Introduction
Acute lung injury (ALI) and its most severe form,
acute respiratory distress syndrome (ARDS), are widespread
inflammatory processes in the lung caused by pneumonia, sepsis,
trauma and aspiration. ALI is characterized by pulmonary
interstitial edema, deteriorated lung compliance and subsequent
severe hypoxemia (1,2). Despite great progress in our
understanding of the pathophysiology of the condition, ALI still
has a relatively high mortality of 30-40% in intensive care units
of the United States and no specific drug has been approved for
clinical treatment. To reduce this mortality rate treatment and
prevention strategies for ALI need to be improved (3-5).
Lipopolysaccharide (LPS), a glycolipid of the outer
membrane of gram-negative bacteria (6), has been determined to be an important
factor in the activation of inflammatory signaling cascades in ALI
development (5). Intraperitoneal
injection of LPS in mice is an effective method to establish an ALI
model.
Protectin D1 (PD1) is a bioactive product generated
from docosahexaenoic acid (DHA) and has been reported to exert
anti-inflammatory effects in various disorders, including acute
kidney injury and neurodegenerative diseases (7,8). A
previous study suggested that PD1 could promote the resolution of
inflammation in a murine model of LPS-induced ALI (9). However, the underlying molecular
mechanism of the anti-inflammatory effect of PD1 in ALI is not well
understood.
Neutrophils are part of the host's first line of
defense against invading pathogens. Activated neutrophils can
release networks of nuclear DNA, enzymes and histones, which are
known as neutrophil extracellular traps (NETs) (10). This process, known as NETosis
(11), was initially considered to
play a host-protective role against infection (12). NETosis is important in the
pathophysiology of many conditions, including rheumatoid arthritis
(13), small vessel vasculitis
(14) and diabetes (15). Recently, NETs formation was
identified in LPS-induced ALI (16)
and further studies revealed that NETs was directly responsible for
alveolar damage and epithelial cell death (17). Targeting NET formation in ALI may
provide an avenue for treatment that has yet to be explored.
However, little is known regarding the beneficial role of PD1 in
inflammatory responses and NETs formation in ALI.
In the present study, the significance of PD1 in
mouse models of LPS induced ALI was explored and the effect of PD1
on NETs formation evaluated.
Materials and methods
Animals
A total of 28 male ICR mice (age, 6-8 weeks; weight,
21-28 g) were obtained from Jinan Pengyue Experimental Animal
Breeding Co. Ltd. All mice were housed at an ambient temperature of
20-22˚C with a relative humidity of 5±5% and on a 12-h light-dark
cycle in specific pathogen-free (SPF) facilities with free access
to food and water. All animal protocols were approved by the
Institutional Animal Care and Use Committee of Qilu Hospital
(Qingdao; China) and were performed in accordance with the
guidelines of the Animal Care and Use Committee (18).
Experimental model and treatments
The mice were randomly divided into four groups (7
mice per group): The control group, control + PD1 group, LPS group
and LPS + PD1 group. The LPS model was induced by intraperitoneal
injection of 10 mg/kg LPS, as previous described (19,20).
Mice in the control group and the control + PD1 group were
administered an equal volume of PBS. At 30 min after LPS/PBS
treatment, PD1 treated mice were injected with PD1 (2 ng/mouse) via
the angular vein. All mice were sacrificed 24 h after the first
administration of LPS. All animals were anesthetized with an
intraperitoneal administration of sodium pentobarbital (50 mg/kg)
before sacrifice. Euthanasia was performed under anesthesia using
cardiac puncture resulting in exsanguination. Death was confirmed
by cervical dislocation. Complete cessation of heartbeat and
breathing and disappearance of reflexes were used as indicators to
judge the death of mice. Lung tissues and blood samples were
obtained for further analyzes.
Histological analysis
The inferior lobe of the right lung was fixed for 24
h at room temperature with 4% formaldehyde, embedded in paraffin
and sectioned at a thickness of 5-µm. The sections were stained
with hematoxylin at 25˚C for 10 min and eosin (H&E) at 25˚C for
3 min. Histopathological scoring analysis of the lung tissues was
evaluated by TC and XW who were blinded to the group information,
with a light microscope at magnifications of x400 according to the
criteria previously described (21). Briefly, 24 areas in the lung
parenchyma were graded on a scale of 0-4 (0, absent and appears
normal; 1, light; 2, moderate; 3, strong; 4, intense) for
congestion, edema, infiltration of inflammatory cells and
hemorrhaging.
Lung wet-to-dry weight ratio
The superior lobe of the right lung was removed and
weighed to measure the wet weight. The lungs were then dried in an
oven at 60˚C for 3 days to obtain the dry lung weight.
Pulmonary wet/dry weight ratios were calculated as
follows: Wet/dry weight ratio=(wet lung weight-dry lung weight)/dry
lung weight.
Measurement of protein concentrations
and cytokines in bronchoalveolar lavage fluid (BALF)
The left lungs were rinsed with 1 ml of PBS through
the tracheal cannula three times to obtain BALF. The BALF was
centrifuged at 850 x g for 10 min at 4˚C. The supernatant was
collected for protein measurement using a bicinchoninic acid
protein assay kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Tumor necrosis factor (TNF-α) and
interleukin-6 (IL-6) levels in BALF were measured using TNF-α ELISA
kit [cat. no. EK282/3; MultiSciences (Lianke) Biotech. Co., Ltd.]
and IL-6 ELISA kit [cat. no. EK206/3; MultiSciences (Lianke)
Biotech. Co., Ltd.].
