1. Introduction
Diabetic retinopathy (DR) is the most common serious
complication of diabetes, mainly manifesting as progressive and
irreversible vision damage. At present, there are ~346 million
individuals with diabetes worldwide, ~10% of which have severe
visual impairment and 2% of them are blind. It is expected that the
number of individuals at risk of vision loss from DR will be double
by 2030(1).
Strategies to prevent or treat DR early have become
a research hotspot. An increasing number of studies have indicated
that the occurrence of retinal neurodegenerative changes in DR may
be earlier than microvascular changes. Furthermore, both the
proliferation of glial cells and the damage of photoreceptor cells
may occur at the beginning of the disease (2,3). Most
previous studies regarded DR as a vascular disease, but now
comprehensive properties such as retinal neurodegeneration should
also be considered (4). The
pathogenesis of DR is highly complex. The changes in the
microenvironment of the retina caused by oxidative stress,
inflammatory response and other factors affect the occurrence and
development of DR (5,6).
The retinal microenvironment homeostasis mediated by
inward rectifying potassium channel 4.1 (Kir4.1) on Müller cells is
a basic system that provides the necessary conditions for retinal
cells to maintain normal physiological functions. The balance of
potassium ions, water and glutamic acid is adjusted through the
bond of Kir4.1(7). In DR, Kir4.1
expression is downregulated, Müller cells exhibit edema and
apoptosis, and glutamate accumulates in the retinal
microenvironment (8). This series
of changes aggravates optical nerve damage. Based on the
destruction of the retinal microenvironment, processes such as
inflammation and oxidative stress occur in sequence to promote the
progression of DR (9,10). As the prime mediator of the entire
retinal microenvironment regulation, Kir4.1 may be considered a
novel target for the treatment of DR in the future.
2. Kir4.1-basics
The Kir channels facilitate potassium ions to enter
cells and may be divided into four groups and seven Kir channel
subfamilies according to their structure and function. Kir2.x is a
classic Kir channel with constitutive activity. Kir3.x is a G
protein-gated Kir channel and Kir6.x is the adenosine triphosphate
(ATP)-sensitive channel in cell metabolism. Furthermore, Kir1.x,
Kir4.x, Kir5.x and Kir7.x are mainly responsible for the
transportation of potassium ions (11).
Kir4.1 is a member of the Kir4.x family and is
widely expressed in the nervous system. Kir4.1 is composed of four
identical Kir4.1 subunits, which mediate potassium ion homeostasis,
and water and glutamate transport inside and outside the cells
(12). The Kir4.1 subunit is
encoded by the KCNJ10 gene (13).
Each subunit contains two transmembrane (TM) domains, TM-1 and
TM-2. One of the extracellular loops contains the characteristic
amino acid sequence GYG, which is involved in the selective ion
filtration of K+ (14).
Kir4.1 has been indicated to be associated with various conditions,
including autism, epilepsy, central nervous system ischemic injury
and inflammation (15). In these
diseases, the expression of the Kir4.1 channel is reduced or
misplaced and the continuous lack of Kir current damages the
buffering capacity of glial cells, leading to the imbalance of ion
homeostasis, which may contribute to pathological processes.
3. Kir4.1 in the retina
The expression of Kir4.1 varies among different
tissues. In the retina, it is mainly distributed on Müller cells.
Penetrating between the inner limiting membrane and the outer
membrane of the retina, Müller cells are a type of astrocytes that
are irreplaceable components of retinal cells. As they constitute
the blood-retinal barrier, Müller cells are responsible for
supporting the function and metabolism of peripheral neurons; they
increase retinal compliance and resistance, secrete a variety of
neuroprotective factors and participate in retinal angiogenesis
(16-18).
Kir4.1 is selectively enriched on the end feet of Müller cells
surrounding microvessels and vitreous bodies, where the expression
of Kir4.1 is significantly higher than that in other regions. It
has been reported that β1-syntrophin and Nogo-A may be involved in
the polar expression of Kir4.1 on Müller cells (19,20).
Kir4.1 has a C-terminal sequence ending with-Ser-Asn-Val. This
sequence may be recognized by β1-syntrophin, which anchors Kir4.1
to the terminal feet of Müller cells and mediates the siphon effect
of K+ to maintain the K+ steady state. The
increase of Nogo-A expression does not lead to any increase in the
total amount of Kir4.1 protein, but selectively affects Kir4.1
expression, causing it to redistribute on Müller cells, which in
turn affects K+ transport.
4. Kir4.1 and the retinal
microenvironment
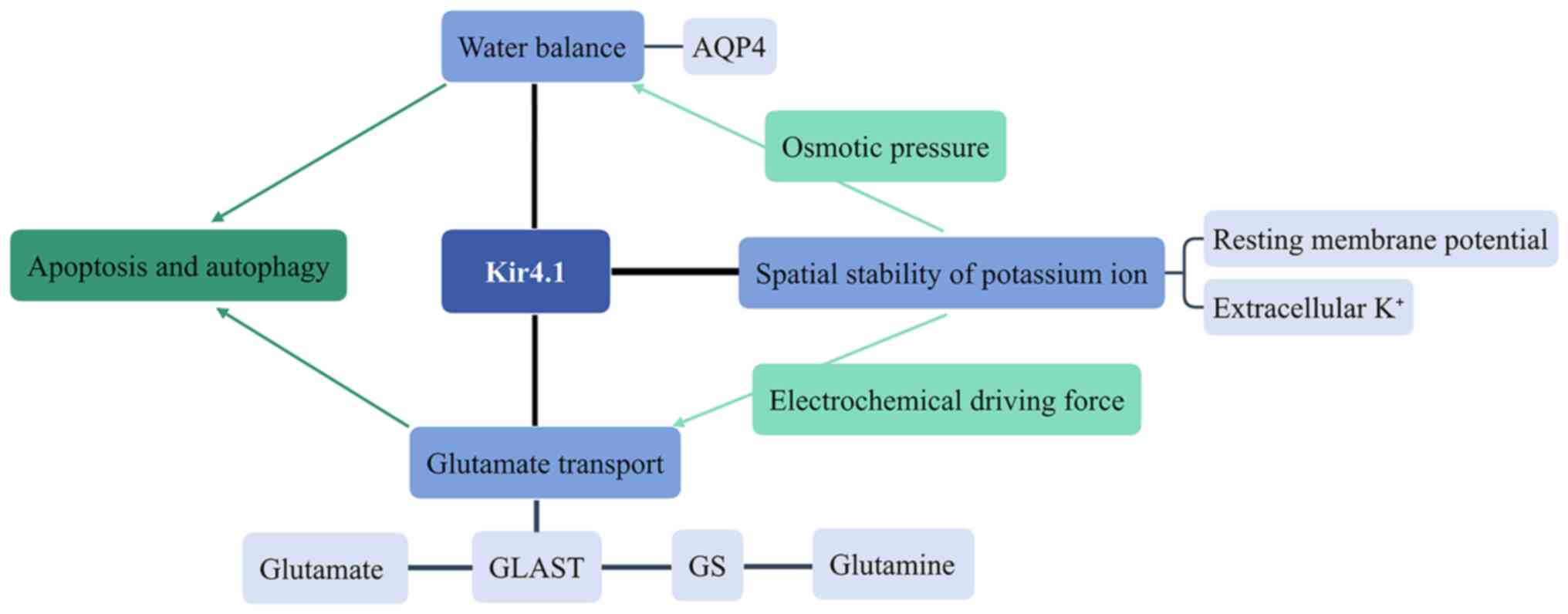
Overview
In the regulation of the retinal microenvironment,
Kir4.1 maintains spatial potassium ion homeostasis, preserves the
water balance and mediates glutamate transport. It is precisely due
to the transport of K+ that the osmotic pressure of the
intracellular fluid of Müller cells changes accordingly and
aquaporin 4 (AQP4) on the membrane of Müller cells thereby
regulates the water balance and maintains the normal cell volume.
Furthermore, the change of K+ conductance also affects
the electrochemical power of glutamate transport, enabling the
function of glutamate/L-aspartate transporter (GLAST). The roles of
Kir4.1 in the retinal microenvironment are presented in Fig. 1.
