30 min at 37˚C and were incubated with caspase-3
(1:100; cat. no. ab13847, Santa Cruz Biotechnology, Inc.) and
caspase-9 (1:100; cat. no. ab52298, Santa Cruz Biotechnology, Inc.)
antibodies at 37˚C for 2 h. EnVision™+/HRP (cat. no. K400011-2;
Dako; Agilent Technologies, Inc.) was added to sections for 1 h at
37˚C. Samples were processed using a peroxide-based substrate
diaminobenzidine kit (Gene Tech Biotechnology Co., Ltd.) and nuclei
were stained using haematoxylin for 1 sec at room temperature.
Finally, heart sections were photographed using a light microscope
(magnification, x200). The results were analyzed using Image-Pro
Plus 6.0 (Media Cybernetics, Inc.).
Statistical analysis
All data were presented as the means ± standard
error of the mean. One-way ANOVA followed by a Dunnett's or
Student-Newman-Keuls post hoc tests were used for data analysis
with GraphPad Prism 5.00 software (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
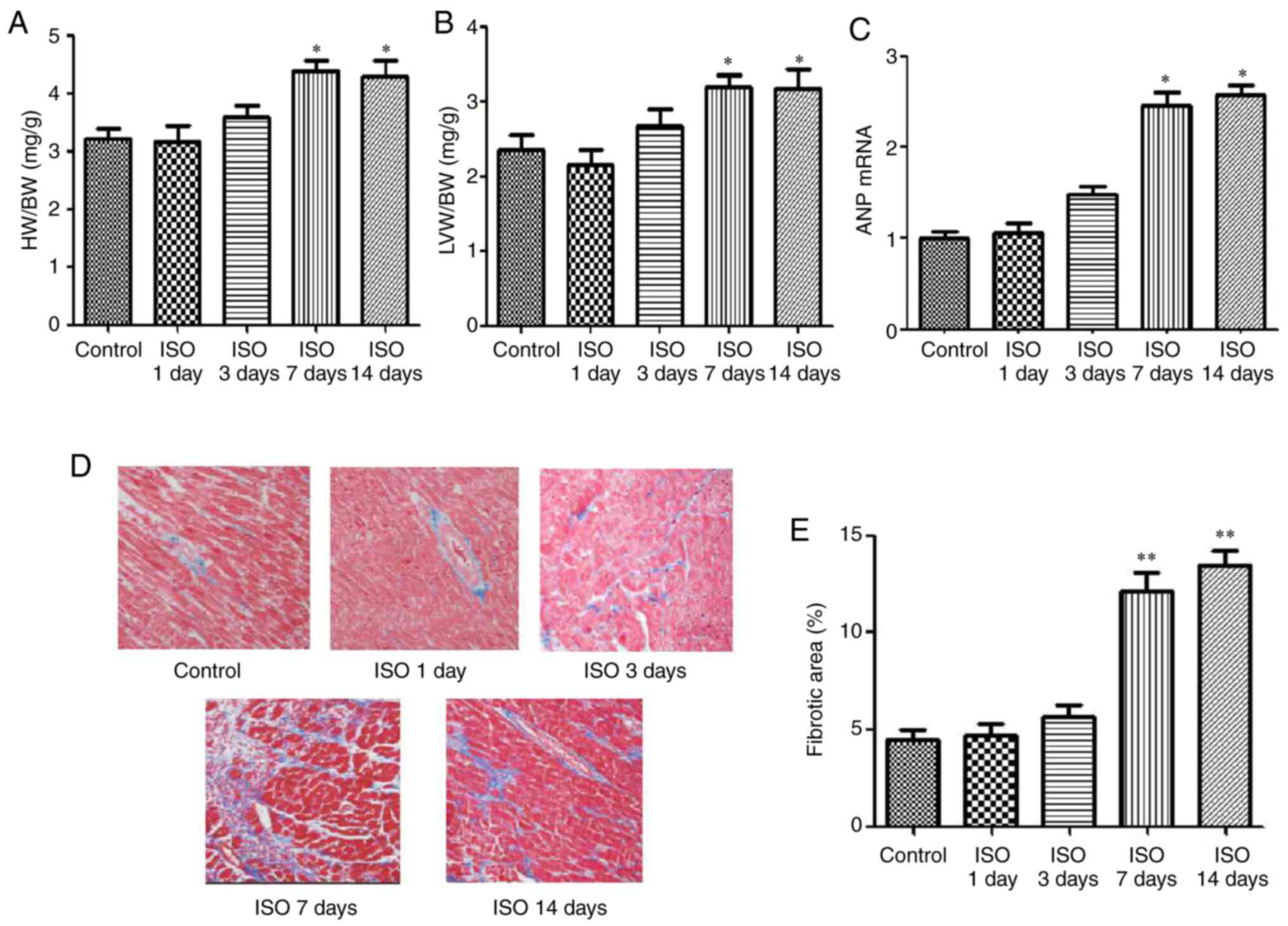
Effect of ISO on cardiac hypertrophy
and cardiac fibrosis
To determine the effects of ISO on the development
of cardiomyocyte hypertrophy, the cardiac hypertrophy model induced
by ISO was established. Parameters of cardiac hypertrophy, such as
HW/BW ratio, LVW/BW ratio, ANP mRNA expression were evaluated at 1,
3, 7 and 14 days following ISO injection. As presented in Fig. 1, the ratios of HW/BW and LVW/BW, the
expression of ANP mRNA (Fig. 1A-C)
and the areas of collagen deposition in the interstitial and
perivascular (Fig. 1D and E) were significantly increased in the
ISO7d and ISO14d groups compared with the control group (P<0.05
and P<0.01).