Measurement of serum TNF-α and
IL-6
The serum levels of TNF-α and IL-6 were determined
using TNF-α ELISA kit [cat. no. EK282/3; MultiSciences (Lianke)
Biotech. Co., Ltd.] and IL-6 ELISA kit [cat. no. EK206/3;
MultiSciences (Lianke) Biotech. Co., Ltd.] according to the
manufacturer's instructions.
Immunohistochemical analysis of
myeloperoxidase (MPO)
Immunohistochemical analysis of MPO was performed
according to previously described methods (22,23).
One portion of the lungs was fixed overnight at 4˚C in freshly
prepared formaldehyde (Sigma-Aldrich; Merck KGaA) in PBS, pH 7.4.
The tissues were then embedded in paraffin. Lung tissue sections
(at a thickness of 5 µm) were deparaffinized in xylene at room
temperature for 15 min, rehydrated using a descending ethanol
gradient and heated in 10 mM sodium citrate buffer (pH 6.0) at 95˚C
for 5 min in a microwave oven for antigen retrieval. Endogenous
peroxidase activity was blocked by immersing the slides in 3%
hydrogen peroxide at 15-25˚C for 10 min. After antigen retrieval,
the tissue sections were immersed in 5% BSA (Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature. The tissue sections were then
incubated with a primary antibody against MPO (1:100 dilution; cat.
no. ab9535; Abcam, Inc.) at 4˚C overnight. The tissue sections were
then rinsed in TBS and incubated with a horseradish peroxidase
(HRP)-conjugated secondary anti-rabbit antibody (1:1,000 dilution;
cat. no. ab97080; Abcam) for 1 h at room temperature. The tissue
sections were again washed in TBS and the bound peroxidase was
detected by incubating the tissue sections in 3,3'-diaminobenzidine
substrate at room temperature for 3 min. The tissue sections were
then washed in distilled water and the nuclei were counterstained
with Mayer's hematoxylin for 1 min at room temperature, followed by
two rinses in distilled water. The sections were dehydrated by
serial immersion, for 1 min each, in 70% ethanol and 95% ethanol,
followed by 2 min each in two changes of 100% ethanol and two
changes of xylene. A coverslip was mounted over each tissue section
using Pertex® mounting medium (Pioneer Research
Chemical, Ltd.). Imaging was performed using a fluorescence
microscope (Nikon MODEL ECLIPSE Ci-L microscope; Nikon Corporation)
at magnifications of x100 and 400.
Immunofluorescence
To detect NET produced by infiltrated neutrophils in
the lung tissue, immunofluorescent staining was performed using
anti-MPO and anti-histone H3 antibodies. Lung tissues were fixed
overnight at 4˚C in freshly prepared formaldehyde (Sigma-Aldrich;
Merck KGaA) in PBS, pH 7.4. The tissues were then embedded in
paraffin. Lung tissue sections (at a thickness of 6 µm) were
deparaffinized and rehydrated in descending concentrations of
ethanol. Antigen retrieval was achieved by treating sections with
10 mM citrate buffer and heating in a microwave oven for 15 min at
95˚C. Samples were then blocked with 20% normal donkey serum (cat.
no. ab7475; Abcam) overnight at 4˚C. After a thorough wash in 0.05%
PBS-Tween-20, the sections were sequentially incubated with rabbit
anti-MPO antibody (1:100 dilution; cat. no. ab9535; Abcam) and
mouse anti-histone H3 antibody (1:200 dilution; cat. no. ab1220;
Abcam) in a humidified chamber at 4˚C overnight. After being washed
again, the samples were incubated with secondary antibodies that
recognize citrullinated histone 3 (CitH3; donkey anti-mouse IgG;
Alexa Fluor® 488-conjugated; 1:500 dilution; cat. no.
ab150105; Abcam) and MPO (donkey anti-rabbit IgG; Alexa
Fluor® 647-conjugated; 1:500 dilution; cat. no.
ab150075; Abcam) for 1 h at room temperature. Following another
wash, the sections were incubated with
4',6-diamidino-2-phenylindole and dihydrochloride (DAPI; Wuhan
Servicebio Technology Co. Ltd.) solution at room temperature for 20
min and then mounted with anti-fade reagent (24,25).
Immunofluorescence images were obtained with a slide scanner
(Panoramic MIDI; 3DHISTECH Ltd.) at magnifications of x100 and 400
and analyzed with ImageJ v1.8.0 software (National Institutes of
Health).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6 software (GraphPad Software Inc.), and the data are
presented as the mean ± standard error (SE). Statistical
significance was assessed using one-way ANOVA followed by Tukey's
post hoc test. A value of P<0.05 was considered statistically
significant.
