Introduction
Doxorubicin (DOX), an anthracycline chemotherapeutic
drug, is one of the most potent, effective and commonly used
anticancer chemotherapeutics (1).
However, its clinical use is hampered due to its acute and chronic
cardiotoxic effects, as long-term use of DOX can result in left
ventricular dysfunction and ultimately heart failure (2,3).
Previous studies have revealed that DOX treatment leads to excess
free radicals and reactive oxygen species (ROS) in the myocardium
(4,5). These radicals induce potential
redox-associated damage through both enzymatic and non-enzymatic
pathways (6). Lipid peroxidation is
induced by DOX-generated free radicals, which eventually leads to
cell membrane damage (7).
Additionally, mitochondrial damage, increase in Ca2+
currents with subsequent sarcoplasmic reticulum dysfunction and
decreased activity of Na/K-ATPase have been implicated in
DOX-induced cardiotoxicity (8,9).
Morphological alterations in DOX-treated myocardium primarily
include myofibrillar loss, dilatation of the sarcoplasmic reticulum
and swollen mitochondria (10).
Destruction of the generated ROS by antioxidants has been
demonstrated to induce protective effects against DOX-induced
cardiomyopathy (11). For example,
antioxidants such as Bombyx mori, benazepril and quercetin
have been used to protect against DOX-induced cardiotoxicity in
previous studies (12,13).
Angiotensin-converting enzyme inhibitors (ACEIs),
including zofenopril (14),
lisinopril (15), enalapril
(16) and dexrazoxane (17), have been proposed as another
potential preventive strategy for DOX-induced cardiac dysfunction
and injury. In rats treated with short- or long-term DOX,
co-administration of ACEIs haa been reported to restore their
cardiac function (18). In
addition, in rats and mice suffering from pathological
cardiovascular conditions (such as spontaneous hypertension,
cardiomyopathy, diastolic heart failure or myocardial infarction)
as a result of experimental autoimmune myocarditis, DOX treatment,
hyperglycemia, inhibition of the renin-angiotensin system by ACEIs
or angiotensin receptor blockers results in a decrease in the
expression levels of NADPH oxidase (Nox) subunits p22phox, Nox2,
p47phox and p67phox (19).
Benazepril (brand name Lotensin) is an ACEI and has
been used for the treatment of congestive heart failure,
hypertension and chronic renal failure (20,21). A
number of studies have confirmed that benazepril protects against
DOX-induced renal dysfunction (22-24).
However, to the best of our knowledge, there are currently no
studies on the role of benazepril in DOX-induced cardiac toxicity.
Since DOX toxicity is mainly associated with the heart, the present
study aims to explore the potential beneficial effects and
mechanisms of benazepril hydrochloride (HCl) for DOX-induced
cardiotoxicity in rat embryonic cardiac myoblast (H9c2) cells,
which may provide the clinical foundation for the use of benazepril
along with DOX to alleviate DOX-induced cardiotoxicity.
Materials and methods
Chemicals and reagents
Benazepril-HCl (cat. no. S1284; Selleck Chemicals)
was dissolved in DMSO to obtain a solution of 10 mM. The purity was
determined to be >99% by high performance liquid chromatography.
DOX (cat. no. 25316-40-9; MilliporeSigma) was dissolved in saline
to generate a stock solution of 100 µM and was diluted to a final
concentration of 2 µM for all experiments unless otherwise
specified. The Akt inhibitor MK2206 was purchased from
MedChemExpress (cat. no. HY-108232).
Cell culture
Rat H9c2 cells were obtained from The Cell Bank of
Type Culture Collection of The Chinese Academy of Sciences. Cells
were cultured in DMEM-high glucose medium supplemented with 10% FBS
(both Gibco; Thermo Fisher Scientific, Inc.) and antibiotics (1%
penicillin/streptomycin) at 37˚C in 5% CO2 and 95%
O2 with saturated humidity.