Kir4.1 and spatial stability of
potassium ions
Kir4.1 moves large amounts of K+ inwards
at hyperpolarized potentials or potentials negative to the
potassium equilibrium potential (EK). During the depolarization
process, only a relatively small outward K+ current may
be observed, which is caused by the temporary blocking of the
potassium ion channel by the positively charged polyamine and
Mg2+. When the membrane potential is close to or more
negative than the EK, these cations will move away from their
binding site, allowing K+ to flow through the channel,
which is conducive to maintaining the cell's resting K+
conductivity (21). However, the
rectification function of Kir4.1 is weak and the current flowing
into and out of the channel has a similar magnitude. Unlike Kir2.1,
which is a strong inward rectifier potassium ion channel
distributed on Müller cells (22),
the weak rectifier characteristic of Kir4.1 may make Kir4.1 more
susceptible to pathological conditions (23).
During neuron excitation, K+ is released
into the extracellular space. Increased extracellular K+
promotes neuronal damage by triggering harmful secondary cascades.
Sibille et al (24)
determined that Kir4.1 is able to promote the fine-tuning of
neuronal synapses, providing astrocytes with another effective
mechanism for regulating neuronal activity. Their study further
elucidated potassium dynamics between neurons, astrocytes and
extracellular space according to a three-compartment model. In the
retina, Kir4.1 has the most important role in maintaining the
resting membrane potential and regulating the extracellular
K+ concentration to prevent the overexcitement of
neurons (25). Specifically, Kir4.1
is able to transport excessive potassium ions in the
microenvironment around the photoreceptor cells into the blood
vessels or vitreous through its polar expression on Müller cells.
Concurrently, Kir4.1 avoids excessive K+ accumulation in
Müller cells and high potassium load on the cells.
Kir4.1 and glutamate transport
Glutamate is the major excitatory neurotransmitter
in the mammalian central nervous system and is involved in neural
development, synaptic plasticity, and learning and memory function
under physiological conditions. When its concentration is
increased, it may cause neurotoxicity, resulting in neuron damage
and degeneration (26). In order to
prevent excessive glutamate levels outside of nerve cells, the body
maintains normal glutamate levels through the glutamate receptor.
Glutamate transporters may be divided into a variety of types, such
as excitatory amino acid transporters (EAAT) and vesicle glutamate
transporters, according to their structural and functional
characteristics (27,28). Glutamate/L-aspartate transporter
(GLAST) is a subtype of EAAT, also known as EAAT1, mainly
distributed on Müller cells in the retina (29). Excessive glutamate in the retina is
mostly transferred to Müller cells through GLAST, where it is
converted to glutamine by glutamine synthetase (GS) to be
eliminated, reducing its neurotoxicity.
Glutamic acid is released from the presynaptic
membrane and diffuses into the intersynaptic space due to an action
potential, bringing about an increase in the extracellular glutamic
acid concentration. GLAST on Müller cells requires energy to take
up glutamate. The stochiometrics of the electrical generation
process are as follows: 3 Na+, 1 H+ and 1
glutamic acid molecule enter the cell and 1 K+ leaves
the cell. Therefore, effective glutamate uptake requires not only
the expression of functional glutamate uptake proteins but also a
certain chemical gradient. The strong resting potential mediated by
Kir4.1 generates an inward-directed electrochemical driving force,
giving GLAST the ability to promote the transfer of glutamate into
Müller cells (30). The decrease of
Kir4.1 channel activity causes the depolarization of the Müller
cell's membrane, which weakens the driving force of the glutamate
transporter that gives rise to the accumulation of extracellular
glutamate and this inhibits GLAST-mediated glutamate transport
(31). It was reported that
pharmacological inhibition and small interfering RNA-mediated
Kir4.1 knockdown in cortical astrocytes reduced glutamate uptake by
33.1 and 57.0%, respectively (32).
Kir4.1 and water balance
The maintenance of water balance in the retina
mainly depends on AQP4 expressed on the surface of Müller cells.
Furthermore, Kir4.1 and AQP4 are co-localized in the perivascular
region of astrocytes (33). The two
have a certain coupling function. Due to the transport of
K+, the osmotic pressure of Müller cells changes with
the K+ concentration and the water balance is then
adjusted by AQP4. Therefore, any abnormal K+ channel
function may lead to impaired retinal water clearance, which
aggravates the development of retinal edema.
In particular, when Kir4.1 suffers functional
impairment, the extracellular potassium ion concentration increases
to cause depolarization of neurons and glia, which leads to
increased glutamate efflux (34).
At this stage, excessive potassium ions cannot be discharged and
the accumulation in Müller cells causes the osmotic pressure to
markedly increase. Glutamate-induced water inflow makes the
swelling of Müller cells more severe, eventually leading to cell