Results
PD1 attenuates LPS-induced lung
histopathological changes and pulmonary edema
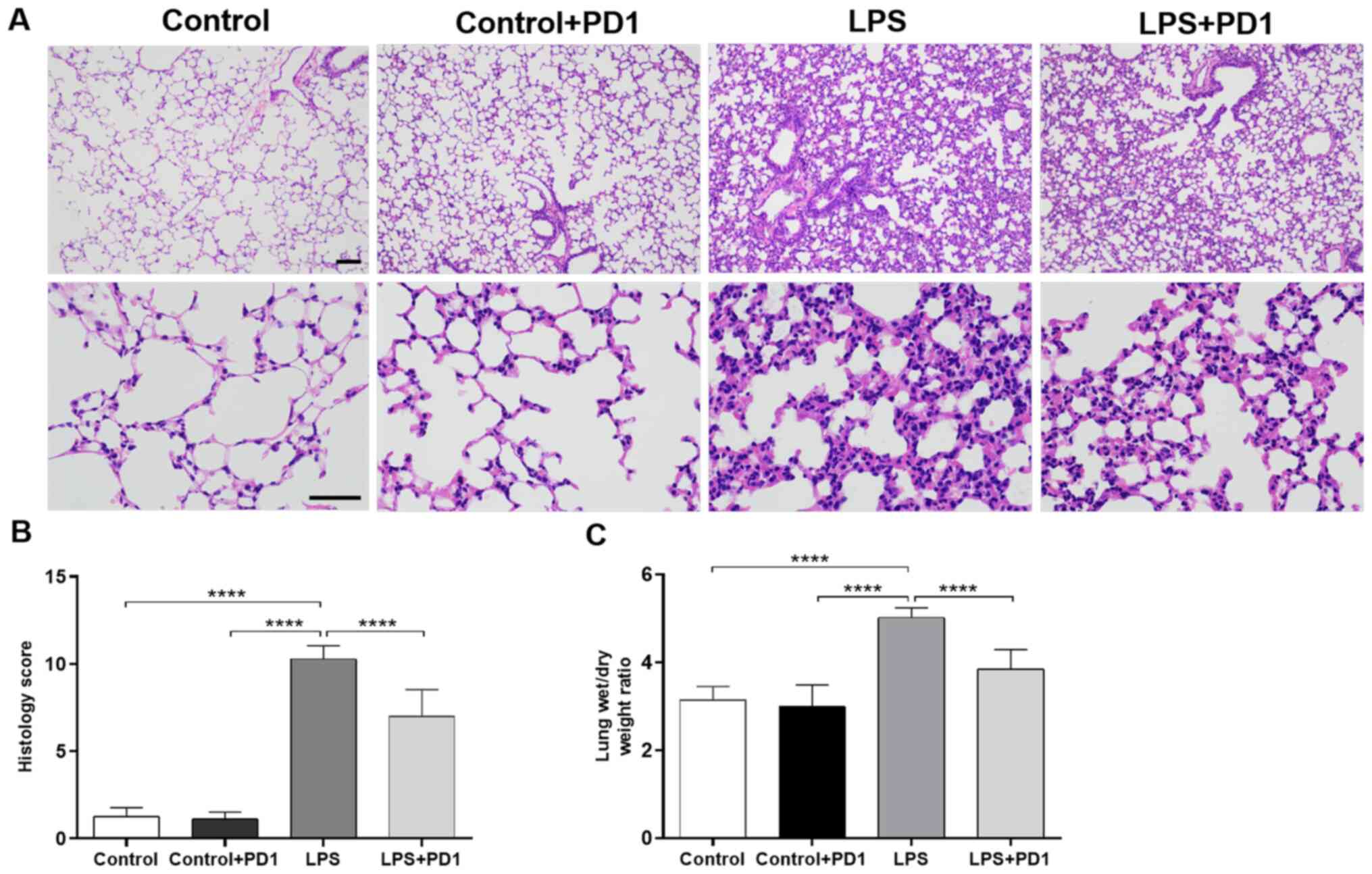
Histopathological analysis was used to determine the
protective effects of PD1 on LPS-induced ALI. As indicated by
Fig. 1A, lungs in the LPS group
showed extensive histopathological changes, including alveolar wall
thickening, interstitial edema, and pulmonary congestion, compared
to those of control group. PD1 attenuated the severity of lung
injuries induced by LPS challenge. As suggested in Fig. 1B, lung injury scores decreased
significantly after PD1 administration. The wet/dry weight ratio of
the lung was examined to estimate lung edema. As demonstrated in
Fig. 1C, the lung wet/dry weight
ratio in the LPS group was significantly increased compared with
that in the control group. However, this increase was significantly
reduced by PD1 treatment.
