Introduction
Premature infants with respiratory disorders usually
require inhalation of high levels of oxygen. Prolonged exposure to
high levels of oxygen may lead to lung injury, which often results
in widespread lung fibrosis (1).
The main manifestation of lung fibrosis is the uncontrolled
proliferation of lung fibroblasts (LFs) that are deposited in the
pulmonary interstitial space or replace the normal lung epithelium
(2). The hyperplasia and
replacement of fibrous tissue after tissue injury are noteworthy
pathophysiological processes of tissue repair. Discovering how to
effectively prevent LF proliferation in order to alleviate lung
fibrosis without affecting tissue repair is the key to curing. To
address this problem, the mechanism that regulates the cell cycle
during LF proliferation needs to be clarified.
A cell divides into two identical cells through the
precisely controlled G1, S, G2 and M phases
of the cell cycle. During cell cycle progression, cells that pass
the G1/S checkpoint of the cycle, characterized by the
initiation of DNA synthesis, are committed to dividing into two
complete cells (3). The
p16INK4a (p16)/cyclin D/CDK4/retinoblastoma-associated
protein (Rb)/E2F1 pathway is recognized as a key pathway in the
regulation of cell proliferation, for which E2F1 is a notable
downstream effector. Deregulated E2F1 activity due to the aberrance
of the upstream components in this pathway, such as inactivation of
p16, confers a growth advantage to cells (4-7).
During the G1 phase of the normal cell cycle, Rb is
inactivated by sequential phosphorylation events that are mediated
by various cyclin-dependent kinases (CDKs), leading to the release
of E2F transcription factors, the activation of numerous genes
(such as p19ARF and p53 et al) and cell cycle progression
(8). p16 is a cyclin-dependent
kinase inhibitor that positively regulates the expression of cyclin
D1 at the post-transcriptional level. p16 and cyclin D1
competitively bind to CDK4 to prevent cells from entering the
corresponding phase of the cell cycle and, therefore, inhibit its
protein kinase activity. This inhibition suppresses the
CDK4-mediated phosphorylation of the Rb protein. When
phosphorylated (p)-Rb binds to E2F1 to form a complex, the
transcriptional activation of E2F1 is inhibited, thereby preventing
the cell from transitioning from the G1 to the S phase
and inhibiting cell proliferation (6,9).
Therefore, together, p16, CDK4, Rb and E2F1 constitute a feedback
loop that directly regulates the cell proliferation cycle. Once the
p16 gene is mutated, transduction through the loop is unable to
proceed.
Our previous study found that LFs isolated from rats
exposed to hyperoxia in vivo showed significantly greater
proliferation compared with those from rats exposed to normoxia. In
addition, using flow cytometry, it was revealed that the percentage
of LFs in the S and G2/M phases increased, and the
proportion of LFs in the G0/G1 phase
decreased simultaneously (10). In
another study, we confirmed that 5-aza-2'-deoxycytidine, a DNA
methylation inhibitor, inhibited hyperoxia-induced LF proliferation
by restoring p16 expression (11).
However, the underlying mechanism has not been investigated and the
results from previous studies are incomplete. In the present study,
the protein expression of p16, cyclin D1, CDK4, Rb and E2F1 was
measured in LFs from rats exposed to hyperoxia or normoxia to
further understand the occurrence and development of the aberrant
LF proliferation induced by hyperoxia.
Materials and methods
Animals and oxygen exposure
The present study was approved by The China Medical
University Animal Research Committee (Shenyang, China; approval no.
2017PS140K). Healthy Sprague-Dawley rats (n=40; female:male, 4:1)
obtained from The Center of Animal Experiments, China Medical
University, were used for this experiment. The higher the
concentration of oxygen inhaled, the more obvious the changes in
lung fibrosis. A fraction of inspired oxygen (FiO2)
>85% (the lethal oxygen concentration) is frequently employed in
experimental studies (12-14).
In the present study, the newborn rats (<12-h old) were randomly
divided into the normoxia (FiO2, 21%) or hyperoxia
(FiO2, 90±2%) groups. The hyperoxia group was monitored
twice daily (MT-01 Gas Monitor). CO2 was absorbed by
soda lime to maintain a concentration of <0.5%. The average
birth weight of the rats was 5.12±0.26 g in the hyperoxia group and
4.98±0.19 g in the normoxia group. No statistically significant
difference was observed. Within 12 h of birth, the newborn rats
(with the mother rats) were housed in standard cages placed inside
425-l capacity plexiglass isolation chambers that received
humidified O2 or room air (30l/min) at ambient pressure
for up to 14 days. Each rat was fed at 20-26˚C and 12 h light/dark
cycle. The cages were opened for 0.5 h every day to add standard
laboratory food and water and change the padding. To avoid oxygen
toxicity, the mother rats were rotated between the normoxia and
hyperoxia environments every day.
Histological examination
On postnatal days 3, 7 and 14, 5 pup rats in each
group were sacrificed with an intraperitoneal injection of
pentobarbital sodium (100 mg/kg) and were exsanguinated by aortic
transection. Right lung tissue samples were washed with PBS, placed
in 4% paraformaldehyde at room temperature for 24 h, then serially
dehydrated with increasing concentrations of ethanol before being
embedded in paraffin. The paraffin-embedded lung tissue samples
were cut into 4-µm sections, stained with hematoxylin and eosin
(H&E; for 5 min) and Masson's trichrome staining (Weigert's
iron hematoxylin solution for 3 min, Ponceau xylidine and fuchsin S
solution for 30 min) at room temperature, examined using light
microscopy and assessed for the presence of intra-alveolar edema,
inflammatory cell infiltration and fibrosis.