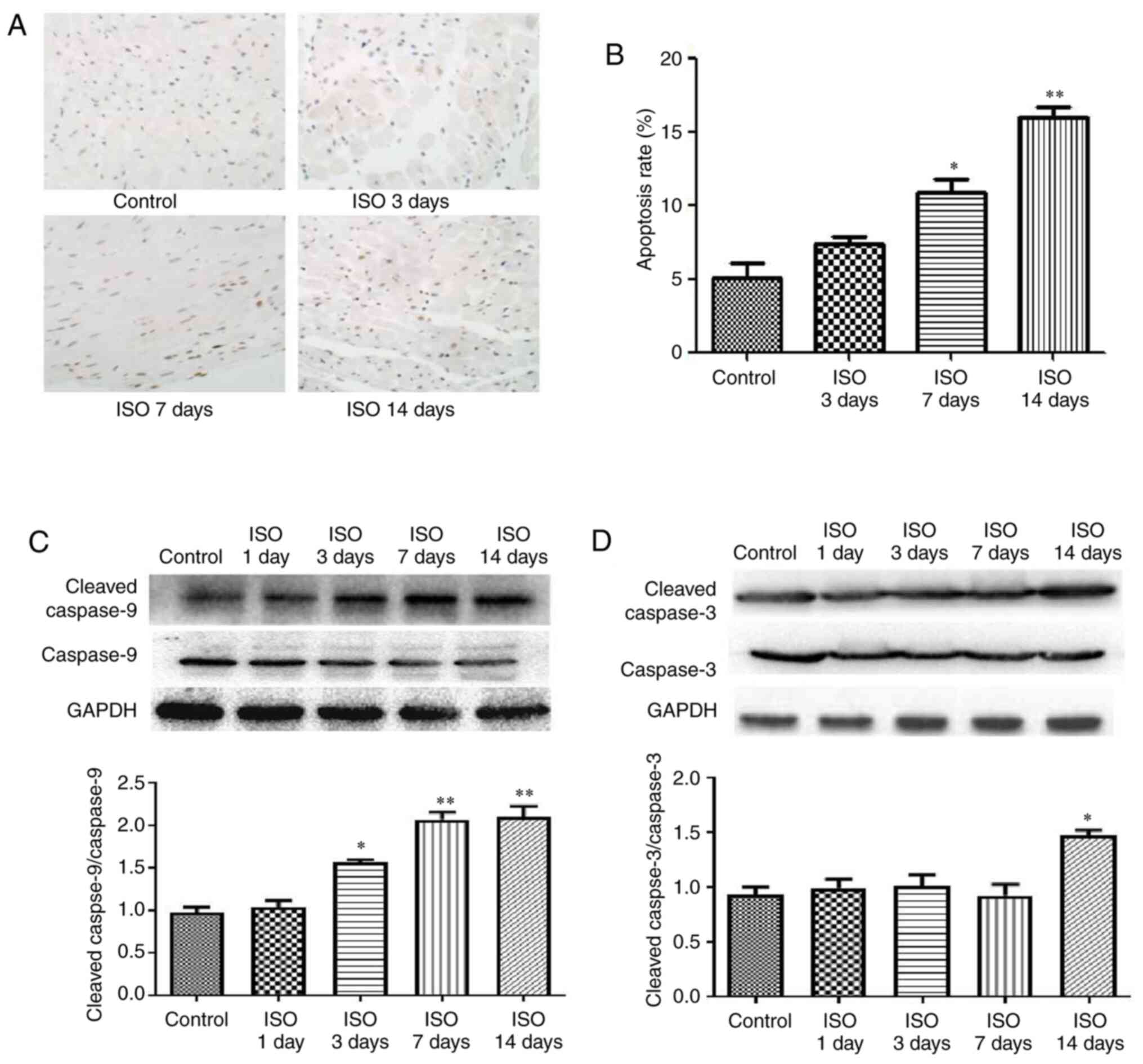
Effect of ISO on myocardial
apoptosis
Cardiomyocyte apoptosis is now considered as a
hallmark of heart failure (24).