PD1 alleviates protein leakage and
reduces proinflammatory cytokine levels in BALF
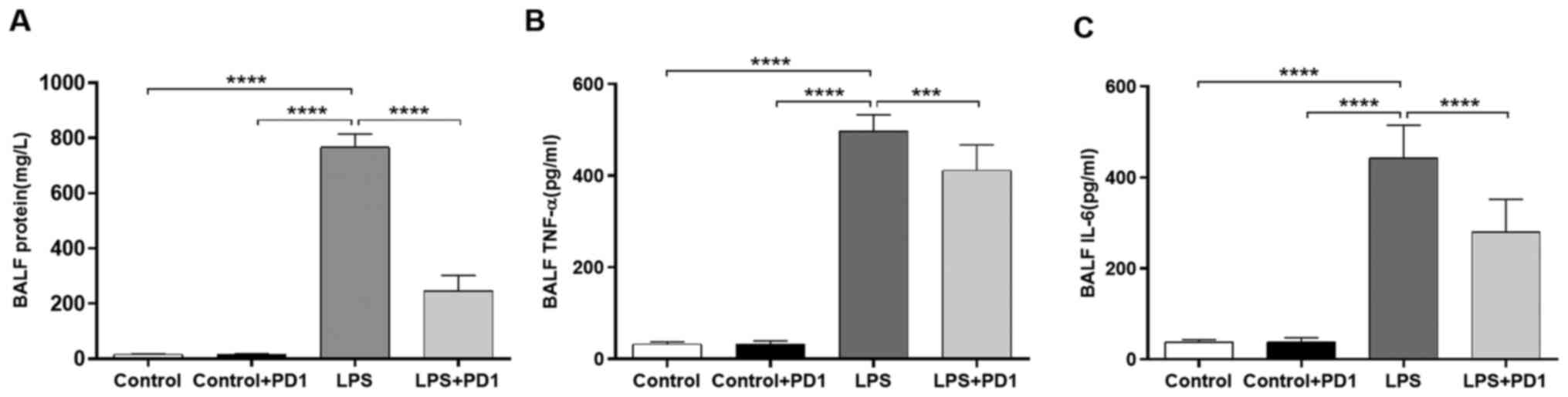
As demonstrated in Fig.
2A, the protein concentration in BALF was increased by LPS
challenge in comparison with control while PD1 administration
alleviated protein leakage in LPS mice. Proinflammatory cytokines
play an important role in the pathogenesis of ALI (26,27).
In the present study, the concentrations of TNF-α (Fig. 2B) and IL-6 (Fig. 2C) in BALF were significantly
increased in LPS-treated mice compared with control mice.
Administration of PD1 decreased LPS-induced inflammatory cytokine
(TNF-α and IL-6) production.
PD1 reduces serum proinflammatory
cytokine levels
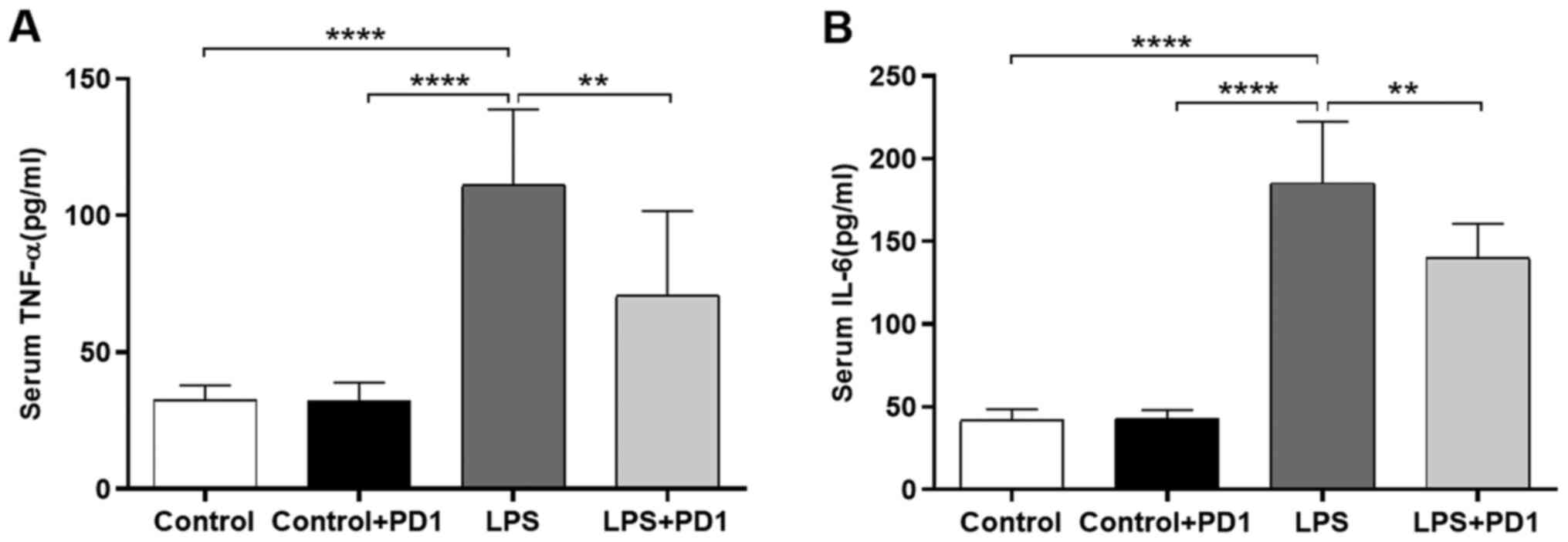
As indicated in Fig.
3, the concentrations of TNF-α and IL-6 in the serum were
significantly increased in the LPS group compared with the control
group. In contrast to that of the LPS group, treatment with PD1
significantly reduced TNF-α and IL-6 release into the serum.