Primary culture of LFs
The rats in both the hyperoxia and normoxia groups
were euthanized on postnatal days 3, 7 and 14. The left lungs were
quickly removed from the chest, minced into 1-mm3 pieces
and digested in 0.2% trypsin and 0.016% deoxyribonuclease I in
minimum essential medium (MEM; Beijing Solarbio Science &
Technology Co., Ltd.) for 2-4 h at 37˚C in a shaking water bath.
Next, 10% FBS (Clark Bioscience) was added to the filtered cell
suspensions to stop further digestion. The harvested cells were
obtained by centrifugation at 400 x g for 3 min at 4˚C, and then
cultured in air and 5% CO2 at 37˚C in MEM with 10% FBS,
10,000 U/ml penicillin and 10,000 µg/ml streptomycin for 72 h. The
LF purity was confirmed at >95% by immunocytochemical staining
for vimentin. Briefly, cultured LFs were fixed in 4%
paraformaldehyde for 30 min at 37˚C. Next, a primary antibody
against vimentin (1:100; cat. no. sc-5565; Santa Cruz
Biotechnology, Inc.) was added to the cells and incubated overnight
at 4˚C. Then, cells were stained with goat anti-rat secondary
antibody (1:100; Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd.) for 30 min at 37˚C. When passaged, the cells were seeded into
150-cm2 culture flasks at a density of
2.5x105 or 5x105 cells/flask. All the
experiments were performed using fibroblasts from passage number
two.
MTT assay
A
3-(4,5-dimethy-lthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay
was performed for cell proliferation. Cells were seeded into
96-well plates at a density of 5x104 cells per well and
cultured at 37˚C in air and 5% CO2 for 24 h. Next, 20 µl
of 5 mg/ml MTT (Sigma-Aldrich; Merck KGaA) was added into each well
and the cells were cultured for an additional 4 h at 37˚C. After
culture, the supernatant was discarded and the resulting formazan
crystals were solubilized by adding 150 µl of DMSO to each well.
The optical density level was measured at a wavelength of 570 nm.
Experiments were performed in quintuplicate.
Methylation-specific polymerase chain
reaction (MSP)
LFs from the hyperoxia and normoxia groups were
collected at 120 h post-incubation in MEM with 10% FBS. A ZR-96
Quick-gDNA™ kit (Zymo Research Corp.) was used to extract the DNA
as recommended by the manufacturer. An EZ DNA Methylation-Gold™ kit
(Zymo Research Corp.) was used to perform the bisulfite
modification of the genomic DNA according to the manufacturer's
instructions. PCR amplification was performed using p16 promoter
gene fragment-specific primers for methylated or unmethylated DNA
(Sangon Biotech Co., Ltd.). The primers used to amplify
unmethylated p16 were as follows: Sense,
5'-TTTTTGGTGTTAAAGGGTGGTGTACT-3' and antisense,
5'-CACAAAAACCCTCACTCACAACAA-3', which yielded a 132-bp fragment.
The primers used to amplify methylated p16 were as follows: Sense,
5'-GTGTTAAAGGGCGGCGTAGC-3' and antisense,
5'-AAAACCCTCACTCGCGACGA-3', which yielded a PCR product of 122 bp.
PCR was performed under the following conditions: 95˚C for 4 min,
followed by 25 cycles of 94˚C for 25 sec, 62˚C for 25 sec and 72˚C
for 30 sec, and a final extension at 72˚C for 5 min. CpGenome
universal methylated DNA (EMD Millipore; Merck KGaA) was used as
the control for methylated DNA. The PCR-amplified products were
separated by electrophoresis on a 2% agarose gel and visualized
using ethidium bromide staining for 20 min under ultraviolet light.
Images were then captured and the DNA methylation of p16 was
determined by MSP as previously described (15).
Western blotting
Western blotting was performed as previously
described (7). The following
specific primary antibodies were used: Anti-cyclin D1 (cat. no.
SC-450; 1:500), anti-CDK6 (cat. no. SC-7961; 1:500), anti-p16 (cat.
no. sc-74400; 1:400), anti-Rb (cat. no. sc-102; 1:500) and
anti-p-Rb (cat. nos. ser-795 and sc-21875; 1:200) obtained from
Santa Cruz Biotechnology, Inc. An anti-CDK4 antibody (cat. no.
12790S; 1:1,000) was obtained from Cell Signaling Technology, Inc.
A β-actin antibody was obtained (cat. no. A-3853 Sigma-Aldrich;
Merck KGaA) and used at a 1:1,000 dilution. The proteins in the
membrane were detected using an enhanced chemiluminescence system
(ECL Advance; Amersham Biosciences; Cytiva) and images were
captured and analyzed using a BioRad ChemiDoc XRS system (BioRad
Laboratories, Inc.).