autophagy and apoptosis (35). As
an important factor in maintaining the retinal microenvironment,
apoptosis of Müller cells increases the severity of the imbalance
of retinal water regulation, the damage of the spatial stability of
potassium ions and the accumulation of glutamic acid, generating a
vicious cycle.
5. Kir4.1 and related biological mechanisms
in DR
Overview
Changes in the retinal microenvironment may affect
inflammation, glutamate toxicity, oxidative stress, apoptosis,
autophagy and biological rhythm. While knowledge regarding the
direct involvement of Kir4.1 in DR remains limited, its roles in
the associated mechanisms of inflammation, glutamate toxicity,
oxidative stress, apoptosis, autophagy and biological rhythm may
provide clues and they are discussed in the remainder of this
chapter. The roles of Kir4.1 in biological mechanisms associated
with DR are presented in Figs. 2
and 3.
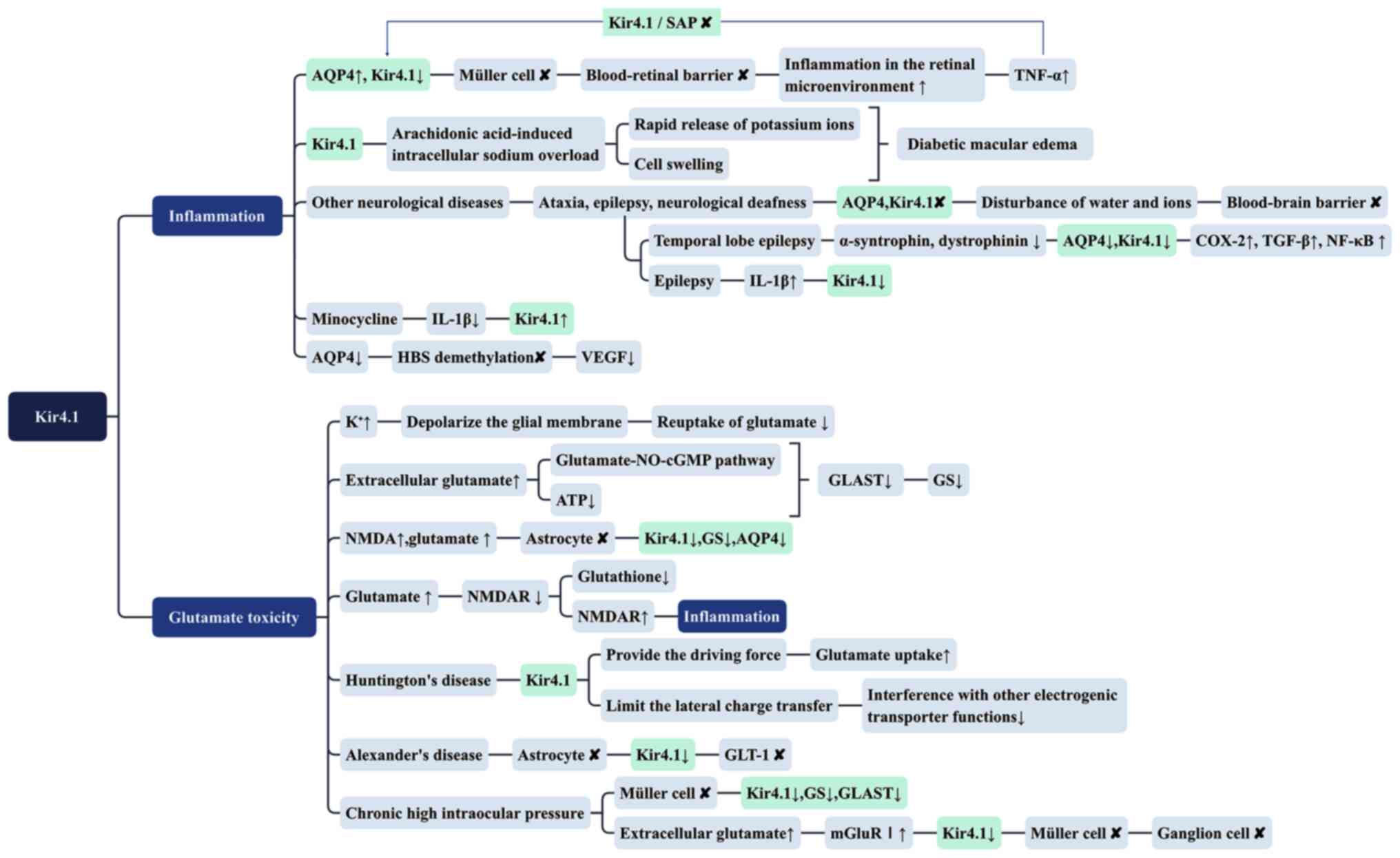 | Figure 2Kir4.1 and related biological
mechanisms (inflammation and glutamate toxicity) in diabetic
retinopathy. Kir4.1, inward rectifying potassium channel 4.1; AQP4,
aquaporin 4; TNF-α, tumor necrosis factor-α; SAP,
synapse-associated protein; COX-2, cyclooxygenase-2; HBS,
hypoxia-inducible factor-1 binding site; NO, nitric oxide; cGMP,
cyclic guanosine monophosphate; GLAST, glutamate/L-aspartate
transporter; GS, glutamine synthetase; NMDA, N-methyl-D-aspartate;
NMDAR, N-methyl-D-aspartate receptor; GLT-1, glutamate
transporter-1; mGluRI, group I metabotropic glutamate receptor. |
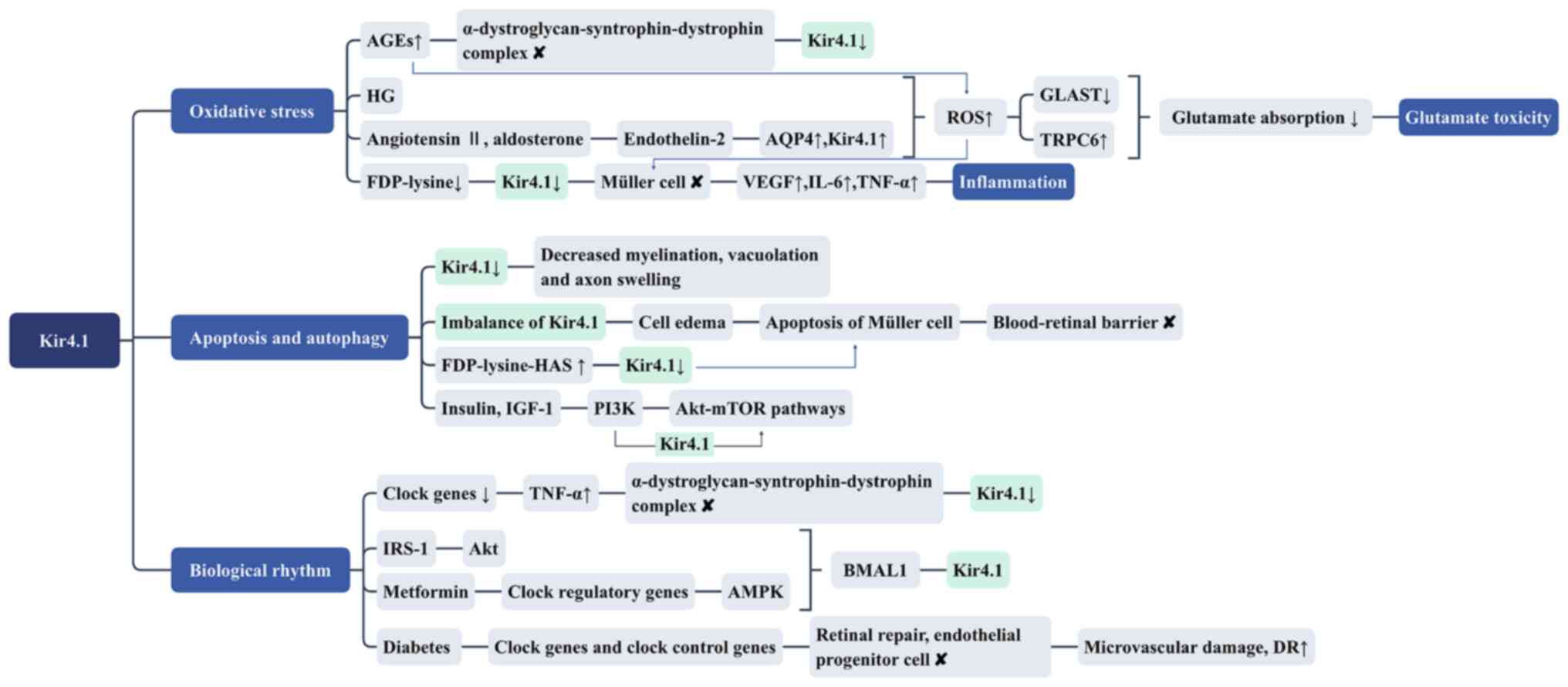 | Figure 3Kir4.1 and related biological
mechanisms (oxidative stress, apoptosis and autophagy and
biological rhythm) in DR. DR, diabetic retinopathy; Kir4.1, inward
rectifying potassium channel 4.1; AGEs, advanced glycation end
products; HG, high glucose; AQP4, aquaporin 4; ROS, reactive oxygen
species; GLAST, glutamate/L-aspartate transporter; TRPC6, transient
receptor potential cation channel 6; FDP-lysine,
Nε-(3-formyl-3,4-dehydropiperidino)lysine; FDP-lysine-HAS,
FDP-lysine-modified human serum albumin; IGF-1, insulin-like growth
factor 1; IRS-1, insulin receptor substrate-1; AMPK, adenosine
5'-monophosphate-activated protein kinase; BMAL1, brain and muscle
arnt-like protein 1. |
Inflammation
A series of studies have suggested that DR is a
chronic inflammatory lesion (36).
Inflammatory changes in the retinal microenvironment are considered
to be the main reason for the development of DR. It has been
detected in both diabetic patients and animal models that the
expression of AQP4 in the retina increases, Kir4.1 expression is
downregulated and Müller cells are dysfunctional, which induces the
destruction of the blood-retinal barrier (37-39).
Furthermore, it has been determined that Kir4.1 may
cause edema of Müller cells by affecting the combined effect of
osmotic pressure and AQP4, triggering the corresponding
inflammatory response. As a key cause of visual impairment in
diabetic patients, the occurrence of macular edema was previously
thought to be due to vascular leakage resulting from the direct
destruction of the blood-retinal barrier. However, failure of the
active potassium transport mechanism mediated by Kir4.1 in the
blood-retinal barrier may be a more important factor leading to
diabetic macular edema of clinical significance (25). This change in the potassium channel
may interfere with arachidonic acid-induced intracellular sodium
overload, contributing to the rapid release of potassium ions and
cell swelling (23). In addition to
DR, AQP4 and Kir4.1 are involved in the inflammatory response of
various neurological diseases such as ataxia, epilepsy and
neurological deafness. Their local expression decrease or content
increase leads to disturbance of the water and ion content of the
glial cells. The volume of the cell and the structure of the
adjacent tissue changes and the corresponding barrier is destroyed
(40). After epileptic seizures,
Kir4.1 transcription of astrocytes is downregulated and accompanied
by upregulation of various inflammatory genes (41). In temporal lobe epilepsy, Kir4.1 and
AQP4 are downregulated simultaneously with α-syntrophin and
dystrophinin, while inflammatory factors, such as cyclooxygenase-2,
TGF-β and NF-κB significantly increase (42). The above studies confirmed again
that Kir4.1 combined with AQP4 to cause edema of Müller cells and
affect the osmotic pressure, leading to the destruction of the
retinal barrier and promoting the subsequent release of
inflammatory factors.
In addition, Kir4.1 and AQP4 may also be directly
involved in the occurrence of inflammation through certain
pathways, rather than indirectly affecting osmotic pressure. In
certain diseases, AQP4 may promote the release of inflammatory
factors through possible pathways (43,44). A
study indicated that upregulation of vascular endothelial growth
factor (VEGF) during retinal hypoxia is related to the methylation
status of the hypoxia-inducible factor-1 binding site (HBS). AQP4
deletion may prevent the upregulation of VEGF by interfering with
the process of hypoxia-induced HBS demethylation (45). Whether Kir4.1 combines with AQP4 to
directly promote inflammation in DR requires further research.