PD1 reduces neutrophil infiltration
into lung tissue
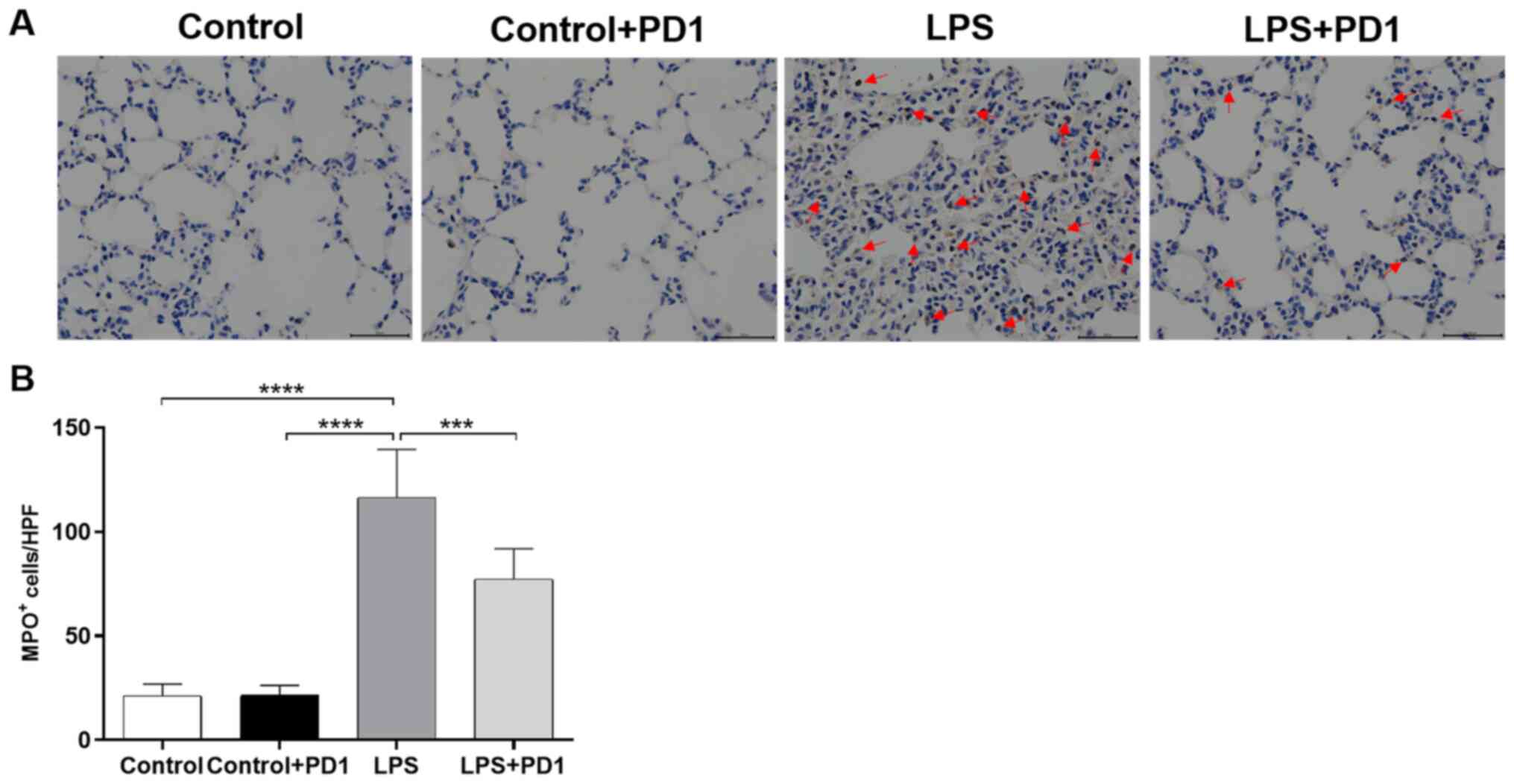
To investigate the infiltration and accumulation of
neutrophils in LPS-induced ALI, immunohistochemistry was carried
out on lung sections using an MPO antibody. The expression of MPO
(Fig. 4A and B) in the lung tissues of the LPS group was
significantly increased compared with that of the control group.
PD1 administration significantly reduced the LPS-induced MPO
increase.