Statistical analysis
The values are expressed as the mean ± standard
deviation. SPSS software (v17.0; SPSS, Inc.) was used for the
statistical analyses. The statistical significance of the
differences was analyzed using an unpaired two-tailed t-test for
comparison between two groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
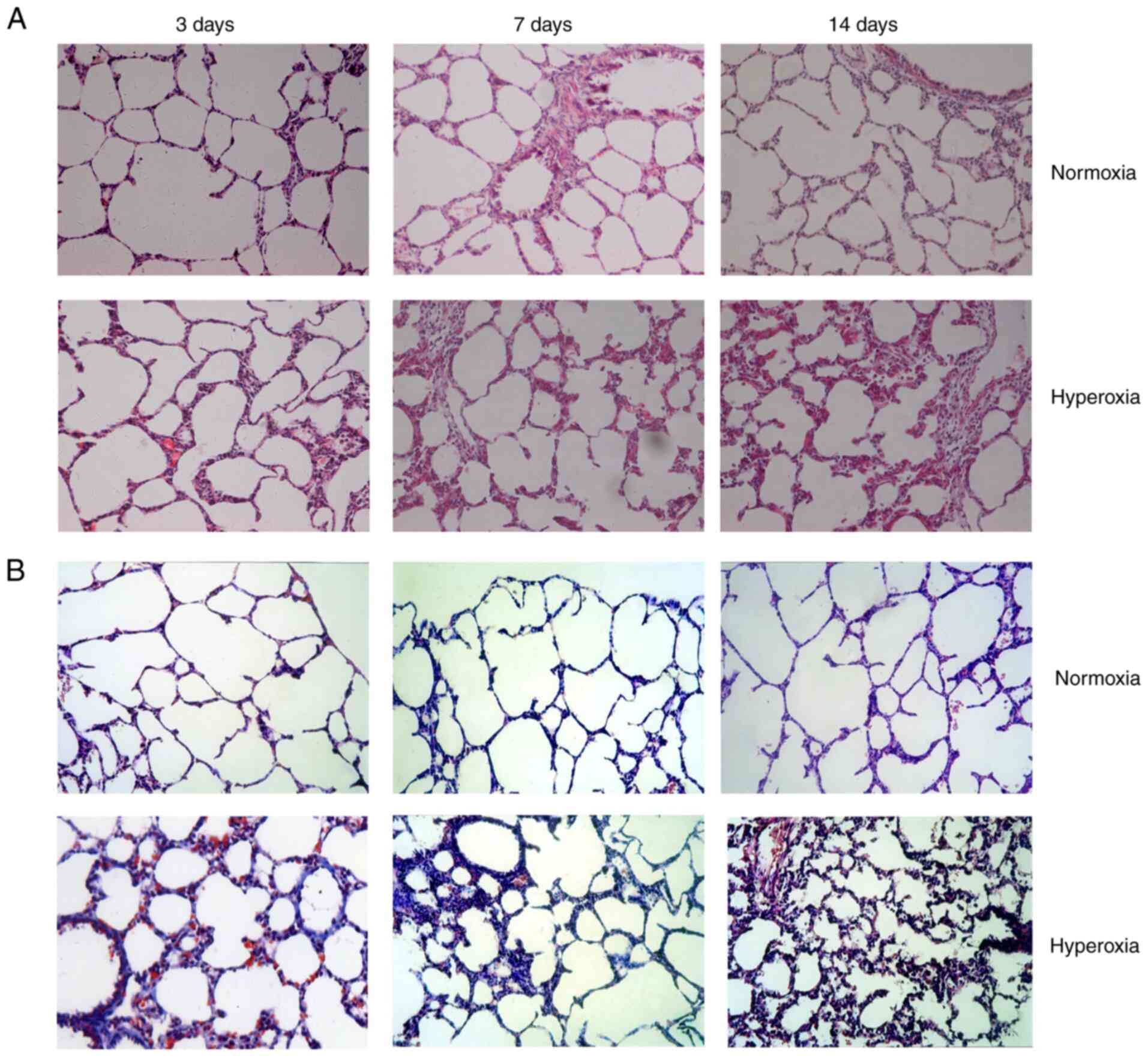
Effect of hyperoxia on lung histology
and morphometrics
Normal structure and alveolarization were observed
in the normoxia group on postnatal days 3, 7 and 14. No obvious
differences in pathology were observed between the groups on
postnatal day 3. Hyperoxia treatment induced the development of a
disordered lung tissue structure, alveolar collapse, obvious
alveolar wall thickening and numerous blue-stained stripes and
flakes, indicating collagen deposition, and these changes were
present on postnatal day 7 and increased over time through
postnatal day 14 (Fig. 1).
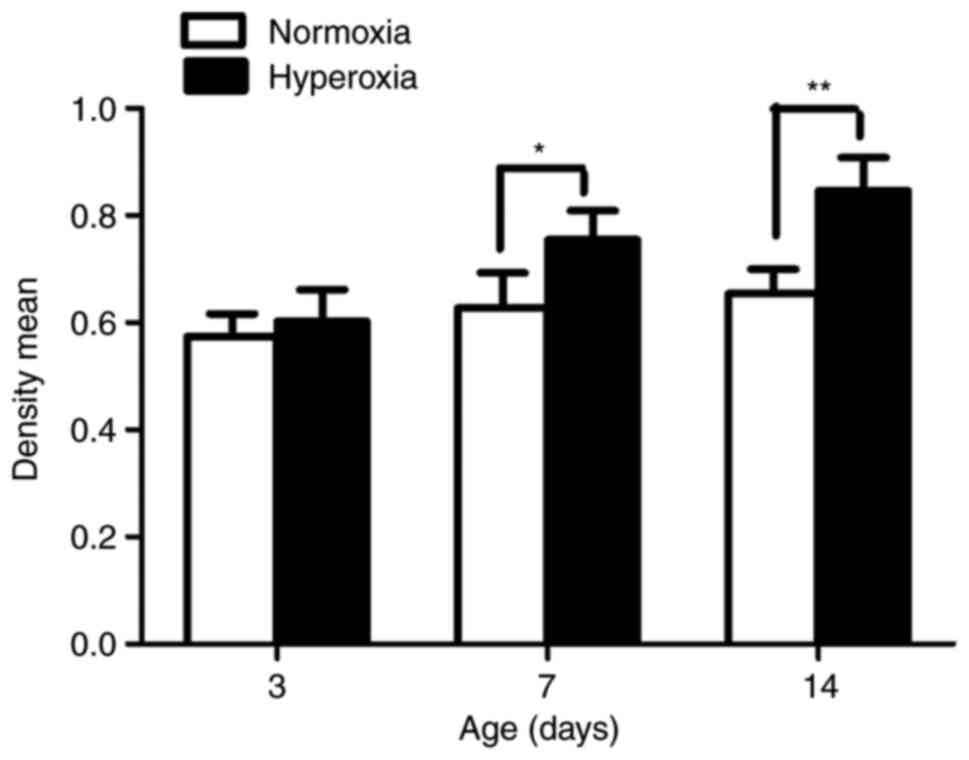
Effect of hyperoxia on cell
proliferation
As seen in Fig. 2,
the LFs isolated from the rats exposed to hyperoxia in vivo
showed significantly greater proliferation compared with the LFs
isolated from normoxia-exposed animals on postnatal day 7 and 14
(P<0.05 and P<0.01, respectively).