By contrast, inflammation may also act on Kir4.1. A
study demonstrated that the expression changes of Kir4.1 in
epilepsy-related lesions may be affected by the local inflammatory
environment, particularly the inflammatory cytokine IL-1β (46). Another study reported that TNF-α is
able to destroy the actin cytoskeleton and dissociation of
Kir4.1/synapse-associated protein (SAP) (47), indicating that inflammation destroys
the attachment skeleton of Kir4.1. Another study suggested that
treatment of the retina of diabetic rats with minocycline increased
the level of Kir4.1 by reducing IL-1β levels (39). Therefore, inflammation may
downregulate the polar expression of Kir4.1, leading to further
deterioration of the retinal microenvironment. The specific
mechanism remains to be elucidated.
Glutamate toxicity
In DR, downregulation of Kir4.1 reduces the ability
of Müller cells to clear glutamate. High concentrations of
potassium ions may reduce the binding force of potassium ions and
depolarize the glial membrane, thereby reducing the reuptake of
glutamate.
Increased extracellular glutamate concentrations may
lead to decreased expression levels of GLAST and GS, resulting in
abnormal synaptic transmission. This may be related to the
glutamate-nitric oxide-cyclic guanosine monophosphate pathway and
the reduction of ATP (48,49). In addition, the expression of Kir4.1
may be mediated by the N-methyl-D-aspartate receptor (NMDAR)
expressed on astrocytes. Continuous exposure of astrocytes to NMDA
and Glu reduces the expression of Kir4.1, GS and AQP4 and also
reduces the activity of GS (50).
Excessive NMDAR activity may increase the incidence of central
nervous system diseases related to glutamatergic tone and
excitotoxicity (51). Due to the
increased glutamate concentration in the protruding gap, NMDAR on
astrocytes cannot drive the intracellular mechanism to exert a
neuroprotective effect, resulting in a reduction in the supply of
antioxidant glutathione induced by this mechanism, which further
strengthens the toxic effect of glutamic acid (52). Furthermore, NMDAR on astrocytes is
also able to increase the secretion of pro-inflammatory cytokines
in cells stimulated by inflammation (53).
Similar observations were made in other
neurodegenerative diseases. In Huntington's disease, glutamate
uptake activity mainly depends on Kir4.1(54). Glutamate transporter-1 (GLT-1) is an
electron transporter that relies on the negative membrane potential
provided by Kir4.1 to clear glutamate released from extracellular
nerves. In Alexander's disease, Kir4.1 expression on astrocytes is
reduced, GLT-1 function is impaired and glutamate toxicity occurs
(55). Despite the limited research
on the glutamate toxicity theory in DR in the past decades, it is
undeniable that the disorder of glutamate transport by
Kir4.1-mediated lack of electrochemical driving force may be one of
the crucial foundations of retinal microenvironment destruction.
One study indicated that chronic high intraocular pressure
downregulated the expression of Kir4.1, GS and GLAST in Müller
cells (56). Wang and Yang
(57) reported that extracellular
glutamate accumulation caused by high intraocular pressure
activates group I metabotropic glutamate receptors on Müller cells,
which induces the activation of Müller cells by downregulation of
Kir4.1 and leads to apoptosis of ganglion cells. This change in the
retinal microenvironment should not be limited to DR but is the
basis of a variety of retinopathies.
Oxidative stress
In the development of DR, oxidative stress
frequently affects the retinal microenvironment along with multiple
mechanisms. Pannicke et al (23) tested the inhibitory effect of
reducing agents on diabetic retina swelling, indicating that acute
oxidative stress is related to the induction of glial cell swelling
under anisotropic penetration conditions. Animal experiments
suggested that the activation of enzymes producing various reactive
oxygen species (ROS) and nitrogen species, as well as the
mitochondrial pathway, are involved in the induction of osmotic
swelling of Müller cells in the retinal tissues of transgenic rats
(58). Furthermore, transient
receptor potential cation channel 6 (TRPC6) is a Ca2+
permeable cation channel sensitive to oxidative stress, which is
easily detected in Müller cells and highly expressed under High
glucose (HG) conditions. HG increases the production of ROS,
reduces the expression of GLAST and activates the TRPC6 channel,
which initiates a reduction in glutamate absorption by Müller cells
(29). Therefore, oxidative stress
may affect the water balance and glutamate transport of Müller
cells and they may have a close relationship with Kir4.1.
Studies have indicated that oxidative stress
products may reduce Kir4.1 expression and interact with
inflammation. The accumulation of
Nε-(3-formyl-3,4-dehydropiperidino)lysine (FDP-lysine, which is an
oxidative stress lipid oxidation end product) causes Müller glial
dysfunction with the protein level of the potassium channel subunit
Kir4.1 decreasing and the downregulation of inwardly rectifying
K+ currents in Müller cells may, at least in part, be
responsible for successive upregulation of VEGF, IL-6 and TNF-α
(59). Endothelin-2 is a potent
vasoconstrictor. Angiotensin II and aldosterone stimulate
endothelin-2 expression, which promotes Müller glial dysfunction
and blood-retinal barrier destruction by generating ROS. In this
process, endothelin-2 was determined to increase AQP4 and Kir4.1 in
Müller cells and their mRNA levels in the retina (60). Apart from this, the accumulation of
advanced glycation end products (AGEs) is the major pathological
event in DR. AGEs promote the formation of ROS, which boost the
production of AGEs, resulting in positive feedback loops that
compromise tissue fitness (61).
AGE-modified laminin causes disorder of the actin cytoskeleton and
destruction of the α-dystroglycan-syntrophin-dystrophin complex,
which in turn reduces Kir4.1 expression and weakens its function
(62). Evidently, oxidative stress
is part of various complex mechanisms involved in Kir4.1.
Apoptosis and autophagy
Autophagy and apoptosis frequently occur as
intermediate processes in the development of DR, which are a result
of the combination of various factors such as inflammation,
oxidative stress, cellular edema and glutamate toxicity. In other
nerve tissues, there is evidence that Kir4.1 is involved in
apoptosis. The loss of Kir4.1 results in decreased myelination,
vacuolation and axon swelling in the spinal cord of mice, which are
all related to apoptosis (63).
Kir4.1 expressed on Müller cells allows neurons that generate
action potentials in the inner layer of the retina to maintain
their function in a potassium buffer environment, particularly
under ischemic conditions. Maintaining the function of Müller cells
may suppress cytotoxic neuron overexcitation and subsequent
neuronal cell loss (64). The cell
edema caused by the imbalance of Kir4.1 in DR may trigger apoptosis
of Müller cells, destroy the retinal barrier and eventually lead to
irreparable visual impairment.
High concentrations of FDP-lysine-modified human
serum albumin cause extensive apoptosis. The accumulation of these
adducts may lead to the apoptosis of Müller cells during long-term
diabetes, while the accumulation of FDP-lysine is associated with a
reduction in the level of potassium channel subunit Kir4.1 protein
(59). Therefore, Kir4.1 may be
related to cell apoptosis. Furthermore, Kir4.1 may prevent the
induction of autophagy via phosphoinositide 3-kinase
(PI3K)-dependent activation through the mechanistic target of
rapamycin (mTOR) pathway. Akt is an important factor involved in
cell survival and apoptosis and mTOR is a downstream target of Akt.
After ischemia and hypoxia, the level of extracellular potassium
ions increases significantly. Subsequently, Akt is also
phosphorylated by recruiting PI3K to activate Kir4.1, which
triggers the mTOR pathway to enable cell survival (65). One study has indicated that the
heteromer Kir4.1/Kir5.1 channel in the renal cortical collecting
duct may be activated in a PI3K-dependent manner by insulin and
insulin-like growth factor-1, indicating that Kir4.1 is likely to
interact with the Akt pathway (66). Despite the limited evidence
available, increasing the activity of Akt may become a novel
strategy to prevent DR. Numerous studies have indicated that
activation of Akt may protect nerve cells from oxidative stress and
inhibition of the function of PTEN has considerable pharmacological
value in ischemia-related diseases and neurodegenerative diseases
such as Alzheimer's disease and Parkinson's disease (67). In summary, the increased expression
and activity of Kir4.1 may effectively antagonize the autophagy and
apoptosis mediated by various mechanisms in DR.