Using a TUNEL assay, cardiac apoptotic cells are shown by
brown-stained nuclei whereas non-apoptotic cells are stained
blue-green or tan shades. In the ISO7d and ISO14d groups, the
apoptosis rates were significantly increased compared with the
control group (P<0.05 and P<0.01, respectively; Fig. 2A and B). Furthermore, the ratios cleaved
caspase-9/caspase-9 was increased in the ISO3d, ISO7d and ISO14d
groups, and the ratio cleaved caspase-3/caspase-3 was significantly
increased in the ISO 14d group compared with the control group
(P<0.05 and P<0.01; Fig. 2C
and D).
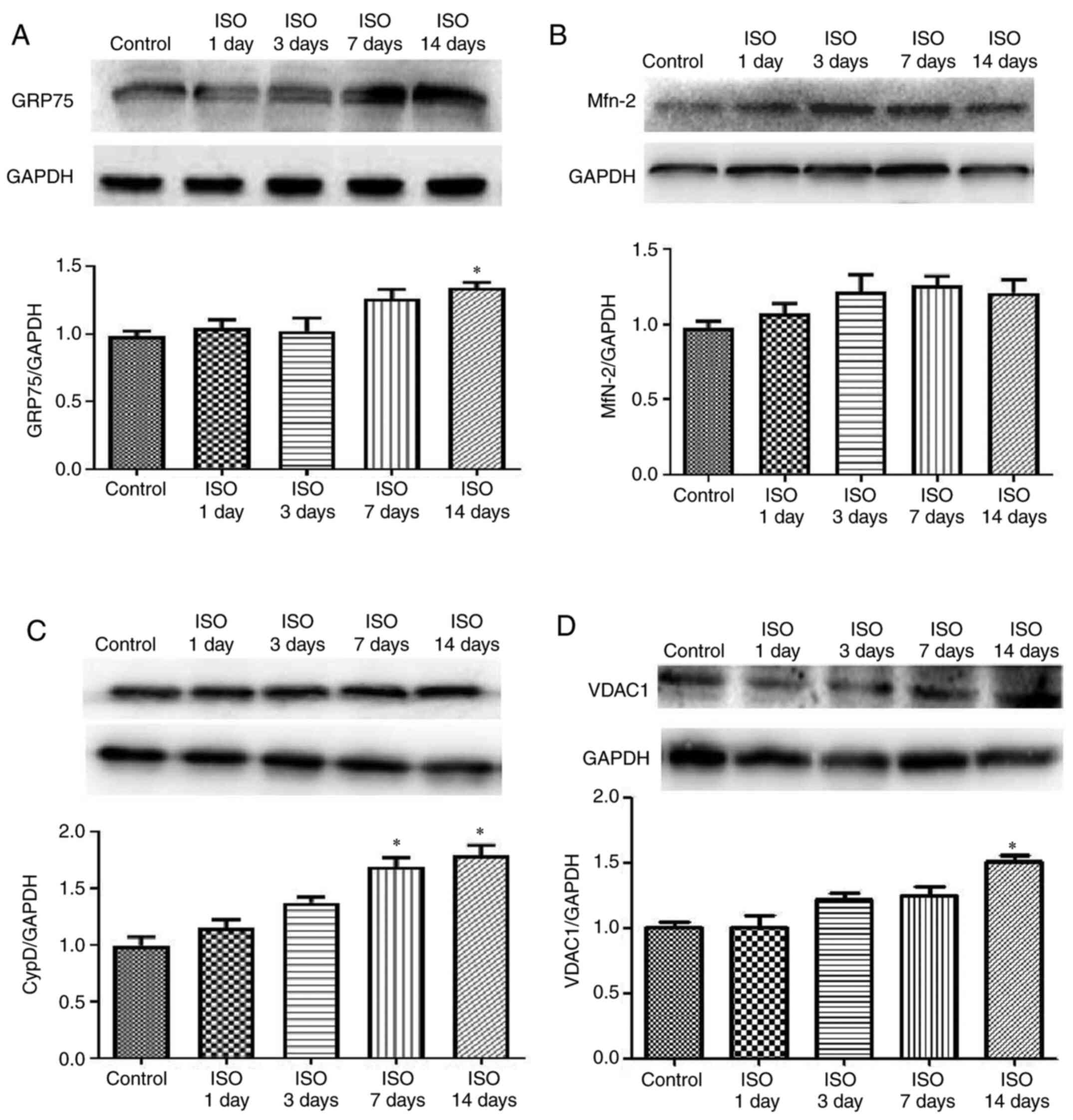
Effect of ISO on MAM pathway
It is well-known that MAMs regulate physiological
functions between ER and mitochondria to maintain Ca2+
flux and lipid and metabolite exchange (25,26).
To investigate the effect of ISO on myocardial cell MAM signal
transduction, the expression of GRP75, VDAC1, Mfn2 and CypD was
determined. The results demonstrated that GRP75 expression was
increased in ISO14d group (P<0.05; Fig. 3A) whereas Mfn2 expression was
unchanged in ISO groups (Fig. 3B).