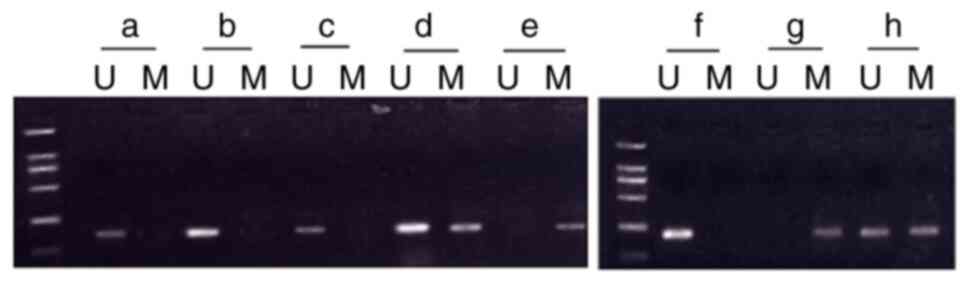
Effect of hyperoxia on p16 methylation
rate and p16 protein expression
No methylation was observed in any of the
normoxia-exposed groups (postnatal days 3, 7 and 14). In the
hyperoxia-exposed group, the rate of p16 promoter methylation on
day 7 was 50% (n=10; the complete methylation rate was 30% and the
partial methylation rate was 20%). The rate of p16 promoter
methylation on day 14 was 80% (n=10; the complete methylation rate
was 70% and the partial methylation rate was 10%). No methylation
was observed on day 3 in the hyperoxia-exposed group (Fig. 3).
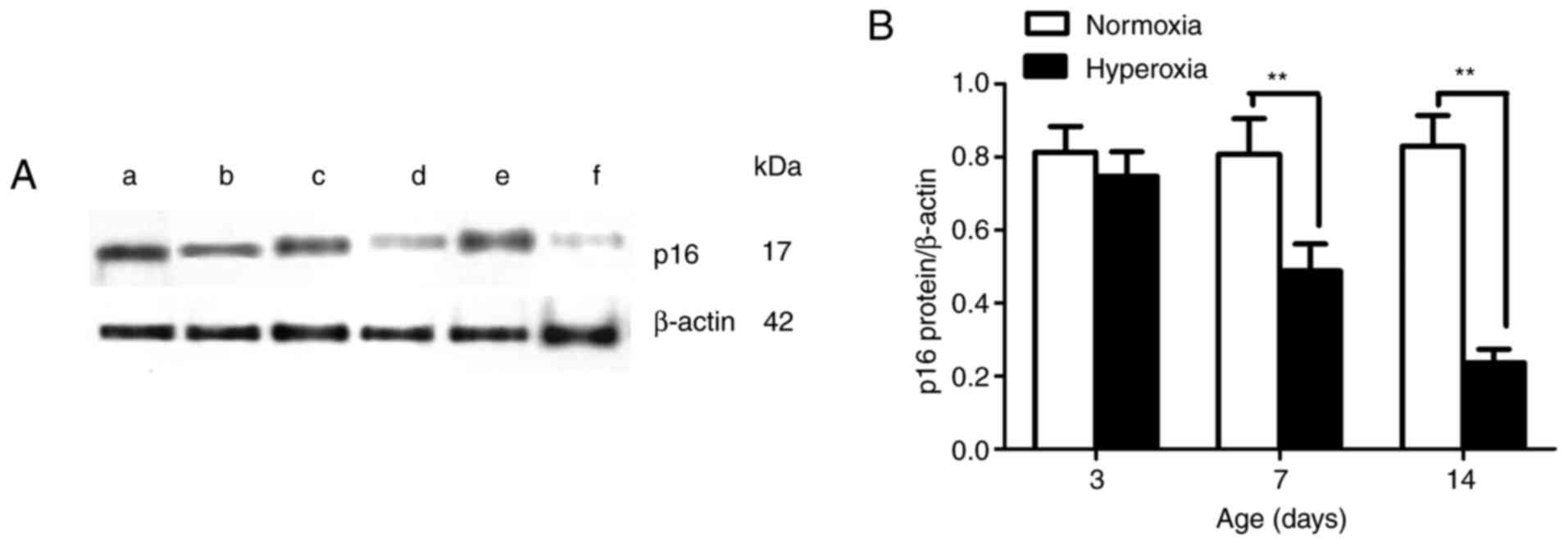
The newborn rats that were exposed to hyperoxia
displayed lower protein expression levels of p16 in LFs on
postnatal days 7 and 14 compared with the newborn rats that were
exposed to normoxia (P<0.01; Fig.
4). No statistically significant differences were found in the
LFs in the two groups on day 3. These results suggest that the
hyperoxia-induced p16 gene promoter hypermethylation in the LFs may
have suppressed or blocked the protein expression of p16.