Total RNA extraction and reverse
transcription-quantitative PCR
Total RNA was isolated from the H9c2 cells using the
TRIzol® reagent (cat. no. 15596026; Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The iScript™ RT Supermix (Bio-Rad Laboratories, Inc.)
was used to produce cDNA. The reverse transcription reaction mix
was added to 2 µg total RNA and incubated at 25˚C for 5 min for
priming, at 46˚C for 20 min for reverse transcription, and then
inactivated for 1 min at 95˚C. SYBR-Green master mix (Bio-Rad
Laboratories, Inc.) was used for Quantitative real-time PCR
reactions, which were performed as follows: 30 sec at 95˚C, and 40
cycles of 95˚C for 5 sec and 60˚C for 30 sec. The mRNA expression
of the ACE gene was determined using the 2-ΔΔCq
method (25). β-actin was used as
an internal reference. The specific primer sequences used were as
follows: ACE forward, 5'-GCCTCCCAACGAGTTAGAAGAG-3' and
reverse, 5'-CGGGACGTGGCCATTATATT-3' (19); β-actin forward,
5'-TGCCTGACGGTCAGGTCA-3' and reverse, 5'-CAGGAAGGAAGGCTGGAAG-3'.
The primers were synthesized by Chengdu TsingKe Biotechnology Co.,
Ltd.
Cell viability assay and lactate
dehydrogenase (LDH) release
The viability of H9c2 cells was measured using Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.).
Briefly, H9c2 cells were seeded into a 96-well plate with each well
containing 5,000 cells/100 µl and pre-incubated at 37˚C with 5%
CO2 for 24 h. Cells were pretreated with benazepril-HCl
at different concentrations (10 and 100 nM, 1, 10 and 100 µM) for 1
h at 37˚C, followed by treatment with DOX (2 µM) for 24 h at 37˚C.
After treatment, 10 µl of CCK-8 solution was added for incubation
at 37˚C for 2 h. Absorbance at 450 nm was read using a microplate
reader (BioTek Instruments, Inc.). The average optical density (OD)
of five wells for each group was measured to determine the
percentage of viable cells: % Of viable cells=mean OD after
treatment/mean OD of controls x100.
H9c2 cells were plated 2x105 cell/ml in a
90-mm dish, treated with 0.1% DMSO (control group) or
benazepril-HCl (1 µM) for 1 h at 37˚C, and cultured in the presence
or absence of DOX (2 µM) for an additional 24 h at 37 ˚C. The
medium was collected by centrifugation at 500 x g for 5 min at room
temperature. The LDH release was measured in the cell medium using
an LDH assay kit (cat. no. A020-2-2; Nanjing Jiancheng
Bioengineering Institute). LDH activity was presented as units per
liter. The level of LDH activity was calculated using the formula:
LDH Activity=[amount (nmol) of NADH generated/min]/[volume of
supernatant used (ml)] x sample dilution factor.
Assessment of H9c2 apoptosis
Apoptosis was evaluated by flow cytometry with an
Annexin V-APC/7-AAD Apoptosis Detection kit (cat. no. 640930;
BioLegend, Inc.). Cells were washed with PBS twice and resuspended
in 100 µl 1X annexin-binding buffer at a concentration of
1x106 cells/ml. Each 100 µl cell solution was incubated
with 5 µl annexin V-APC and 1 µl 7-AAD (1 µM) at 37˚C in a
CO2 incubator for 15 min. Following the addition of 400
µl 1X annexin-binding buffer into each solution, cells were
analyzed by a FACS Aria SORP (BD Biosciences). The average early
and late apoptotic percentage of cells was determined with three
repeats, which were analyzed using BD CellQuest Pro software
version 5.2.1 (Becton-Dickinson and Company).