Biological rhythm
Biological rhythms are not only able to regulate the
sleep-wake cycle of an organism but are also related to the
secretion and release of hormones. Among the core genes relevant to
biological rhythms, the clock circadian regulator gene, brain and
muscle aryl hydrocarbon receptor nuclear translocator-like 1 gene
(BMAL1, also known as ARNTL) and period circadian clock gene have
gained widespread attention. A series of recent studies suggested
that biological rhythms have a certain impact on diabetes, mainly
through metabolism (68,69). The vision of patients with diabetes
changes regularly with day and night and their electroretinograms
also indicate that the a and b waves of the retina have a
biological rhythm (70).
The expression of Kir4.1 in the retina also exhibits
specific biological rhythms. Disturbances in the daily expression
pattern of clock genes and clock control genes caused by diabetes
may lead to loss of synchronization of retinal repair as well as
the release and migration of endothelial progenitor cells required
for effective repair processes, contributing to increased
microvascular damage and DR (71).
Hassan et al (47) indicated
that silencing of clock genes upregulated the expression of TNF-α,
leading to the destruction of the actin cytoskeleton and
dissociation of Kir4.1/SAP. Another study reported that insulin
receptor substrate-1 (IRS-1) is able to regulate Kir4.1 on Müller
cells through the BMAL1 gene and IRS-1-mediated dysfunction signals
may downregulate Kir4.1 expression. IRS-1 blockade indirectly
inhibits Kir4.1 by downregulating the IRS1-Akt-BMAL1 axis and the
Kir4.1 rhythm is synchronized with insulin-mediated signaling
(72). Metformin, as a traditional
hypoglycemic agent, may indirectly activate 5'adenosine
monophosphate (AMP)-activated protein kinase (AMPK) by inhibiting
the mitochondrial complex (73). A
recent animal study indicated that metformin may increase the
expression of BMAL1 protein and Kir4.1 by controlling clock
regulatory genes to activate the AMPK pathway (74). For the retina with considerable
circulation, the expression of Kir4.1 is regulated by the BMAL1
gene through the influence of light and dark cycles, which has a
close effect on retinal metabolism and inflammation. The circadian
rhythm of Kir4.1 is synchronized with the rhythm of IRS-1 and the
tyrosine phosphorylation of IRS-1 provides binding sites for
multiple signaling molecules, particularly the activation of Akt.
At the same time, Akt activation involves mechanisms such as cell
survival, metabolism and apoptosis. AMPK is a sensor of energy
availability within the cell, which is activated at low energy
levels and regulates cellular processes accordingly. In addition,
AMPK activity supports systemic glucose homeostasis and improves
insulin sensitivity by promoting processes such as glucose uptake
and energy expenditure. Glucose deficiency, hypoxia, oxidative
stress and other changes may reduce cell energy levels and increase
the AMP/ATP ratio, thereby activating AMPK. Activated AMPK is able
to indirectly inhibit the mechanistic target of rapamycin complex 1
(mTORC1) by counteracting the inhibitory effects of mTORC1 and
IRS-1(75). In addition, the
inflammatory environment may be involved in influencing
IRS-1-Kir4.1 signaling. Since biological rhythms may interact with
various mechanisms such as autophagy and oxidative stress to affect
Kir4.1, Akt and AMPK may be important targets for drug design to
prevent Müller cell dysfunction in DR.
6. Prospects
DR is a chronic inflammatory neurodegenerative
disease with multiple mechanisms, which ultimately damages vision
and even leads to permanent loss of sight. This type of vision
impairment is irreversible. In the early stages of DR, the major
pathological manifestations are changes in microvascular function,
including retinal vasodilation and microaneurysms, while in the
later stage, vitreous integrity is lost, new blood vessels are
ruptured and bleed and normal physiological functions of the retina
are lost (76). However, the
current clinical treatment of DR mostly focuses on the period of
vascular hyperplasia with obvious symptoms, which cannot reverse
the loss of vision. Therefore, it is necessary to explore a path
for early prevention and treatment of DR.
To pursue the new therapeutic direction involving
Kir4.1, its molecular biological mechanisms require to be
thoroughly investigated. There are three approaches worth
exploring, as discussed below: i) Containment of inflammatory
response; ii) in-depth study of Akt- and AMPK-targeted treatments;
and iii) further exploration of therapies targeting the Kir4.1
cytoskeleton.
DR is a chronic inflammatory disease and the core
part of the treatment lies in exerting anti-inflammatory effects in
the retinal microenvironment. In DR, disorder of Kir4.1 induces
changes in the retinal microenvironment, accompanied by
upregulation of inflammatory factors. Inflammation and oxidative
stress products destroy the attachment framework of Kir4.1 to cause
further imbalance, aggravating the destruction of the retinal
microenvironment. It is necessary to further explore the specific
pathways of the arachidonic acid pathway that Kir4.1 may
participate in. Furthermore, certain inflammation-related factors
are also worthy of attention from researchers, particularly VEGF.
Various clinical trials have proved that anti-VEGF treatment in the
vitreous is an important therapeutic method for diabetic macular
edema (77). VEGF is an important
mediator of the inflammation release pathway and AQP4 has been
indicated to have a regulatory relationship with VEGF. Kir4.1 may
also participate in this process together with AQP4.
Akt and AMPK may be targets for DR treatment. On the
one hand, PI3K-Akt-mTOR is a classic pathway that responds to
insulin signaling. The Kir4.1 rhythm is synchronized with
insulin-mediated signaling and Kir4.1 may activate Akt indirectly
in a PI3K-dependent manner. Furthermore, the PI3K/Akt pathway may
be regulated by GluN2A subunit-containing NMDAR and the expression
of Kir4.1 may be related to NMDAR (78). A number of animal experiments have
indicated that NMDAR inhibitors are able to reduce the death of
retinal neurons in DR caused by glutamate toxicity (79-81).
The design of drugs targeting Kir4.1 to activate Akt may be a novel
treatment approach, but this is required to be verified by further
research. On the other hand, AMPK is also crucial for the survival
of Müller cells. In the high-glucose and hypoxia environment of DR,
ensuring sufficient levels of Müller cells is needed in order to
maintain the integrity of the blood-eye barrier, which may enhance
the stability of the retinal microenvironment (82-84).
A previous study has indicated that AMPK activation protects
astrocytes from hypoxia-induced cell death (85). Upregulation of Kir4.1 is related to
the activation of the AMPK pathway. Furthermore, a novel study in
the field of biological rhythms has indicated that therapies
targeting IRS-1-Kir4.1 signaling may be of great clinical
significance (72).
Protecting the normal structure of Kir4.1's attached
cytoskeleton may become another novel therapeutic aspect. Improving
the dislocation of Kir4.1 may have a more important role than
increasing its expression level. A study indicated that
proliferative gliosis in the retina is related to the inactivation
of glial Kir channels, which is not caused by the downregulation of
channel proteins, but is related to their dislocation in the cell
membrane (86). Dystrophin (Dp)71
is a membrane-associated cytoskeletal protein on Müller cells,
which is the core anchor for the polar expression of AQP4 and
Kir4.1(87). From the perspective
of membrane cytoskeletal proteins, a study has indicated that
AAV-mediated gene therapy has an effect on the blood-retinal
barrier of dystrophin-Dp71-deficient mice. The distribution of
Kir4.1 and AQP4 has a restorative effect to ameliorate the
condition of retinal edema (88).
It has also been reported that the adrenal cortex hormone
dexamethasone is able to inhibit the downregulation of Dp71 by
inhibiting the downregulation of heat shock factor 1, thereby
inhibiting the downregulation of AQP4 and Kir4.1 in Müller cells to
prevent the occurrence and development of blood-retinal barrier
destruction (89).
Furthermore, Kir4.1 and AQP4 are related to each
other in positional expression and function and they may have a
synergistic effect via related mechanisms. However, at present,
relevant studies are limited. In the future, the mechanisms
mediated by Kir4.1 and AQP4 will become the focus of research.