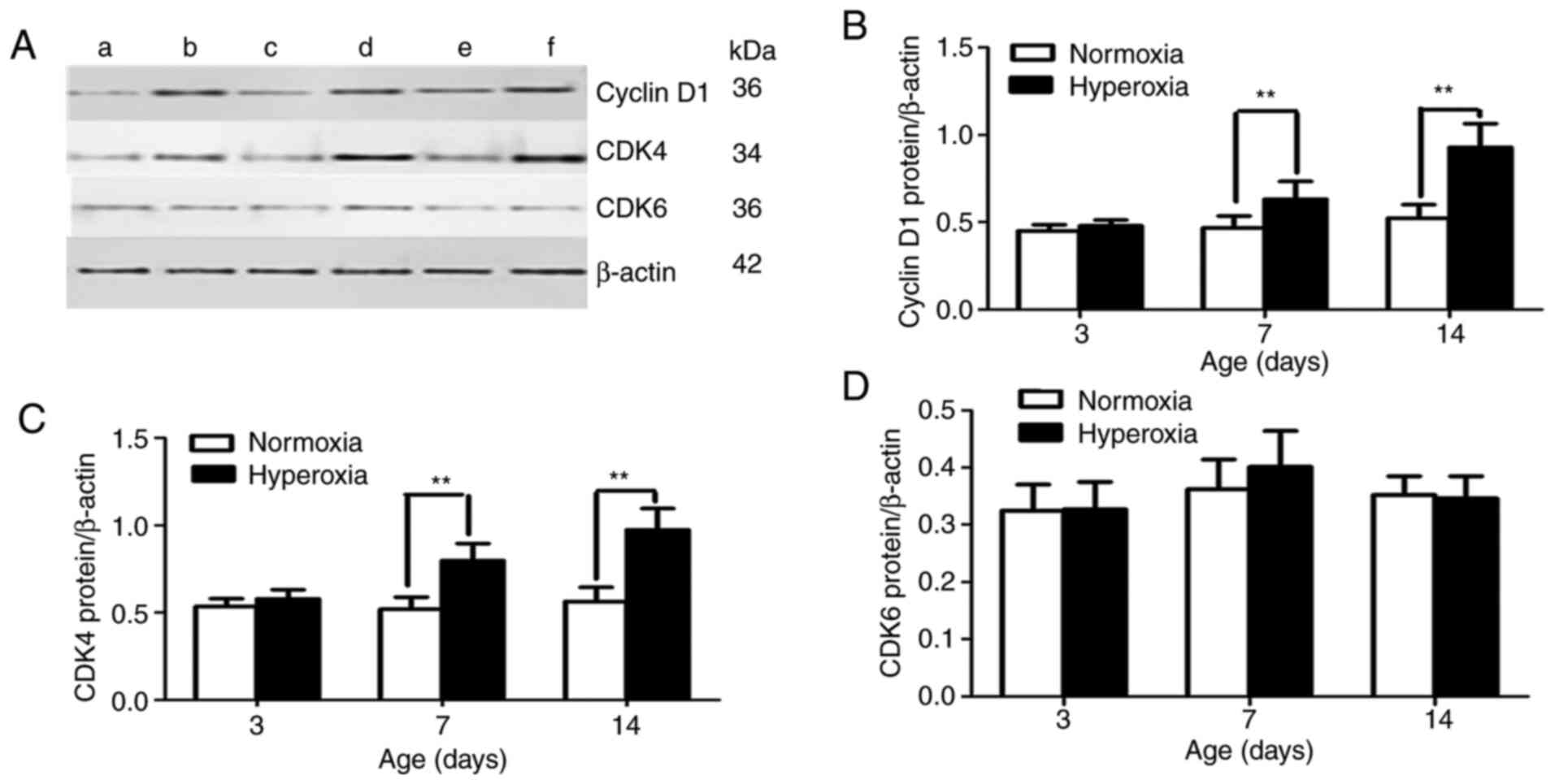
Effect of hyperoxia on cyclin D1, CDK4
and CDK6 protein expression
By controlling cell proliferation in the
G1 phase, p16 inhibits the ability of the cyclin D/CDK4
complex to phosphorylate p-Rb. Therefore, p16 loss is correlated
with cyclin D1 upregulation. Protein levels of cyclin D1 and CDK4
were examined in cultured lung cells in the present study. On
postnatal day 3, no statistically significant difference was
observed between the cyclin D1 and CDK4 protein expression levels
in the LFs in the two groups (Fig.
5). The cyclin D1 and CDK4 protein expression levels were
significantly greater in the hyperoxia-exposed group compared with
those in the normoxia-exposed group on postnatal days 7 and 14 (all
P<0.01; Fig. 5B and C). However, no statistically significant
difference was found in CDK6 protein expression between the two
groups at the different time points (P>0.05; Fig. 5D).