Measurement of mitochondrial ROS
The ROS level was detected with a fluorescent probe
2',7'-dichlorofluorescein diacetate (DCFH-DA; cat. no. C6827;
Thermo Fisher Scientific, Inc.) in H9c2 cells as previously
described (23). Briefly, H9c2
cells were treated with vehicle or DOX (2 µM) in the presence or
absence of benazepril-HCl (1 µM for 1 h prior to DOX exposure) for
24 h at 37˚C and rinsed with PBS. Subsequently, the cells were
incubated for 1 h at 37˚C with 10 µM DCFH-DA in PBS. After washing
twice with PBS to remove extracellular DCFH-DA, images were
captured at room temperature using a Zeiss OBSERVER D1/AX10 cam
(Carl Zeiss AG). Fluorescence intensity was measured using a
microplate reader (Synergy Mx; BioTek Instruments, Inc.) at 488 nm
excitation and 525 nm emission wavelengths.
Analysis of malondialdehyde (MDA)
content and superoxide dismutase (SOD), catalase (CAT) and
glutathione peroxidase (GSH-Px) activities
H9c2 cells were harvested using a RIPA lysis buffer
(cat. no. P0013K; Beyotime Institute of Biotechnology) and
centrifuged at 12,000 x g for 10 min at 4˚C. The supernatants were
used to assess the concentration of MDA (cat. no. S0131S; Beyotime
Biotechnology) and the activities of SOD (cat. no. S0101), CAT
(cat. no. S0051) and GSH-Px (cat. no. S0056) (all from Beyotime
Institute of Biotechnology) using the corresponding detection kits.
All samples were immediately analyzed or frozen at -80˚C for up to
one month. The total protein amount was determined by a BCA protein
assay kit (Beyotime Institute of Biotechnology). The level of MDA
was expressed as nmol/mg protein. The activity levels of SOD, CAT
and GSH-Px were expressed as U/mg protein by normalizing against
the total protein concentration in each sample.
Western blotting
H9c2 cells were seeded in 6-cm dishes preincubated
in a humidified incubator at 37˚C with 5% CO2 for 24 h.
The cells were pretreated with an Akt inhibitor MK2206 (1 µM) for
30 min and incubated with benazepril-HCl (1 µM) for 1 h at 37˚C,
followed by DOX (2 µM) for 24 h at 37˚C. Cells treated with DOX
and/or benazepril-HCl were obtained, washed with PBS and lysed in
RIPA lysis buffer containing a 1X phosphatase and protease
inhibitor cocktail (Roche Molecular Diagnostics) for 30 min on ice.
Protein concentrations were determined using a BCA kit (cat. no.
P0009; Beyotime Institute of Biotechnology). Samples containing 40
µg protein were boiled in 1X loading buffer (0.05 M Tris-HCl, 0.1 M
DTT, 70 mM SDS, 1.5 mM bromophenol blue and 1.0 M glycerol) for 5
min. Then, equal amounts of protein (40 µg/lane protein) were
separated by 12% SDS-PAGE, and electroblotted onto PVDF membranes
(MilliporeSigma). The membranes were blocked with a blocking buffer
[5% (w/v) non-fat milk powder in Tris-buffered saline with 0.1%
Tween-20 (Bio-Rad Laboratories, Inc.) (TBST)] for 1 h at room
temperature, and incubated overnight with primary antibodies at
4˚C. Following washing by TBST, the membranes were incubated with
secondary antibodies for 1 h at room temperature, followed by three
additional TBST washes. Primary antibodies against GAPDH (1:2,000;
cat. no. sc-32233), total Akt (1:1,000; cat. no. sc-81434) and
phosphorylated (p)-Akt (Ser473; 1:1,000; cat. no. sc-293125) were
obtained from Santa Cruz Biotechnology, Inc., and the anti-mouse
(1:5,000; TA130003) or anti-rabbit (1:5,000; TA130024)
HRP-conjugated secondary antibodies were supplied by OriGene
Technologies, Inc.. The protein bands were visualized using an
enhanced chemiluminescence reagent (MilliporeSigma) using Image Lab
Software 6.0.1 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± SEM and were
analyzed using GraphPad Prism 6 (GraphPad Software, Inc.).