PD1 significantly reduces NETs
formation in mouse lungs
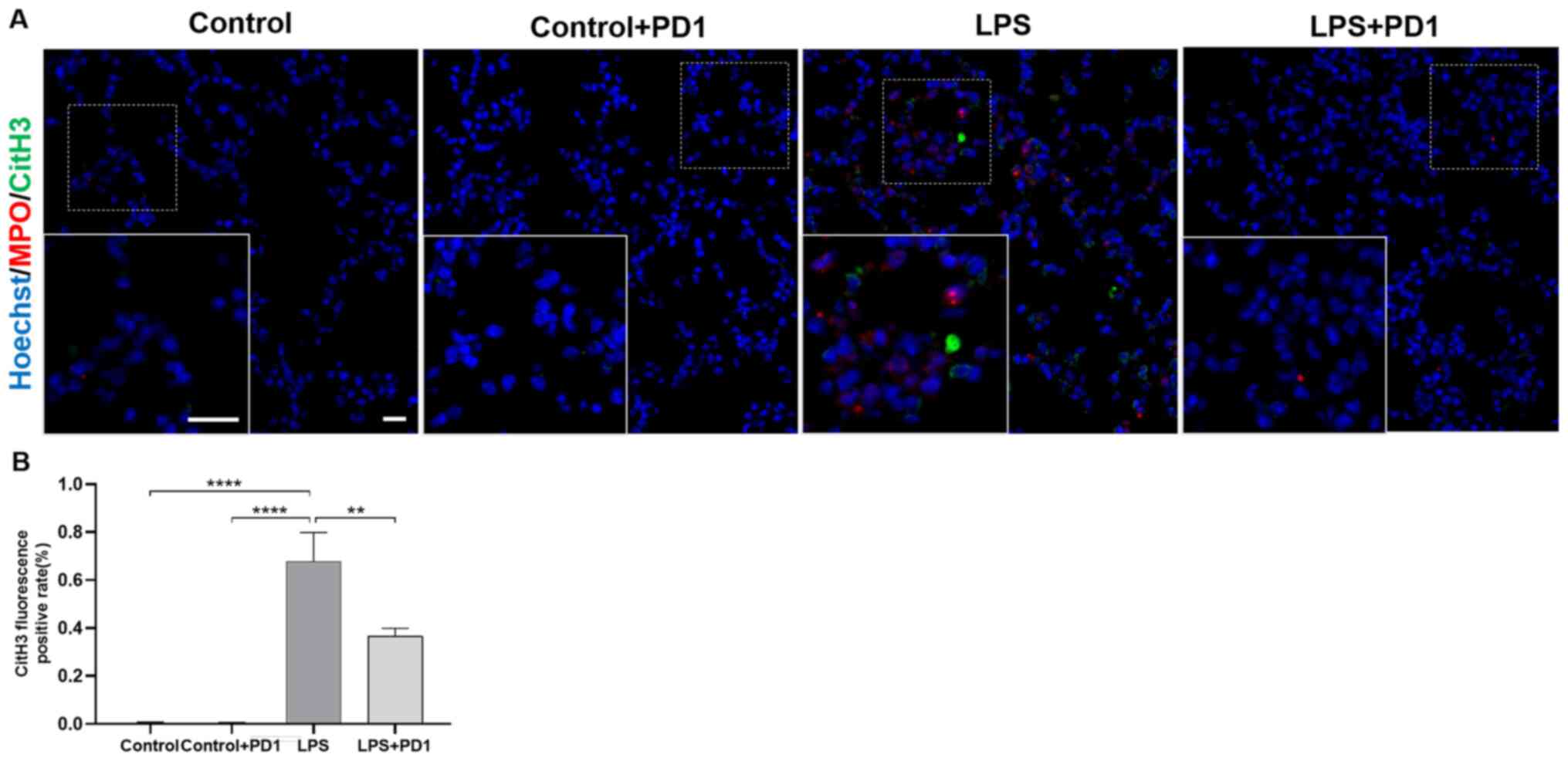
CitH3 is found in the extracellular space of
neutrophils along with DNA as components of NETs. CitH3 is widely
accepted as a marker of NETs formation (28,29).
NETs were identified on the basis of co-localization of CitH3 with
MPO using immunofluorescence. As demonstrated in Fig. 5, neutrophil infiltration, as
assessed by MPO and CitH3 colocalization, was significantly
increased in the LPS group in comparison with the control group.
Treatment with PD1 reduced neutrophil infiltration and NETs
formation in lung tissue of LPS mice, as demonstrated by reduced
CitH3/MPO co-localization.
Discussion
In the present study, the protective effect of PD1
on LPS-induced ALI in mice was evaluated. The data indicated that
PD1 significantly attenuated LPS-induced histopathological changes
and pulmonary edema and alleviated protein leakage in BALF in
comparison with an LPS control. Moreover, administration of PD1
reduced the BALF and serum levels of proinflammatory cytokines
TNF-α and IL-6, reduced the infiltration of inflammatory cells and
suppressed the formation of NETs in LPS-induced lung tissue.
ALI and ARDS, the major causes of respiratory
failure in intensive care units worldwide, are characterized by
uncontrolled inflammatory responses (30). Currently, clinical treatment is
limited to supportive care. Novel therapeutic methods or preventive
strategies are urgently needed (31). PD1, a bioactive product generated
from docosahexaenoic acid (DHA), has been reported to exert
anti-inflammatory effects in a murine model of LPS-induced ALI
(9). In the present study, the
protective effect of PD1 on LPS-induced ALI was further explored.
The present data revealed that intraperitoneal injection of LPS
resulted in the destruction of alveolar structures and pulmonary
edema and that PD1 exhibited substantial protective effects against
LPS-induced ALI, as evidenced by alleviated pathological injuries
and pulmonary edema. However, the underlying molecular mechanism of
the anti-inflammatory effect of PD1 on LPS-induced ALI is still not
well understood.
A key feature of LPS-induced ALI is an early, robust
neutrophilic response. Infiltration of neutrophils and release of
proinflammatory cytokines play important roles in ALI (32). A previous study suggested that LPS
may promote the production of inflammatory cytokines, which may
lead to neutrophil infiltration into lung tissues and initiate
focal inflammatory responses (33,34).
Excessive accumulation of neutrophils contributes to disturbances
in the alveolar-capillary barrier, resulting in the efflux of
protein-rich fluid and ultimately pulmonary edema (35). In the present study, the
concentrations of protein, TNF-α and IL-6 in BALF were
significantly increased in LPS-treated mice compared with control
mice. Furthermore, this increase was significantly suppressed by
PD1 treatment. Consistent with the BALF results, serum TNF-α and
IL-6 levels in the LPS group were increased significantly compared
with those of the control group and were significantly reduced by
PD1 administration.