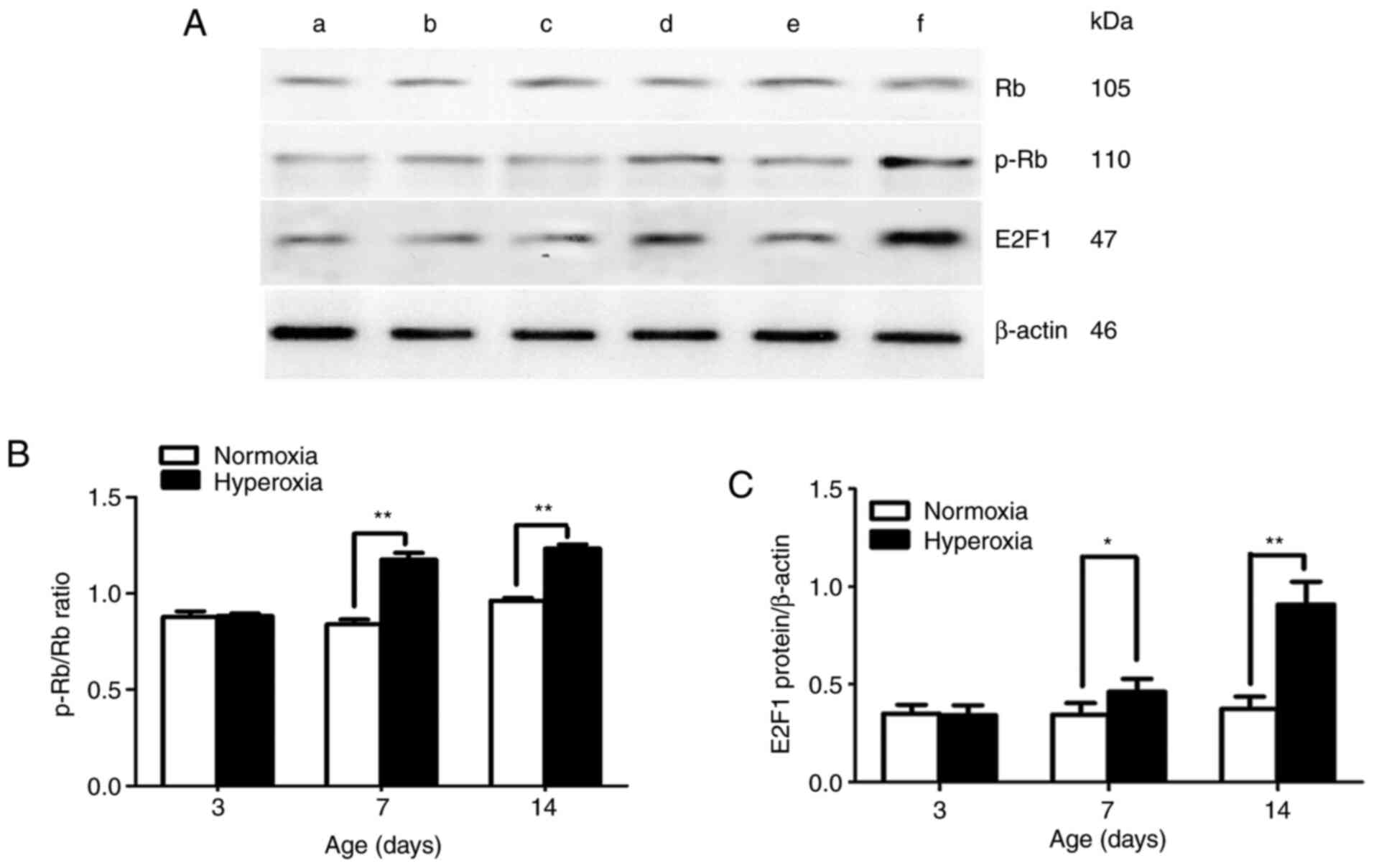
Effect of hyperoxia on Rb, p-Rb and
E2F1 protein expression
In the present study, no statistically significant
difference was found in Rb protein expression between the two
groups at the different time points (days 3, 7 and 14). p-Rb/Rb
ratio and E2F1 protein expression were significantly greater in the
hyperoxia-exposed group compared with that in the normoxia-exposed
group on postnatal days 7 and 14 (P<0.05 and P<0.01,
respectively; Fig. 6B and C).
Discussion
Excessive LF proliferation is a key process in
pulmonary fibrosis, and abnormal regulation of the cell cycle at
the G1/S phase is necessary for hyperoxia-induced LF
overproliferation (10). Our
previous study found that exposure to hyperoxia weakened the
G1/S checkpoint, enabling more LFs to pass this
checkpoint and progress from the G1 phase to the S and
G2/M phases to complete DNA replication; this phenomenon
promotes eventual LF division and proliferation (10). In the transition from the
G1 phase to the S phase, the activity of cyclin
D1-CDK4/6 plays a decisive role. Once activated, the Rb protein is
phosphorylated by cyclin D1-CDK4 to become p-Rb, which releases
sequestered E2F transcription factors, thereby promoting the
transcription of genes required for cell cycle progression
(16). Cell cycle progression is
under the control of CDK inhibitors, such as p16, that
competitively bind to CDK4/6 kinases and prevent them from binding
to their regulator cyclin D1; this blocks Rb protein
phosphorylation, which sequesters the E2F transcription factors
that control the transcription of S-phase genes (17). Therefore, the p16/cyclin
D/CDK4/Rb/E2F pathway is a critical regulator of the important
G1 phase to S phase transition of the cell cycle
(18-20).
Yue et al (21) first showed that the p16 promoter was
methylated in the lung tissues of hyperoxia-exposed mice, and that
p16 promoter methylation increased as the duration of hyperoxia
exposure increased. We previously repeated this experiment at the
cellular level and reached the same conclusion, namely, that
exposure to hyperoxia inhibited p16 gene expression in LFs and that
high levels of p16 methylation in the promoter region may be the
main cause of p16 transcriptional inactivation (11). The loss of p16 function may promote
cellular proliferation and impair cell cycle arrest or senescence,
allowing the survival of genetically damaged cells (22). The p16 protein acts as a cell
proliferation inhibitor by competitively binding to CDK4/6 kinases,
which prevents them from binding to their regulator cyclin D1 and
blocks Rb protein phosphorylation, thus leading to cell cycle
arrest (23). The loss of p16
during G1/S phase cell cycle regulation is closely
associated with the upregulation of cyclin D1 and CDK4, which act
synergistically with p16 loss to subvert the control of the
G1 phase in Rb-positive normal cells (24,25).