However, it is worth noting that Kir4.1 and AQP4 are not completely
coupled. One study indicated that Kir4.1 and AQP4 clusters were
co-localized in the perivascular area, but not in parenchyma
(33). An animal experiment
indicated that after intravitreal injection of lipopolysaccharide
during rat uveitis, the expression of Kir4.1 in the retina
decreased significantly at the protein and gene levels, but the
expression of AQP4 remained almost unchanged (90). Evidently, the relationship between
Kir4.1 and AQP4 is exceedingly complex and further research still
needs a long way to go.
Kir4.1-targeted treatment has been effectively
explored, although the relevant mechanisms have remained to be
elucidated. Sun et al (91)
observed in rat model of DR that after the application of the
potassium channel opener pinadil, the expression of Kir4.1 was
upregulated, the function of Müller cells was improved and the
symptoms of retinal macular edema were alleviated. Another study
suggested that aloe vera may help protect from retinal damage
associated with liver failure by normalizing Kir4.1 and AQP4
through reducing oxidative stress and inflammation (92).
In conclusion, Kir4.1 is likely to represent a novel
alternative therapeutic target for DR through affecting the retinal
microenvironment. It is thought that the mechanisms of Kir4.1 will
be ulteriorly clarified with the deepening of future research.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Natural Science
Foundation of Liaoning Province (grant no. 2020-BS-189), the
National Natural Science Foundation of China (grant no. 31371218),
the Basic Scientific Research Projects of Liaoning Provincial
Education Department (grant no. LQ2017005) and the Liaoning
Provincial Program for Top Discipline of Basic Medical Sciences
(grant nos. 2020-BS-189 and LQ2017005).
Availability of data and materials
Not applicable.
Authors' contributions
XYL conceived the topic of the review. XYL and JJL
retrieved and analyzed relevant documents. JJL was responsible for
drawing the figures. XYL wrote the manuscript. JZL and XR revised
the manuscript. All authors read and approved the final manuscript.
Data sharing is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mohammad HMF, Sami MM, Makary S, Toraih
EA, Mohamed AO and El-Ghaiesh SH: Neuroprotective effect of
levetiracetam in mouse diabetic retinopathy: Effect on glucose
transporter-1 and GAP43 expression. Life Sci.
232(116588)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hafner J, Zadrazil M, Grisold A, Ricken G,
Krenn M, Kitzmantl D, Pollreisz A, Gleiss A and Schmidt-Erfurth U:
Retinal and corneal Neurodegeneration and their association with
systemic signs of peripheral neuropathy in type 2 diabetes. Am J
Ophthalmol. 209:197–205. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lynch SK and Abramoff MD: Diabetic
retinopathy is a neurodegenerative disorder. Vision Res.
139:101–107. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang W and Lo ACY: Diabetic retinopathy:
Pathophysiology and treatments. Int J Mol Sci.
19(1816)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dehdashtian E, Mehrzadi S, Yousefi B,
Hosseinzadeh A, Reiter RJ, Safa M, Ghaznavi H and Naseripour M:
Diabetic retinopathy pathogenesis and the ameliorating effects of
melatonin; involvement of autophagy, inflammation and oxidative
stress. Life Sci. 193:20–33. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
He M, Long P, Guo L, Zhang M, Wang S and
He H: Fushiming capsule attenuates diabetic rat retina damage via
antioxidation and anti-inflammation. Evid Based Complement Alternat
Med. 2019(5376439)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Noël G, Belda M, Guadagno E, Micoud J,
Klöcker N and Moukhles H: Dystroglycan and Kir4.1 coclustering in
retinal Müller glia is regulated by laminin-1 and requires the
PDZ-ligand domain of Kir4.1. J Neurochem. 94:691–702.
2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Coughlin BA, Feenstra DJ and Mohr S:
Müller cells and diabetic retinopathy. Vision Res. 139:93–100.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Curtis TM, Hamilton R, Yong PH, McVicar
CM, Berner A, Pringle R, Uchida K, Nagai R, Brockbank S and Stitt
AW: Müller glial dysfunction during diabetic retinopathy in rats is
linked to accumulation of advanced glycation end-products and
advanced lipoxidation end-products. Diabetologia. 54:690–698.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bringmann A, Pannicke T, Grosche J,
Francke M, Wiedemann P, Skatchkov SN, Osborne NN and Reichenbach A:
Müller cells in the healthy and diseased retina. Prog Retin Eye
Res. 25:397–424. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hibino H, Inanobe A, Furutani K, Murakami
S, Findlay I and Kurachi Y: Inwardly rectifying potassium channels:
Their structure, function, and physiological roles. Physiol Rev.
90:291–366. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mendez-Gonzalez MP, Kucheryavykh YV,
Zayas-Santiago A, Vélez-Carrasco W, Maldonado-Martínez G, Cubano
LA, Nichols CG, Skatchkov SN and Eaton MJ: Novel KCNJ10 gene
variations compromise function of inwardly rectifying potassium
channel 4.1. J Biol Chem. 291:7716–7726. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nwaobi SE and Olsen ML: Correlating
Gene-specific DNA methylation changes with expression and
transcriptional activity of astrocytic KCNJ10 (Kir4.1). J Vis Exp.
(52406)2015.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Ohno Y, Kinboshi M and Shimizu S: Inwardly
rectifying potassium channel Kir4.1 as a novel modulator of BDNF
expression in astrocytes. Int J Mol Sci. 19(3313)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Thuringer D, Chanteloup G, Boucher J,
Pernet N, Boudesco C, Jego G, Chatelier A, Bois P, Gobbo J, Cronier
L, et al: Modulation of the inwardly rectifying potassium channel
Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells.
Oncotarget. 8:37681–37693. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Govetto A, Hubschman JP, Sarraf D,
Figueroa MS, Bottoni F, dell'Omo R, Curcio CA, Seidenari P,
Delledonne G, Gunzenhauser R, et al: The role of Müller cells in
tractional macular disorders: An optical coherence tomography study
and physical model of mechanical force transmission. Br J
Ophthalmol. 104:466–472. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Eastlake K, Luis J and Limb GA: Potential
of Müller glia for retina neuroprotection. Curr Eye Res.
45:339–348. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li X and Liu J, Hoh J and Liu J: Müller
cells in pathological retinal angiogenesis. Transl Res. 207:96–106.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rao SB, Katoozi S, Skauli N, Froehner SC,
Ottersen OP, Adams ME and Amiry-Moghaddam M: Targeted deletion of
β1-syntrophin causes a loss of Kir 4.1 from Müller cell endfeet in
mouse retina. Glia. 67:1138–1149. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Joly S, Dodd DA, Grewe BF and Pernet V:
Reticulon 4A/Nogo-A influences the distribution of Kir4.1 but is
not essential for potassium conductance in retinal Müller glia.
Neurosci Lett. 627:168–177. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nwaobi SE, Cuddapah VA, Patterson KC,
Randolph AC and Olsen ML: The role of glial-specific Kir4.1 in
normal and pathological states of the CNS. Acta Neuropathol.
132:1–21. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kofuji P, Biedermann B, Siddharthan V,
Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A and
Reichenbach A: Kir potassium channel subunit expression in retinal
glial cells: Implications for spatial potassium buffering. Glia.
39:292–303. 2002.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pannicke T, Iandiev I, Wurm A, Uckermann
O, vom Hagen F, Reichenbach A, Wiedemann P, Hammes HP and Bringmann
A: Diabetes alters osmotic swelling characteristics and membrane
conductance of glial cells in rat retina. Diabetes. 55:633–639.
2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sibille J, Dao Duc K, Holcman D and Rouach
N: The neuroglial potassium cycle during neurotransmission: Role of
Kir4.1 channels. PLoS Comput Biol. 11(e1004137)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mori F, Hikichi T, Takahashi J, Nagaoka T
and Yoshida A: Dysfunction of active transport of blood-retinal
barrier in patients with clinically significant macular edema in
type 2 diabetes. Diabetes Care. 25:1248–1249. 2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang Y and Qin ZH: Molecular and cellular
mechanisms of excitotoxic neuronal death. Apoptosis. 15:1382–1402.
2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li F, Eriksen J, Finer-Moore J, Chang R,
Nguyen P, Bowen A, Myasnikov A, Yu Z, Bulkley D, Cheng Y, et al:
Ion transport and regulation in a synaptic vesicle glutamate
transporter. Science. 368:893–897. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pavić A, Holmes AOM, Postis VLG and
Goldman A: Glutamate transporters: A broad review of the most
recent archaeal and human structures. Biochem Soc Trans.