The protein expression of CypD was increased in ISO7d and ISO14d
groups (P<0.05; Fig. 3C) and the
protein expression of VDAC 1 was increased in ISO14d group
(P<0.05; Fig. 3D) compared with
control group.
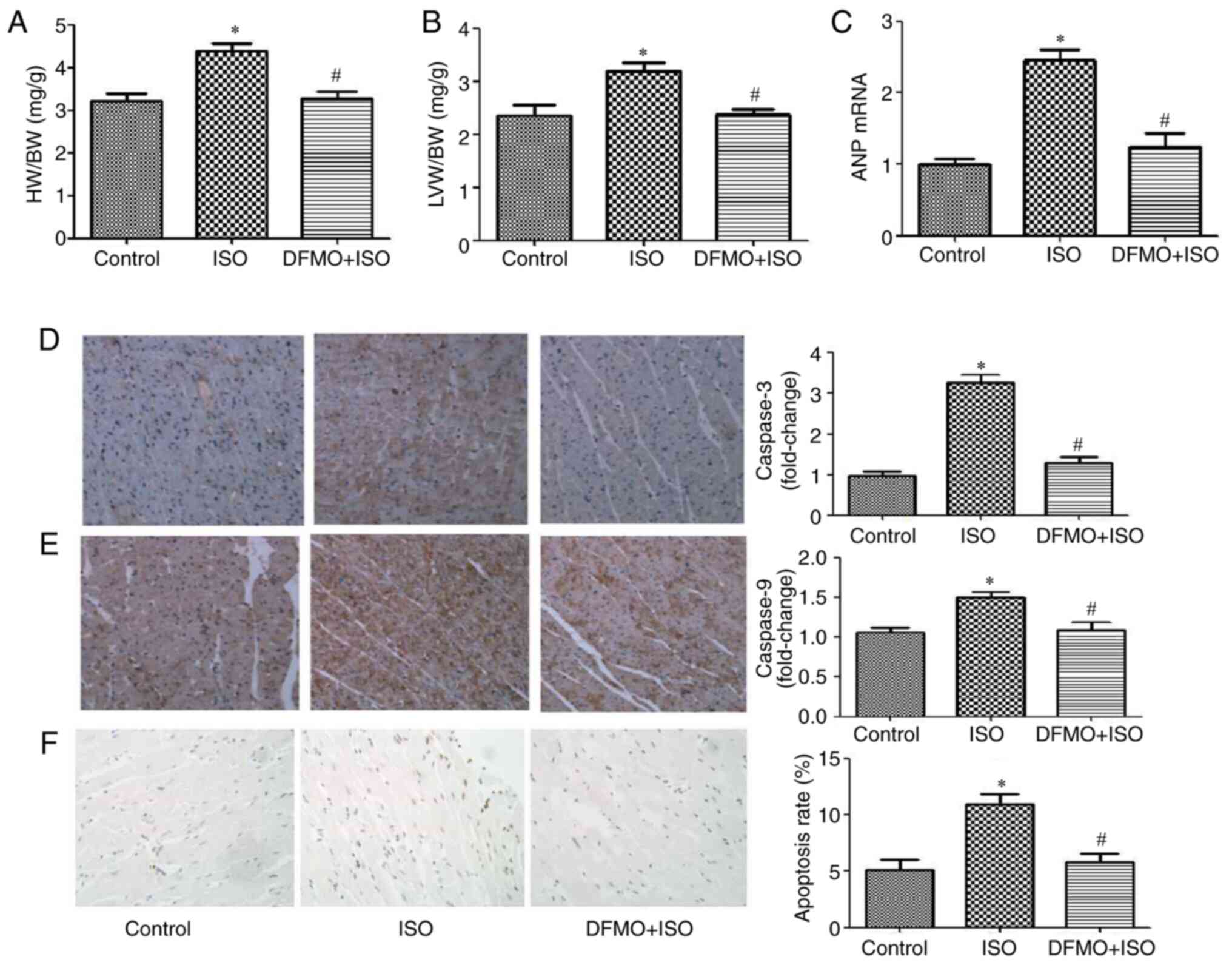
DFMO alleviates myocardial hypertrophy
and apoptosis induced by ISO
Cardiac hypertrophy and apoptosis were measured to
assess the cytoprotective effect of DFMO. The ratios HW/BW and
LVW/BW and the mRNA expression of ANP were significantly decreased
in the DFMO treatment group compared with the ISO14d group
(P<0.05; Fig. 4A-C).
Furthermore, caspase-3 and caspase-9 protein expression was
increased in ISO group compared with control group, which was
reversed following DFMO treatment (Fig.
4D and E). In addition, the
apoptosis rate was significantly decreased following DFMO treatment
compared with the ISO group (P<0.05; Fig. 4F). These findings indicated that
DFMO may alleviate ISO-induced myocardial hypertrophy and
myocardial apoptosis.