In the present study, cyclin D1 and CDK4 were
expressed at significantly higher levels in the LFs of
hyperoxia-exposed rats compared with the levels in the LFs from
normoxia-exposed rats on postnatal days 7 and 14. However, no
statistically significant difference was observed in CDK6 protein
expression between the two groups at the different time points.
This finding indicated that the loss of p16 expression was an early
event in abnormal LF proliferation that activated the cyclin
D1-CDK4 complex. The Rb protein was phosphorylated by the cyclin
D1-CDK4 complex to become p-Rb.
Rb is the link between extracellular signals and
nuclear signals (26). Rb exhibits
two phosphorylation states. The non-phosphorylated form of Rb is
expressed in the G0/G1 phase of the cell
cycle, and it is phosphorylated in the S/G2 phase, which
suggests that Rb is a key regulatory gene (27). In its hypo-phosphorylated form, the
Rb proteins acts as a cell cycle regulator to induce G1
arrest. Dysfunction of the proteins involved in the p16 pathway,
such as p16 gene deletion and cyclin D1 upregulation, leads to Rb
phosphorylation, subsequent G1/S phase transition and
uncontrolled cell proliferation (28-30).
The present study first determined the overall
expression level of Rb in LFs and found no significant difference
between the Rb expression levels in the hyperoxia-exposed and
normoxia-exposed groups. This finding showed that hyperoxia did not
change the overall expression level of Rb. The Rb protein contains
16 cyclin-dependent kinase phosphorylation sites. The functional
state of the Rb protein depends on its phosphorylation at these
different sites. Ser-795 phosphorylation indicates that Rb has
transitioned from an active non-phosphorylated state to an inactive
phosphorylated state. Ser-795 is phosphorylated by cyclin D1/CDK4,
which arrests the Rb-mediated cell cycle (31). The current study next examined the
expression levels of p-Rb (Ser-795) in LFs and found that the
p-Rb/Rb ratio was significantly higher in the hyperoxia-exposed
group compared with that in the normoxia-exposed group on postnatal
days 7 and 14. This finding indicated that hyperoxia did not change
the overall expression levels of Rb in LFs, but did change its
phosphorylation state.
Once Rb is phosphorylated, it induces the release of
E2F in the late G1 phase, which in turn enhances the
expression of genes that encode the regulatory proteins that are
necessary for cell cycle progression (27). E2F family members play a noteworthy
role during the G1/S transition in the mammalian cell
cycle. The E2F family is generally divided into two groups by
function: The transcriptional activators and the transcriptional
repressors. Activators such as E2F1, E2F2 and E2F3a promote and
help perpetuate the cell cycle, while repressors inhibit the cell
cycle (32). E2F1 upregulation
stimulates cellular proliferation. In numerous types of human
tumors (such as breast cancer, lung tumors, et al), E2F1
overexpression accelerates the transition of cells from the
G1 phase to the S phase, resulting in excessive cell
proliferation, malignant transformation and tumor formation
(33-35).
In the present study, a high level of E2F1 expression was observed
in primary cultured LFs from hyperoxia-exposed rats.
Our previous study determined that the p16 promoter
methylation induced by hyperoxia plays a role in the excessive
proliferation of LFs (11). The
current study found that the loss of p16 expression activated the
cyclin D1-CDK4 complex, and that the Rb protein was phosphorylated
by cyclin D1-CDK4 to become p-Rb, after which E2F1 binding on it
could be released. E2F1 upregulation accelerated the transition of
cells, resulting in excessive LF proliferation. In summary, the
disruption of the p16/cyclin D/CDK4/Rb/E2F1 pathway may play a key
role in the aberrant LF proliferation induced by hyperoxia.
Acknowledgements
The authors would like to thank Dr Xindong Xue
(Shengjing Hospital of China Medical University, Shenyang,
Liaoning, China) for advice on experimental design and support of
the study.
Funding
Funding: No funding was received.
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ performed the experiments. ZC and SH collected
and analyzed the data. HW designed the study and was the major
contributor in writing the manuscript. All authors read and
approved the final manuscript. ZC and SH confirmed the authenticity
of all the raw data.
Ethics approval and consent to
participate
The current study was approved by the Medical Ethics
Committee at the China Medical University (grant no.
2017PS140K).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen CM: Therapy for neonatal
hyperoxia-induced lung injury. Pediatr Neonatol. 55:329–330.
2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Crapo JD, Barry BE, Foscue HA and
Shelburne J: Structural and biochemical changes in rat lungs
occurring during exposures to lethal and adaptive dose of oxygen.
Am Rev Respir Dis. 122:123–143. 1980.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Reddy GP: Cell cycle: Regulatory events in
G1->S transition of mammalian cells. J Cell Biochem. 54:379–386.
1994.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Al-Khalaf HH, Colak D, Al-Saif M,
Al-Bakheet A, Hendrayani SF, Al-Yousef N, Kaya N, Khabar KS and
Aboussekhra A: p16(INK4a) positively regulates cyclin D1 and E2F1
through negative control of AUF1. PLoS One.
6(e21111)2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Haller F, Löbke C, Ruschhaupt M, Cameron
S, Schulten HJ, Schwager S, von Heydebreck A, Gunawan B, Langer C,
Ramadori G, et al: Loss of 9p leads to p16(INK4A) down-regulation
and enables RB/E2F1-dependent cell cycle promotion in
gastrointestinal stromal tumours (GISTs). J Pathol. 215:253–262.
2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wu Z and Yu Q: E2F1-mediated apoptosis as
a target of cancer therapy. Curr Mol Pharmacol. 2:149–160.