It is well known that neutrophils are crucial in the
initiation and exacerbation of pathological processes and
inflammatory responses (32).
Inhibition of neutrophil recruitment has been demonstrated to
attenuate LPS-induced ALI by reducing the production of
proinflammatory cytokines (36). In
the present study, administration of PD1 significantly inhibited
neutrophil infiltration and accumulation in the lungs.
NETs have been identified as critical mediators of
pulmonary vascular dysfunction and the pathogenesis of lethal
endotoxemia (29,37). Inhibition of NET formation was
demonstrated to alleviate LPS-induced pulmonary dysfunction and
improve survival in a mouse model of lethal endotoxemia (29). Targeted intervention in NETs
formation may be a new strategy for the clinical treatment of AP.
In the present study, CitH3 released from infiltrating neutrophils
was measured to assess NETs formation in LPS-induced lung tissue.
CitH3 expression was colocalized with MPO, suggesting that CitH3
was released from infiltrating neutrophils. PD1 administration
significantly reduced neutrophil infiltration and NETs formation in
LPS-induced lung tissue.
In conclusion, the present study demonstrated that
PD1 protects against LPS-induced ALI by inhibiting the infiltration
of neutrophils and the formation of NETs in lung tissue. PD1 may be
a promising therapeutic candidate for the clinical treatment of
ALI.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
Scientific Research Foundation of Qilu Hospital (grant no.
QDKY2019RX11) and The Youth Program of the National Natural Science
Foundation of China (grant no. 81704164).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZW and DW designed the study. LZ, HL and ZW
performed the experiments and collected the data. XZ, ZL and CB
contributed to the interpretation of data for the work. RW, TC and
XW analyzed the data, and ZW and LZ prepared the manuscript and
assessed the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the
Institutional Animal Care and Use Committee of Qilu Hospital
(Qingdao), Shandong University (Qingdao, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferguson ND, Frutos-Vivar F, Esteban A,
Fernández-Segoviano P, Aramburu JA, Nájera L and Stewart TE: Acute
respiratory distress syndrome: Underrecognition by clinicians and
diagnostic accuracy of three clinical definitions. Crit Care Med.
33:2228–2234. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mokra D and Kosutova P: Biomarkers in
acute lung injury. Respir Physiol Neurobiol. 209:52–58.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Goss CH, Brower RG, Hudson LD and
Rubenfeld GD: Incidence of acute lung injury in the United States.
Crit Care Med. 31:1607–1611. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693.
2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rossol M, Heine H, Meusch U, Quandt D,
Klein C, Sweet MJ and Hauschildt S: LPS-induced cytokine production
in human monocytes and macrophages. Crit Rev Immunol. 31:379–446.
2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xu ZZ, Liu XJ, Berta T, Park CK, Lü N,
Serhan CN and Ji RR: Neuroprotectin/protectin D1 protects against
neuropathic pain in mice after nerve trauma. Ann Neurol.
74:490–495. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Duffield JS, Hong S, Vaidya VS, Lu Y,
Fredman G, Serhan CN and Bonventre JV: Resolvin D series and
protectin D1 mitigate acute kidney injury. J Immunol.
177:5902–5911. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li X, Li C, Liang W, Bi Y, Chen M and Dong
S: Protectin D1 promotes resolution of inflammation in a murine
model of lipopolysaccharide-induced acute lung injury via enhancing
neutrophil apoptosis. Chinese Med J (Engl). 127:810–814.
2014.PubMed/NCBI
|
|
10
|
Brinkmann V, Reichard U, Goosmann C,
Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y and Zychlinsky A:
Neutrophil extracellular traps kill bacteria. Science.
303:1532–1535. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kaplan MJ and Radic M: Neutrophil
extracellular traps: Double-edged swords of innate immunity. J
Immunol. 189:2689–2695. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fuchs TA, Abed U, Goosmann C, Hurwitz R,
Schulze I, Wahn V, Weinrauch Y, Brinkmann V and Zychlinsky A: Novel
cell death program leads to neutrophil extracellular traps. J Cell
Biol. 176:231–241. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sur Chowdhury C, Giaglis S, Walker UA,
Buser A, Hahn S and Hasler P: Enhanced neutrophil extracellular
trap generation in rheumatoid arthritis: Analysis of underlying
signal transduction pathways and potential diagnostic utility.
Arthritis Res Ther. 16(R122)2014.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Kessenbrock K, Krumbholz M, Schönermarck
U, Back W, Gross WL, Werb Z, Gröne HJ, Brinkmann V and Jenne DE:
Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med.
15:623–625. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Wong SL, Demers M, Martinod K, Gallant M,
Wang Y, Goldfine AB, Kahn CR and Wagner DD: Diabetes primes
neutrophils to undergo NETosis, which impairs wound healing. Nat
Med. 21:815–819. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S,
Dai M, Li Y, Li Q, Mao Z, et al: Neutrophil extracellular traps
contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS.
Oncotarget. 9:1772–1784. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Saffarzadeh M, Juenemann C, Queisser MA,
Lochnit G, Barreto G, Galuska SP, Lohmeyer J and Preissner KT:
Neutrophil extracellular traps directly induce epithelial and
endothelial cell death: A predominant role of histones. PLoS One.