DFMO downregulates GRP75, VDAC1 and
CypD and upregulates cardiomyocyte autophagy
The effects of DFMO on the MAM pathway and autophagy
were evaluated in ISO-treated rats. The results demonstrated that
in rats with ISO-induced myocardial hypertrophy, DFMO could
downregulate the expression of GRP75, VDAC1 and CypD and upregulate
the expression of the autophagy-associated proteins Atg5 and
Beclin1, leading to an attenuation of ISO effects (P<0.05 and
P<0.01; Fig. 5A-E).
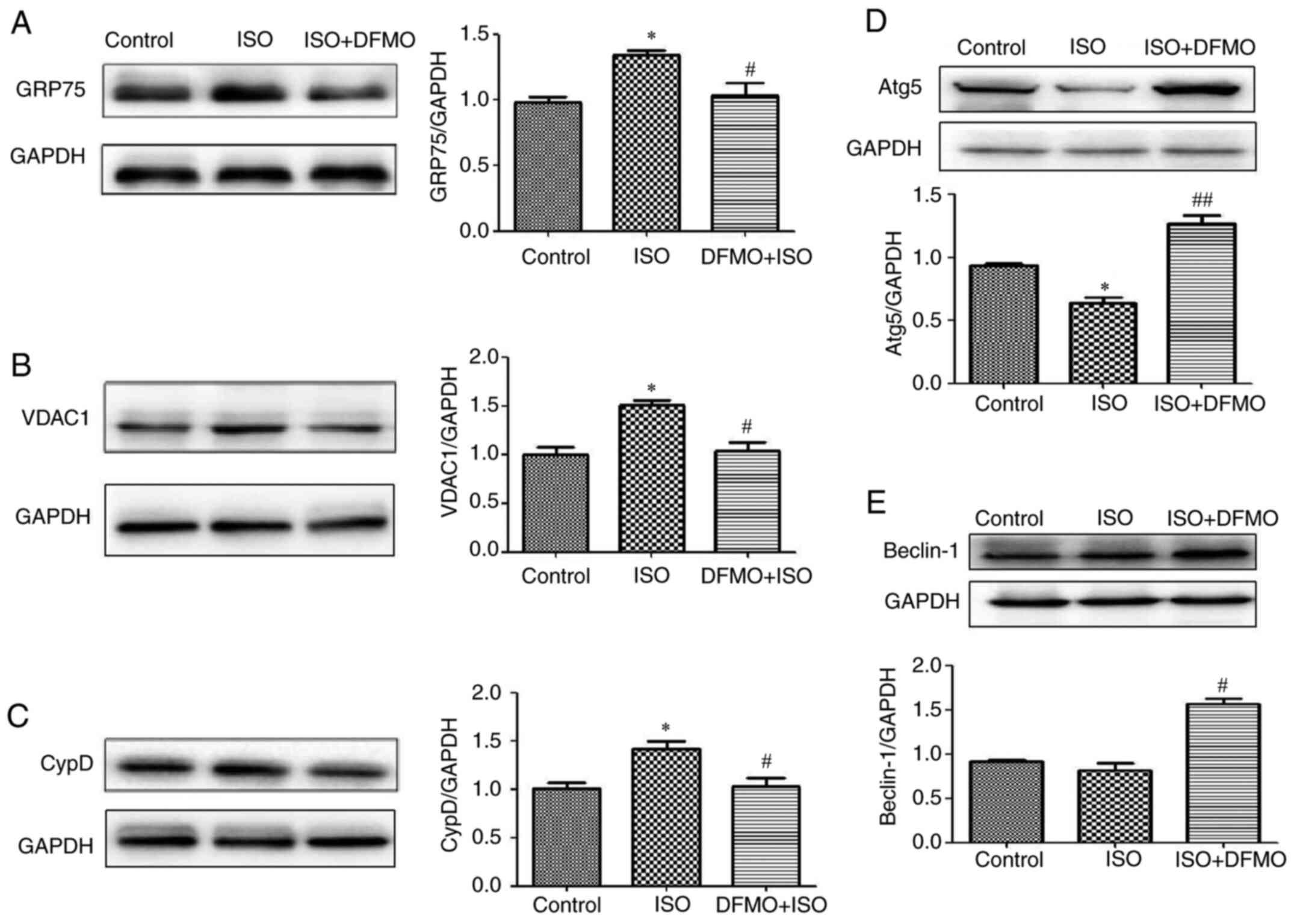 | Figure 5Effect of DFMO on
mitochondria-associated membranes pathway proteins and autophagy
proteins following treatment with ISO in control, ISO and DFMO
pretreatment groups. Protein expression of (A) GRP75, (B) VDAC1,
(C) CypD, (D) Atg5 and (E) Beclin-1 was detected by western
blotting. *P<0.05 vs. control group;
#P<0.05 and ##P<0.01 vs. ISO group.