2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tyagi E, Liu B, Li C, Liu T, Rutter J and
Grossman D: Loss of p16 INK4A stimulates aberrant mitochondrial
biogenesis through a CDK4/Rb-independent pathway. Oncotarget.
8:55848–55862. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhao SM, Wu HM, Cao ML and Hna D: Primary
culture of lung fibroblasts from hyperoxia-exposed rats and a
proliferative characteristics study. Cytotechnology. 70:751–760.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhao S, Cao M, Wu H, Hu Y and Xue X:
5-aza-2'-deoxycytidine inhibits the proliferation of lung
fibroblasts in neonatal rats exposed to hyperoxia. Pediatr
Neonatol. 58:122–127. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen X, Zhang X and Pan J: Effect of
montelukast on bronchopulmonary dysplasia (BPD) and related
mechanisms. Med Sci Monit. 25:1886–1893. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Qi XJ, Ning W, Xu F, Dang HX, Fang F and
Li J: Fasudil, an inhibitor of Rho-associated coiled-coil kinase,
attenuates hyperoxia-induced pulmonary fibrosis in neonatal rats.
Int J Clin Exp Pathol. 8:12140–12150. 2015.PubMed/NCBI
|
|
14
|
Hu Y, Fu J and Xue X: Association of the
proliferation of lung fibroblasts with the ERK1/2 signaling pathway
in neonatal rats with hyperoxia-induced lung fibrosis. Exp Ther
Med. 17:701–708. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation specific PCR: A novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Huang Y, Wu G, Fan H, Ye J and Liu X:
Electroacupuncture promotes chondrocyte proliferation via
accelerated G1/S transition in the cell cycle. Int JMol Med.
31:1443–1448. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sharpless NE and DePinho RA: The INK4A/ARF
locus and its two gene products. Curr Opin Genet Dev. 9:22–30.
1999.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Williams RT, Barnhill LM, Kuo HH, Lin WD,
Batova A, Yu AL and Diccianni MB: Chimeras of p14ARF and p16:
Functional hybrids with the ability to arrest growth. PLoS One.
9(e88219)2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gil J and Peters G: Regulation of the
INK4b-ARF-INK4a tumour suppressor locus: All for one or one for
all. Nat Rev Mol Cell Biol. 7:667–677. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Sharpless NE: INK4a/ARF: A multifunctional
tumor suppressor locus. Mutat Res. 576:22–38. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yue X, Fu J, Xue X, Gao H, Liu D, Zong Z,
Wang W and Yuan Z: Detection of p16 promoter methylation in
premature rats with chronic lung disease induced by hyperoxia.
Pediatr Int. 52:520–526. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rayess H, Wang MB and Srivatsan ES:
Cellular senescence and tumor suppressor gene p16. Int J Cancer.
130:1715–1725. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Thompson JF, Scolyer RA and Kefford RF:
Cutaneous melanoma. Lancet. 365:687–701. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Myong NH: Cyclin D1 overexpression, p16
loss, and pRb inactivation play a key role in pulmonary
carcinogenesis and have a prognostic implication for the long-term
survival in non-small cell lung carcinoma patients. Cancer Res
Treat. 40:45–52. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Dobashi Y, Goto A, Fukayama M, Abe A and
Ooi A: Overexpression of cdk4/cyclin D1, a possible mediator of
apoptosis and an indicator of prognosis in human primary lung
carcinoma. Int J Cancer. 110:532–541. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mittnacht S: The retinoblastoma
protein-from bench to bedside. Eur J Cell Biol. 84:97–107.
2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang X, Huang K and Xu LN: Interaction
among Rb/p16, Rb/E2F1 and HDAC1 proteins in gallbladder carcinoma.
J Huazhong Univ Sci Technolog Med Sci. 29:729–731. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Haluska FG, Tsao H, Wu H, Haluska FS,
Lazar A and Goel V: Genetic alterations in signaling pathways in
melanoma. Clin Cancer Res. 12:2301s–2307s. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Palmieri G, Capone M, Ascierto ML,
Gentilcore G, Stroncek DF, Casula M, Sini MC, Palla M, Mozzillo N
and Ascierto PA: Main roads to melanoma. J Transl Med.
7(86)2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wikman H and Kettunen E: Regulation of the
G1/S phase of the cell cycle and alterations in the RB pathway in
human lung cancer. Expert Rev Anticancer Ther. 6:515–530.
2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Thomas NS, Pizzey AR, Tiwari S, Williams
CD and Yang J: p130, p107, and pRb are differentially regulated in
proliferating cells and during cell cycle arrest by
alpha-interferon. J Biol Chem. 273:23659–23667. 1998.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Clijsters L, Hoencamp C, Calis JJA, Marzio
A, Handgraaf SM, Cuitino MC, Rosenberg BR, Leone G and Pagano M:
Cyclin F controls cell-cycle transcriptional outputs by directing
the degradation of the three activator E2Fs. Mol Cell.
74:1264–1277.e7. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Stanelle J, Stiewe T, Theseling CC, Peter
M and Pützer BM: Gene expression changes in response to E2F1
activition. Nucleic Acides Res. 30:1859–1867. 2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Han S, Park K, Bae BN, Kim KH, Kim HJ, Kim
YD and Kim HY: E2F1expression is related with the poor survival of
lymph node-positive breast cancer patients treated with
fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res
Treat. 82:11–16. 2003.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Eymin B, Gazzeri S, Brambilla C and
Brambilla E: Distinct pattern of E2F1 expression in human lung
tumours: E2F1 is upregulated in small cell lung carcinomal.
Oncogene. 20:1678–1687. 2001.PubMed/NCBI View Article : Google Scholar
|