47:1197–1207. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ma M, Zhao S, Zhang J, Sun T, Fan Y and
Zheng Z: High glucose-induced TRPC6 channel activation decreases
glutamate uptake in rat retinal Müller cells. Front Pharmacol.
10(1668)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kharade SV, Kurata H, Bender AM, Blobaum
AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, et
al: Discovery, characterization, and effects on renal fluid and
electrolyte excretion of the Kir4.1 potassium channel pore blocker,
VU0134992. Mol Pharmacol. 94:926–937. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Frizzo ME: Can a selective serotonin
reuptake inhibitor act as a glutamatergic modulator? Curr Ther Res
Clin Exp. 87:9–12. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kucheryavykh YV, Kucheryavykh LY, Nichols
CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN and Eaton
MJ: Downregulation of Kir4.1 inward rectifying potassium channel
subunits by RNAi impairs potassium transfer and glutamate uptake by
cultured cortical astrocytes. Glia. 55:274–281. 2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Smith AJ and Verkman AS: Superresolution
imaging of Aquaporin-4 cluster size in antibody-stained paraffin
brain sections. Biophys J. 109:2511–2522. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Djukic B, Casper KB, Philpot BD, Chin LS
and McCarthy KD: Conditional knock-out of Kir4.1 leads to glial
membrane depolarization, inhibition of potassium and glutamate
uptake, and enhanced short-term synaptic potentiation. J Neurosci.
27:11354–11365. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Reichenbach A and Bringmann A: New
functions of Müller cells. Glia. 61:651–678. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Rübsam A, Parikh S and Fort PE: Role of
inflammation in diabetic retinopathy. Int J Mol Sci.
19(942)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Vujosevic S, Micera A, Bini S, Berton M,
Esposito G and Midena E: Aqueous humor biomarkers of Müller cell
activation in diabetic eyes. Invest Ophthalmol Vis Sci.
56:3913–3918. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li XM, Wendu RL, Yao J, Ren Y, Zhao YX,
Cao GF, Qin J and Yan B: Abnormal glutamate metabolism in the
retina of aquaporin 4 (AQP4) knockout mice upon light damage.
Neurol Sci. 35:847–853. 2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang Y, Xu G, Ling Q and Da C: Expression
of aquaporin 4 and Kir4.1 in diabetic rat retina: Treatment with
minocycline. J Int Med Res. 39:464–479. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Setkowicz Z, Kosonowska E and Janeczko K:
Inflammation in the developing rat modulates astroglial reactivity
to seizures in the mature brain. J Anat. 231:366–379.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Frigerio F, Frasca A, Weissberg I,
Parrella S, Friedman A, Vezzani A and Noé FM: Long-lasting
pro-ictogenic effects induced in vivo by rat brain exposure to
serum albumin in the absence of concomitant pathology. Epilepsia.
53:1887–1897. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Das A, Wallace GC IV, Holmes C, McDowell
ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray
SK and Banik NL: Hippocampal tissue of patients with refractory
temporal lobe epilepsy is associated with astrocyte activation,
inflammation, and altered expression of channels and receptors.
Neuroscience. 220:237–246. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wu J, Ding D, Wang X, Li Q, Sun Y, Li L
and Wang Y: Regulation of aquaporin 4 expression by lipoxin A4 in
astrocytes stimulated by lipopolysaccharide. Cell Immunol.
344(103959)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Li Y, Lu H, Lv X, Tang Q, Li W, Zhu H and
Long Y: Blockade of aquaporin 4 inhibits irradiation-induced
pulmonary inflammation and modulates macrophage polarization in
mice. Inflammation. 41:2196–2205. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Pisani F, Cammalleri M, Dal Monte M, Locri
F, Mola MG, Nicchia GP, Frigeri A, Bagnoli P and Svelto M:
Potential role of the methylation of VEGF gene promoter in response
to hypoxia in oxygen-induced retinopathy: Beneficial effect of the
absence of AQP4. J Cell Mol Med. 22:613–627. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zurolo E, de Groot M, Iyer A, Anink J, van
Vliet EA, Heimans JJ, Reijneveld JC, Gorter JA and Aronica E:
Regulation of Kir4.1 expression in astrocytes and astrocytic
tumors: A role for interleukin-1 β. J Neuroinflammation.
9(280)2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hassan I, Luo Q, Majumdar S, Dominguez JM
II, Busik JV and Bhatwadekar AD: Tumor necrosis factor Alpha
(TNF-α) disrupts Kir4.1 channel expression resulting in Müller cell
dysfunction in the retina. Invest Ophthalmol Vis Sci. 58:2473–2482.
2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin Z, Huang P, Huang S, Guo L, Xu X, Shen
X, Xie B and Zhong Y: Effect of adenosine and adenosine receptor
antagonists on retinal Müller cell inwardly rectifying potassium
channels under exogenous glutamate stimulation. Biomed Res Int.
2018(2749257)2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Saeed Dar M: Functional role for mouse
cerebellar NO/cGMP/KATP pathway in ethanol-induced ataxia. Alcohol
Clin Exp Res. 38:100–107. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Skowrońska K, Obara-Michlewska M,
Zielińska M and Albrecht J: NMDA receptors in astrocytes: In search
for roles in neurotransmission and astrocytic homeostasis. Int J
Mol Sci. 20(309)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gonzalez J, Jurado-Coronel JC, Ávila MF,
Sabogal A, Capani F and Barreto GE: NMDARs in neurological
diseases: A potential therapeutic target. Int J Neurosci.
125:315–327. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Jimenez-Blasco D, Santofimia-Castaño P,
Gonzalez A, Almeida A and Bolaños JP: Astrocyte NMDA receptors'
activity sustains neuronal survival through a Cdk5-Nrf2 pathway.
Cell Death Differ. 22:1877–1889. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Skowrońska K, Obara-Michlewska M,
Czarnecka A, Dąbrowska K, Zielińska M and Albrecht J: Persistent
overexposure to N-Methyl-D-Aspartate (NMDA) calcium-dependently
downregulates glutamine synthetase, aquaporin 4, and Kir4.1 channel
in mouse cortical astrocytes. Neurotox Res. 35:271–280.
2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Dvorzhak A, Vagner T, Kirmse K and Grantyn
R: Functional indicators of glutamate transport in single striatal
astrocytes and the influence of Kir4.1 in normal and huntington
mice. J Neurosci. 36:4959–4975. 2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Minkel HR, Anwer TZ, Arps KM, Brenner M
and Olsen ML: Elevated GFAP induces astrocyte dysfunction in caudal
brain regions: A potential mechanism for hindbrain involved
symptoms in type II Alexander disease. Glia. 63:2285–2297.
2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yang Z, Huang P, Liu X, Huang S, Deng L,
Jin Z, Xu S, Shen X, Luo X and Zhong Y: Effect of adenosine and
adenosine receptor antagonist on Müller cell potassium channel in
Rat chronic ocular hypertension models. Sci Rep.
5(11294)2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wang ZF and Yang XL: Glutamate
receptor-mediated retinal neuronal injury in experimental glaucoma.
Sheng Li Xue Bao. 68:483–491. 2016.PubMed/NCBI(In Chinese).
|
|
58
|
Vogler S, Pannicke T, Hollborn M, Grosche
A, Busch S, Hoffmann S, Wiedemann P, Reichenbach A, Hammes HP and
Bringmann A: Müller cell reactivity in response to photoreceptor
degeneration in rats with defective polycystin-2. PLoS One.
8(e61631)2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Yong PH, Zong H, Medina RJ, Limb GA,
Uchida K, Stitt AW and Curtis TM: Evidence supporting a role for
N-(3-formyl-3,4-dehydropiperidino)lysine accumulation in Müller
glia dysfunction and death in diabetic retinopathy. Mol Vis.
16:2524–2538. 2010.PubMed/NCBI
|
|
60
|
Alrashdi SF, Deliyanti D, Talia DM and
Wilkinson-Berka JL: Endothelin-2 injures the blood-retinal barrier
and macroglial Müller cells: Interactions with angiotensin ii,
aldosterone, and NADPH oxidase. Am J Pathol. 188:805–817.