GRP75, glucose regulated protein 75; Mfn2, mitofusin2; CypD,
cyclophilin D; VDAC1, voltage-dependent anion channels 1; ISO,
isoproterenol; DFMO, difluoromethylornithine. |
Discussion
The present study demonstrated that DFMO could
significantly attenuate ISO-induced cardiac hypertrophy and
apoptosis and downregulate the MAM pathway while increasing
autophagy. It is widely known that β-adrenergic receptor activation
maintains cardiac output (27), but
that sustained stimulation results in heart failure (5,28). The
present study demonstrated that cardiac hypertrophy, fibrosis and
apoptosis were induced following ISO treatment. Cardiomyocyte
apoptosis is involved in the pathogenesis and progression of
cardiac dysfunction during heart failures, such as myocardial
infarction, HF and ischemia reperfusion (26,27).
Previous studies have suggested that increased cardiomyocyte
apoptosis is implicated in the pathogenesis and development of
cardiovascular complications (28-30).
Accumulating evidence showed that MAMs, including
VDAC, GRP75 and CypD, are fundamental for cellular homeostasis to
support mitochondrial Ca2+ signaling (31-33).
Previous studies have reported that mitochondria are critical
integrators of signal transduction during the development of
cardiac hypertrophy (34,35). It has been demonstrated that VDAC,
which is associated with the outer mitochondrial membrane,
interacts with IP3R on the ER through the molecular chaperone GRP75
that is thought to participate in the refolding of proteins
translocated into this organelle (36). VDAC1 may be associated with both
mPTP opening and mitochondrial membrane depolarization.
Importantly, VDAC1 is upregulated during cardiac hypertrophy
(37,38). The results from the present study
indicated that MAM pathway may play a key role during cardiac
hypertrophy, and that downregulation of the MAM pathway may be
cardioprotective.
Polyamines (Pas) levels are tightly regulated by
ODC, which is the key enzyme controlling the biosynthesis of PAs. A
previous study from our group has demonstrated that DFMO could
deplete cellular polyamines via inhibition of ODC (19) and attenuate cardiomyocyte
hypertrophy by inhibiting the NO/cGMP-dependent protein kinase-1
pathway and ERS pathway in vivo (20). The findings from the present study
indicated DFMO treatment may prevent ISO-induced cardiac
hypertrophy, fibrosis and cardiomyocyte apoptosis, and downregulate
the MAM pathway.
Autophagy maintains cellular homeostasis and
regulates apoptosis by degrading proteins with long half-lives or
damaging organelles (39,40). The complexes formed by ATG5 and
Beclin-1 proteins are the core molecular machinery for activation
of the autophagy pathway (41).
Previous studies have reported that MAMs serve a crucial role in
autophagy. Beclin1 is a proautophagic protein that forms at
specific regions of MAM in starvation-induced autophagy (42,43).
To explore the effect of DFMO on autophagy, the present study
evaluated the expression of Atg5 and Beclin1. The results
demonstrated that Atg5 expression was significantly decreased in
ISO-induced hypertrophic rat hearts, and that DFMO could
significantly increase the expression of Atg5 and Beclin1. We
therefore hypothesized that Atg5 and Beclin1 may promote the
survival of cardiomyocytes and inhibit hypertrophy.
In summary, the present study used an animal model
of ISO-induced cardiac hypertrophy to assess the protective effect
of DFMO. In ISO-treated rats, the expression of MAM pathway
proteins was upregulated and associated with ISO-induced cardiac
hypertrophy. Importantly, DFMO was demonstrated to ameliorate
cardiac hypertrophy via inhibition of the MAM pathway and
activation of the autophagy pathway (Fig. 6) One potential limitation of this
study would be that the effect of DFMO on heart function, including
ejection fraction and fractional shortening using echocardiography,
was not evaluated. Further investigation is therefore needed to
understand the relationship between MAM and the cardiac function
following treatment with DFMO. The findings from the present study
suggested that DFMO may be considered as a potential therapeutic
agent to treat cardiac hypertrophy.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the National Natural
Science Foundation of China (grant nos. 82074148, 31751004), the
Natural Science Foundation of Heilongjiang Province (grant no.
H2017080) and the Postdoctoral Scientific Research Foundation of
Heilongjiang Province (grant no. LBH-Q18128).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and YL designed the experiments and wrote and
revised the manuscript. WWJ, SR and WX performed the experiments
and analyzed the data. GWL and LJ carried out the experiments and
revised the manuscript. All authors read and approved the final
version. YL and GWL confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The experimental protocols were approved by the
Committee on the Ethics of Animal Experiments of Qiqihar Medical
University, China (approval no. QMU-AECC-2019-53).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Branco AF, Sampaio SF, Wieckowski MR,
Sardão VA and Oliveira PJ: Mitochondrial disruption occurs
downstream from - adrenergic overactivation by isoproterenol in
differentiated, but not undifferentiated H9c2 cardiomyoblasts:
Differential activation of stress and survival pathways. Int J
Biochem Cell Biol. 45:2379–2391. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Berenji K, Drazner MH, Rothermel BA and
Hill JA: Does load-induced ventricular hypertrophy progress to
systolic heart failure? Am J Physiol Heart Circ Physiol.