7(e32366)2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kertz AF: Animal care and use: An issue
now and in the future. J Anim Sci. 74:257–261. 1996.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yu D, Fang X, Xu Y, Xiao H, Huang T, Zhang
Y, Ge Y, Li Y, Zong L and Gao J: Rev-erbα can regulate the
NF-κB/NALP3 pathway to modulate lipopolysaccharide-induced acute
lung injury and inflammation. Int Immunopharmacol. 73:312–320.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xiao Z, Jia B, Zhao X, Bi S and Meng W:
Attenuation of lipopolysaccharide-induced acute lung injury by
Cyclosporine-A via suppression of mitochondrial DNA. Med Sci Monit.
24:7682–7688. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hagiwara S, Iwasaka H, Maeda H and Noguchi
T: Landiolol, an ultrashort-acting beta1-adrenoceptor antagonist,
has protective effects in an LPS-induced systemic inflammation
model. Shock. 31:515–520. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhai KF, Duan H, Cui CY, Cao YY, Si JL,
Yang HJ, Wang YC, Cao WG, Gao GZ and Wei ZJ: Liquiritin from
glycyrrhiza uralensis attenuating rheumatoid arthritis via reducing
inflammation, suppressing angiogenesis, and inhibiting MAPK
signaling pathway. J Agric Food Chem. 67:2856–2864. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhai KF, Duan H, Chen Y, Khan GJ, Cao WG,
Gao GZ, Shan LL and Wei ZJ: Apoptosis effects of imperatorin on
synoviocytes in rheumatoid arthritis through
mitochondrial/caspase-mediated pathways. Food Funct. 9:2070–2079.
2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhai KF, Duan H, Khan GJ, Xu H, Han FK,
Cao WG, Gao GZ, Shan LL and Wei ZJ: Salicin from alangium Chinense
ameliorates rheumatoid arthritis by modulating the Nrf2-HO-1-ROS
pathways. J Agric Food Chem. 66:6073–6082. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhai KF, Zheng JR, Tang YM, Li F, Lv YN,
Zhang YY, Gao Z, Qi J, Yu BY and Kou JP: The saponin D39 blocks
dissociation of non-muscular myosin heavy chain IIA from TNF
receptor 2, suppressing tissue factor expression and venous
thrombosis. Br J Pharmacol. 174:2818–2831. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kurundkar D, Kurundkar AR, Bone NB, Becker
EJ Jr, Liu W, Chacko B, Darley-Usmar V, Zmijewski JW and Thannickal
VJ: SIRT3 diminishes inflammation and mitigates endotoxin-induced
acute lung injury. JCI insight. 4(e120722)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dolinay T, Kim YS, Howrylak J, Hunninghake
GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L,
Nakahira K, et al: Inflammasome-regulated cytokines are critical
mediators of acute lung injury. Am J Respir Crit Care Med.
185:1225–1234. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Deng Q, Pan B, Alam HB, Liang Y, Wu Z, Liu
B, Mor-Vaknin N, Duan X, Williams AM, Tian Y, et al: Citrullinated
histone H3 as a therapeutic target for endotoxic shock in mice.
Front Immunol. 10(2957)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liang Y, Pan B, Alam HB, Deng Q, Wang Y,
Chen E, Liu B, Tian Y, Williams AM, Duan X, et al: Inhibition of
peptidylarginine deiminase alleviates LPS-induced pulmonary
dysfunction and improves survival in a mouse model of lethal
endotoxemia. Eur J Pharmacol. 833:432–440. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Jiang W, Luo F, Lu Q, Liu J, Li P, Wang X,
Fu Y, Hao K, Yan T and Ding X: The protective effect of Trillin
LPS-induced acute lung injury by the regulations of inflammation
and oxidative state. Chem Biol Interact. 243:127–134.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Patel VJ, Biswas Roy S, Mehta HJ, Joo M
and Sadikot RT: Alternative and natural therapies for acute lung
injury and acute respiratory distress syndrome. Biomed Res Int.
2018(2476824)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Grommes J and Soehnlein O: Contribution of
neutrophils to acute lung injury. Mol Med. 17:293–307.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Strieter RM, Belperio JA and Keane MP:
Cytokines in innate host defense in the lung. J Clin Invest.
109:699–705. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Jiang C, Ting AT and Seed B: PPAR-gamma
agonists inhibit production of monocyte inflammatory cytokines.
Nature. 391:82–86. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Pugin J, Verghese G, Widmer MC and Matthay
MA: The alveolar space is the site of intense inflammatory and
profibrotic reactions in the early phase of acute respiratory
distress syndrome. Crit Care Med. 27:304–312. 1999.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lee JM, Yeo CD, Lee HY, Rhee CK, Kim IK,
Lee DG, Lee SH and Kim JW: Inhibition of neutrophil elastase
contributes to attenuation of lipopolysaccharide-induced acute lung
injury during neutropenia recovery in mice. J Anesth. 31:397–404.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H,
Wu D, Liu B, Li H, Li H, et al: Neutrophil extracellular traps are
indirectly triggered by lipopolysaccharide and contribute to acute
lung injury. Sci Rep. 6(37252)2016.PubMed/NCBI View Article : Google Scholar
|