2018.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Aragonès G, Rowan S, G Francisco S, Yang
W, Weinberg J, Taylor A and Bejarano E: Glyoxalase system as a
therapeutic target against diabetic retinopathy. Antioxidants
(Basel). 9(1062)2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Thompson K, Chen J, Luo Q, Xiao Y, Cummins
TR and Bhatwadekar AD: Advanced glycation end (AGE) product
modification of laminin downregulates Kir4.1 in retinal Müller
cells. PLoS One. 13(e0193280)2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Neusch C, Rozengurt N, Jacobs RE, Lester
HA and Kofuji P: Kir4.1 potassium channel subunit is crucial for
oligodendrocyte development and in vivo myelination. J Neurosci.
21:5429–5438. 2001.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Pannicke T, Frommherz I, Biedermann B,
Wagner L, Sauer K, Ulbricht E, Härtig W, Krügel U, Ueberham U,
Arendt T, et al: Differential effects of P2Y1 deletion on glial
activation and survival of photoreceptors and amacrine cells in the
ischemic mouse retina. Cell Death Dis. 5(e1353)2014.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Milton M and Smith PD: It's all about
timing: The involvement of Kir4.1 channel regulation in acute
ischemic stroke pathology. Front Cell Neurosci.
12(36)2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Zaika O, Palygin O, Tomilin V, Mamenko M,
Staruschenko A and Pochynyuk O: Insulin and IGF-1 activate
Kir4.1/5.1 channels in cortical collecting duct principal cells to
control basolateral membrane voltage. Am J Physiol Renal Physiol.
310:F311–F321. 2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Nitulescu GM, Van De Venter M, Nitulescu
G, Ungurianu A, Juzenas P, Peng Q, Olaru OT, Grădinaru D, Tsatsakis
A, Tsoukalas D, et al: The Akt pathway in oncology therapy and
beyond (Review). Int J Oncol. 53:2319–2331. 2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Serin Y and Acar Tek N: Effect of
circadian rhythm on metabolic processes and the regulation of
energy balance. Ann Nutr Metab. 74:322–330. 2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Lemmer B and Oster H: The role of
circadian rhythms in the hypertension of diabetes mellitus and the
metabolic syndrome. Curr Hypertens Rep. 20(43)2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Di R, Luo Q, Mathew D and Bhatwadekar AD:
Diabetes alters diurnal rhythm of electroretinogram in db/db mice.
Yale J Biol Med. 92:155–167. 2019.PubMed/NCBI
|
|
71
|
Wang Q, Tikhonenko M, Bozack SN, Lydic TA,
Yan L, Panchy NL, McSorley KM, Faber MS, Yan Y, Boulton ME, et al:
Changes in the daily rhythm of lipid metabolism in the diabetic
retina. PLoS One. 9(e95028)2014.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Luo Q, Xiao Y, Alex A, Cummins TR and
Bhatwadekar AD: The diurnal rhythm of insulin receptor substrate-1
(IRS-1) and Kir4.1 in diabetes: Implications for a clock gene
Bmal1. Invest Ophthalmol Vis Sci. 60:1928–1936. 2019.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wang Y, An H, Liu T, Qin C, Sesaki H, Guo
S, Radovick S, Hussain M, Maheshwari A, Wondisford FE, et al:
Metformin improves mitochondrial respiratory activity through
activation of AMPK. Cell Rep. 29:1511–1523.e5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Alex A, Luo Q, Mathew D, Di R and
Bhatwadekar AD: Metformin corrects abnormal circadian rhythm and
Kir4.1 channels in diabetes. Invest Ophthalmol Vis Sci.
61(46)2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Schultze SM, Hemmings BA, Niessen M and
Tschopp O: PI3K/AKT, MAPK and AMPK signalling: Protein kinases in
glucose homeostasis. Expert Rev Mol Med. 14(e1)2012.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Lechner J, O'Leary OE and Stitt AW: The
pathology associated with diabetic retinopathy. Vision Res.
139:7–14. 2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Stefanini FR, Badaró E, Falabella P, Koss
M, Farah ME and Maia M: Anti-VEGF for the management of diabetic
macular edema. J Immunol Res. 2014(632307)2014.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Lai TW, Zhang S and Wang YT:
Excitotoxicity and stroke: Identifying novel targets for
neuroprotection. Prog Neurobiol. 115:157–188. 2014.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Welters A, Klüppel C, Mrugala J, Wörmeyer
L, Meissner T, Mayatepek E, Heiss C, Eberhard D and Lammert E:
NMDAR antagonists for the treatment of diabetes mellitus-Current
status and future directions. Diabetes Obes Metab. 19 (Suppl
1):S95–S106. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Bai N, Aida T, Yanagisawa M, Katou S,
Sakimura K, Mishina M and Tanaka K: NMDA receptor subunits have
different roles in NMDA-induced neurotoxicity in the retina. Mol
Brain. 6(34)2013.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Fuwa M, Kageyama M, Ohashi K, Sasaoka M,
Sato R, Tanaka M and Tashiro K: Nafamostat and sepimostat
identified as novel neuroprotective agents via NR2B
N-methyl-D-aspartate receptor antagonism using a rat retinal
excitotoxicity model. Sci Rep. 9(20409)2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Han N, Yu L, Song Z, Luo L and Wu Y:
Agmatine protects Müller cells from high-concentration
glucose-induced cell damage via N-methyl-D-aspartic acid receptor
inhibition. Mol Med Rep. 12:1098–1106. 2015.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Ozaki H, Inoue R, Matsushima T, Sasahara
M, Hayashi A and Mori H: Serine racemase deletion attenuates
neurodegeneration and microvascular damage in diabetic retinopathy.
PLoS One. 13(e0190864)2018.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Chen H, Ji Y, Yan X, Su G, Chen L and Xiao
J: Berberine attenuates apoptosis in rat retinal Müller cells
stimulated with high glucose via enhancing autophagy and the
AMPK/mTOR signaling. Biomed Pharmacother. 108:1201–1207.
2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Barialai L, Strecker MI, Luger AL, Jäger
M, Bruns I, Sittig ACM, Mildenberger IC, Heller SM, Delaidelli A,
Lorenz NI, et al: AMPK activation protects astrocytes from
hypoxia-induced cell death. Int J Mol Med. 45:1385–1396.
2020.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Ulbricht E, Pannicke T, Hollborn M, Raap
M, Goczalik I, Iandiev I, Härtig W, Uhlmann S, Wiedemann P,
Reichenbach A, et al: Proliferative gliosis causes mislocation and
inactivation of inwardly rectifying K(+) (Kir) channels in rabbit
retinal glial cells. Exp Eye Res. 86:305–313. 2008.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Sene A, Tadayoni R, Pannicke T, Wurm A, El
Mathari B, Benard R, Roux MJ, Yaffe D, Mornet D, Reichenbach A, et
al: Functional implication of Dp71 in osmoregulation and vascular
permeability of the retina. PLoS One. 4(e7329)2009.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Vacca O, Charles-Messance H, El Mathari B,
Sene A, Barbe P, Fouquet S, Aragón J, Darche M, Giocanti-Aurégan A,
Paques M, et al: AAV-mediated gene therapy in Dystrophin-Dp71
deficient mouse leads to blood-retinal barrier restoration and
oedema reabsorption. Hum Mol Genet. 25:3070–3079. 2016.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Siqueiros-Marquez L, Bénard R, Vacca O,
Charles-Messance H, Bolaños-Jimenez R, Guilloneau X, Sennlaub F,
Montañez C, Sahel JA, Rendon A, et al: Protection of glial Müller
cells by dexamethasone in a mouse model of surgically induced
blood-retinal barrier breakdown. Invest Ophthalmol Vis Sci.
58:876–886. 2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Liu XQ, Kobayashi H, Jin ZB, Wada A and
Nao IN: Differential expression of Kir4.1 and aquaporin 4 in the
retina from endotoxin-induced uveitis rat. Mol Vis. 13:309–317.
2007.PubMed/NCBI
|
|
91
|
Sun W, Li T, Ma H, Lin S, Xie M, Luo Y,
Tian R and Tang S: The effect of K+ channel opener
pinacidil on the transmembrane potassi channel protein Kir4.1 of
retinal Müller cells in vitro and diabetic rats. Panminerva Med.
62:268–270. 2020.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Jung E and Kim J: Aloin inhibits Müller
cells swelling in a rat model of thioacetamide-induced hepatic
retinopathy. Molecules. 23(2806)2018.PubMed/NCBI View Article : Google Scholar
|