289:H8–H16. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Forte M, Schirone L, Ameri P, Basso C,
Catalucci D, Modica J, Chimenti C, Crotti L, Frati G, Rubattu S, et
al: The role of mitochondrial dynamics in cardiovascular diseases.
Br J Pharmacol. 15:1–17. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wyss RK, Méndez-Carmona N, Sanz MN, Arnold
M, Segiser A, Fiedler GM, Carrel TP, Djafarzadeh S, Tevaearai
Stahel HT and Longnus SL: Mitochondrial integrity during early
reperfusion in an isolated rat heart model of donation after
circulatory death-consequences of ischemic duration. J Heart Lung
Transplant. 38:647–657. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dudek J, Hartmann M and Rehling P: The
role of mitochondrial cardiolipin in heart function and its
implication in cardiac disease. Biochim Biophys Acta Mol Basis Dis.
1865:810–821. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Raut GK, Manchineela S, Chakrabarti M,
Bhukya CK, Naini R, Venkateshwari A, Reddy VD, Mendonza JJ, Suresh
Y, Nallari P, et al: Imine stilbene analog ameliorate
isoproterenol-induced cardiac hypertrophy and hydrogen
peroxide-induced apoptosis. Free Radic Biol Med. 153:80–88.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang S, Binder P, Fang Q, Wang Z, Xiao W,
Liu W and Wang X: Endoplasmic reticulum stress in the heart:
Insights into mechanisms and drug targets. Br J Pharmacol.
175:1293–1304. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rieusset J: Mitochondria and endoplasmic
reticulum: Mitochondria-endoplasmic reticulum interplay in type 2
diabetes pathophysiology. Int J Biochem Cell Biol. 43:1257–1262.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Monteiro JP, Oliveira PJ and Jurado AS:
Mitochondrial membrane lipid remodeling in pathophysiology: A new
target for diet and therapeutic interventions. Prog Lipid Res.
52:513–528. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tang LL, Wang JD, Xu TT, Zhao Z, Zheng JJ,
Ge RS and Zhu DY: Mitochondrial toxicity of perfluorooctane
sulfonate in mouse embryonic stem cell-derived cardiomyocytes.
Toxicology. 382:108–116. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sironi L, Restelli LM, Tolnay M, Neutzner
A and Frank S: Dysregulated interorganellar crosstalk of
mitochondria in the pathogenesis of Parkinson's disease. Cells.
9(233)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Murata D, Arai K, Iijima M and Sesaki H:
Mitochondrial division, fusion and degradation. J Biochem.
167:233–241. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ou Y, Wang SJ, Li D, Chu B and Gu W:
Activation of SAT1 engages polyamine metabolism with p53-mediated
ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Pegg AE: Functions of polyamines in
mammals. J Biol Chem. 291:14904–14912. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Murray Stewart T, Dunston TT, Woster PM
and Casero RA Jr: Polyamine catabolism and oxidative damage. J Biol
Chem. 293:18736–18745. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen M, Xin J, Liu B, Luo L, Li J, Yin W
and Li M: Mitogen-activated protein kinase and intracellular
polyamine signaling is involved in TRPV1 activation-induced cardiac
hypertrophy. J Am Heart Assoc. 5(e003718)2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lin Y, Zhang X, Wang L, Zhao Y, Li H, Xiao
W, Xu C and Liu J: Polyamine depletion attenuates
isoproterenol-induced hypertrophy and endoplasmic reticulum stress
in cardiomyocytes. Cell Physiol Biochem. 34:1455–1465.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Casero RA Jr, Murray Stewart T and Pegg
AE: Polyamine metabolism and cancer: Treatments, challenges and
opportunities. Nat Rev Cancer. 18:681–695. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lin Y, Liu JC, Zhang XJ, Li GW, Wang LN,
Xi YH, Li HZ, Zhao YJ and Xu CQ: Downregulation of the ornithine
decarboxylase/polyamine system inhibits angiotensin-induced
hypertrophy of cardiomyocytes through the NO/cGMP-dependent protein
kinase type-I pathway. Cell Physiol Biochem. 25:443–450.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pegg AE: Regulation of ornithine
decarboxylase. J Biol Chem. 281:14529–14532. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lin Y, Zhang X, Xiao W, Li B, Wang J, Jin
L, Lian J, Zhou L and Liu J: Endoplasmic reticulum stress is
involved in DFMO attenuating isoproterenol-induced cardiac
hypertrophy in rats. Cell Physiol Biochem. 38:1553–1562.
2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jiang G, Gong H, Niu Y, Yang C, Wang S,
Chen Z, Ye Y, Zhou N, Zhang G, Ge J, et al: Identification of amino
acid residues in angiotensin ii type 1 receptor sensing mechanical
stretch and function in cardiomyocyte hypertrophy. Cell Physiol
Biochem. 37:105–116. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang W, Zhang H, Xue G, Zhang L, Zhang W,
Wang L, Lu F, Li H, Bai S, Lin Y, et al: Exercise training
preserves ischemic preconditioning in aged rat hearts by restoring
the myocardial polyamine pool. Oxid Med Cell Longev.
2014(457429)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Braunwald E: Heart failure. JACC Heart
Fail. 1:1–20. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Perrone M, Caroccia N, Genovese I,
Missiroli S, Modesti L, Pedriali G, Vezzani B, Vitto VAM, Antenori
M, Lebiedzinska-Arciszewska M, et al: The role of
mitochondria-associated membranes in cellular homeostasis and
diseases. Int Rev Cell Mol Biol. 350:119–196. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Imaeda A, Tanaka S, Tonegawa K, Fuchigami
S, Obana M, Maeda M, Kihara M, Kiyonari H, Conway SJ, Fujio Y, et
al: Myofibroblast β2 adrenergic signaling amplifies cardiac
hypertrophy in mice. Biochem Biophys Res Commun. 510:149–155.
2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
de Lucia C, Eguchi A and Koch WJ: New
insights in cardiac β-adrenergic signaling during heart failure and
aging. Front Pharmacol. 9(904)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ferrara N, Komici K, Corbi G, Pagano G,
Furgi G, Rengo C, Femminella GD, Leosco D and Bonaduce D:
β-adrenergic receptor responsiveness in aging heart and clinical
implications. Front Physiol. 4(396)2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ramani D, De Bandt JP and Cynober L:
Aliphatic polyamines in physiology and diseases. Clin Nutr.
33:14–22. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li Y and Liu X: The inhibitory role of
Chinese materia medica in cardiomyocyte apoptosis and underlying
molecular mechanism. Biomed Pharmacother.
118(109372)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Huang P, Fu J, Chen L, Ju C, Wu K, Liu H,
Liu Y, Qi B, Qi B and Liu L: Redd1 protects against post infarction
cardiac dysfunction by targeting apoptosis and autophagy. Int J Mol
Med. 44:2065–2076. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Takeshima H, Venturi E and Sitsapesan R:
Takeshima1 H, Venturi E and Sitsapesan R: New and notable
ion-channels in the sarcoplasmic/endoplasmic reticulum: Do they
support the process of intracellular Ca2+ release? J
Physiol. 593:3241–3251. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sun D, Chen X, Gu G, Wang J and Zhang J:
Potential roles of mitochondria-associated ER membranes (MAMs) in
traumatic brain injury. Cell Mol Neurobiol. 37:1349–1357.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dingreville F, Panthu B, Thivolet C,
Ducreux S, Gouriou Y, Pesenti S, Chauvin MA, Chikh K,
Errazuriz-Cerda E, Van Coppenolle F, et al: Differential effect of
glucose on ER-mitochondria Ca2+ exchange participates in
insulin secretion and glucotoxicity-mediated dysfunction of
β-cells. Diabetes. 68:1778–1794. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Betz C, Stracka D, Prescianotto-Baschong
C, Frieden M, Demaurex N and Hall MN: mTOR complex 2-Akt signaling
at mitochondria-associated endoplasmic reticulum membranes (MAM)
regulates mitochondrial physiology. Proc Natl Acad Sci USA.
110:12526–12534. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Roman B, Kaur P, Ashok D, Kohr M, Biswas
R, O'Rourke B, Steenbergen C and Das sS: Nuclear-mitochondrial
communication involving miR-181c plays an important role in cardiac
dysfunction during obesity. J Mol Cell Cardiol. 144:87–96.
2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911.
2006.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mitra A, Basak T, Datta K, Naskar S,
Sengupta S and Sarkar S: Role of α-crystallin B as a regulatory
switch in modulating cardiomyocyte apoptosis by mitochondria or
endoplasmic reticulum during cardiac hypertrophy and myocardial
infarction. Cell Death Dis. 4(e582)2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Javadov S, Rajapurohitam V, Kilić A,
Zeidan A, Choi A and Karmazyn M: Anti-hypertrophic effect of NHE-1
inhibition involves GSK-3beta-dependent attenuation of
mitochondrial dysfunction. J Mol Cell Cardiol. 46:998–1007.
2009.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li X, Lu J, Xu Y, Wang J, Qiu X, Fan L, Li
B, Liu W, Mao F, Zhu J, et al: Discovery of nitazoxanide-based
derivatives as autophagy activators for the treatment of
Alzheimer's disease. Acta Pharm Sin B. 10:646–666. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Cheon SY, Kim H, Rubinsztein DC and Lee
JE: Autophagy, cellular aging and age-related human diseases. Exp
Neurobiol. 28:643–657. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gelmetti V, De Rosa P, Torosantucci L,
Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM
and Valente EM: PINK1 and BECN1 relocalize at
mitochondria-associated membranes during mitophagy and promote
ER-mitochondria tethering and autophagosome formation. Autophagy.
13:654–669. 2017.PubMed/NCBI View Article : Google Scholar
|