Comparisons among different groups were performed using one-way
ANOVA followed by Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
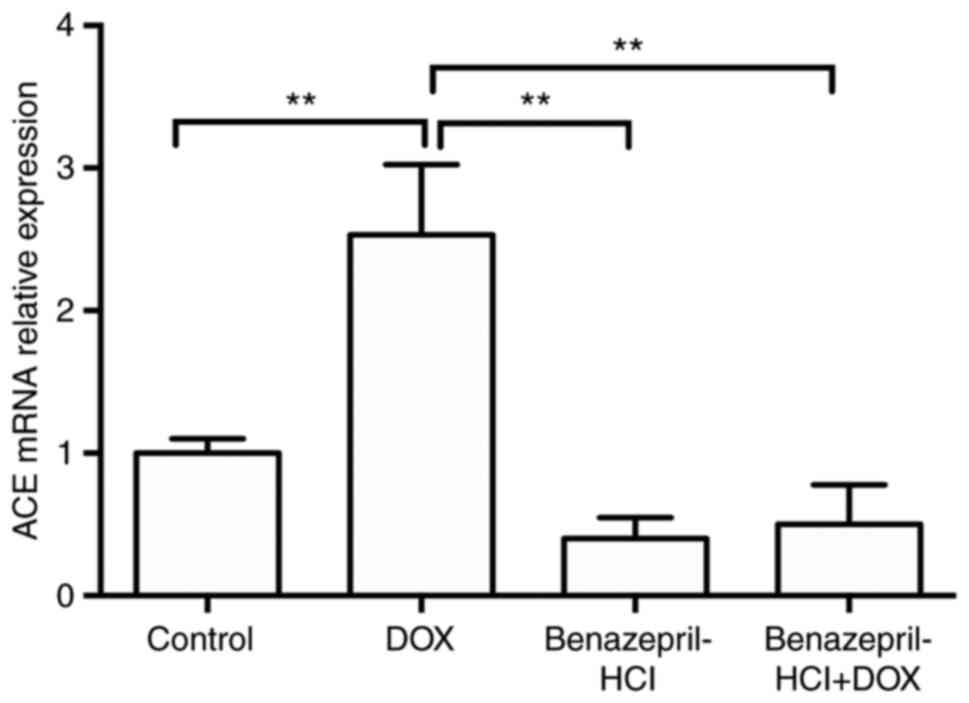
DOX increases ACE mRNA expression
levels
The expression levels of ACE mRNA were significantly
increased in DOX-treated H9c2 cardiomyocytes compared with those in
the control cells (P<0.01). However, treatment with
benazepril-HCl and benazepril-HCl + DOX resulted in a significant
decrease in the ACE mRNA expression levels compared with those in
the DOX-treated group (P<0.01; Fig.
1).
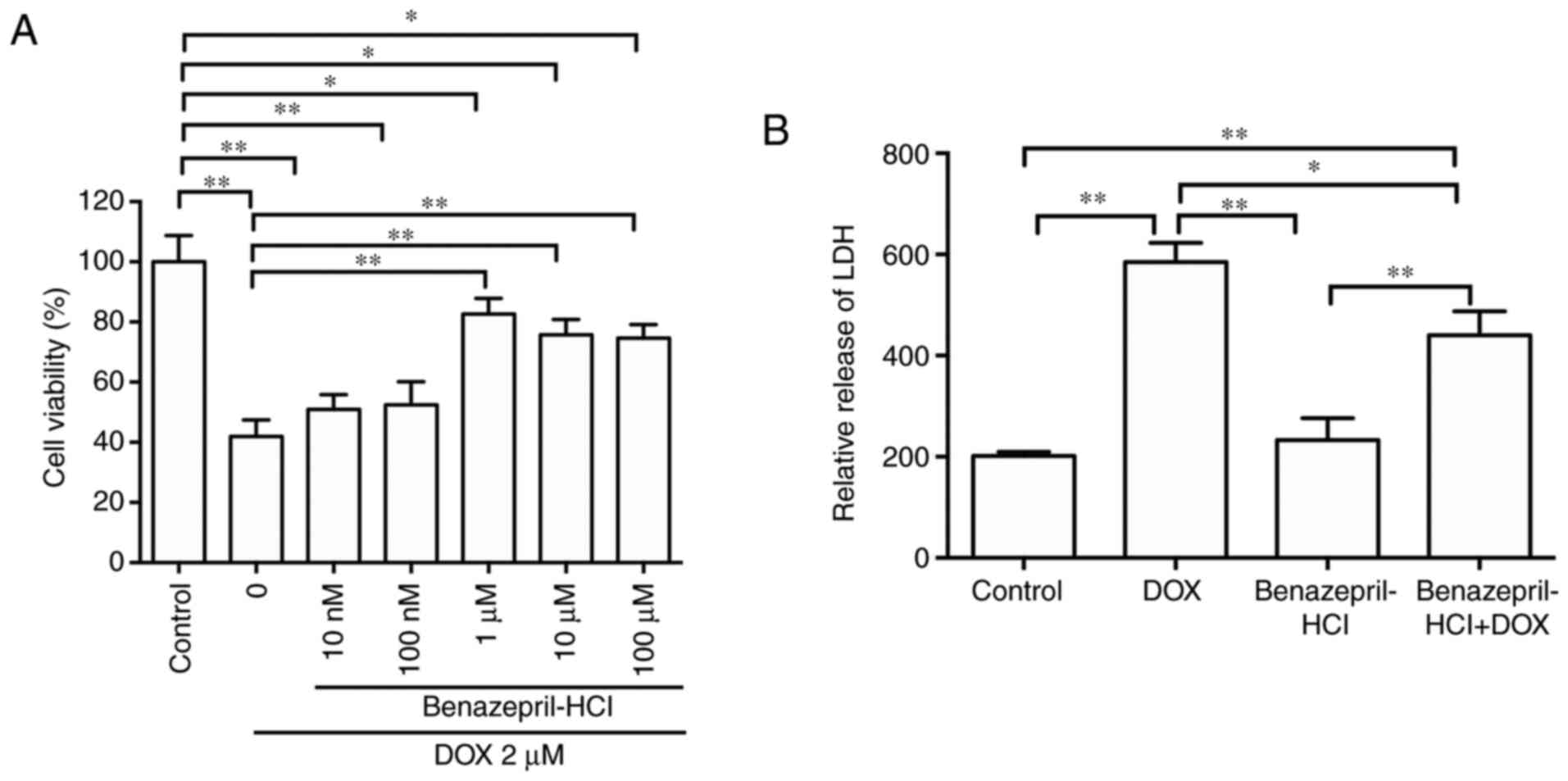
Effects of DOX and benazepril-HCl on
H9c2 cell viability and LDH release
The CCK-8 assay results revealed that DOX treatment
(2 µM; 24 h) significantly decreased H9c2 cell viability compared
with that of the control group by 56.3±3.6% (P<0.01; Fig. 2A). Pretreatment with benazepril-HCl
at 10 and 100 nM did not increase the viability of DOX-treated
cells (P>0.05), whereas pretreatment with 1 µM benazepril-HCl
significantly enhanced cell viability by 34.2±4.2% (P<0.01)
compared with that in the DOX-treated group. The concentrations of
benazepril-HCl >1 µM did not provide an additional
cytoprotective effect compared with that observed with 1 µM
(P>0.05). These results suggested that benazepril-HCl at 1 µM
may protect cardiomyocytes from DOX-induced proliferation
inhibition. Consequently, 1 µM benazepril-HCl was used in
subsequent experiments.
As presented in Fig.
2B, the level of LDH release in the supernatants of DOX-treated
H9c2 cells was significantly increased (~189%) compared with that
in the control group (P<0.01), and that of the benazepril-HCL +
DOX treatment group was significantly decreased 24.6% (P<0.05)
compared with the DOX treatment group.
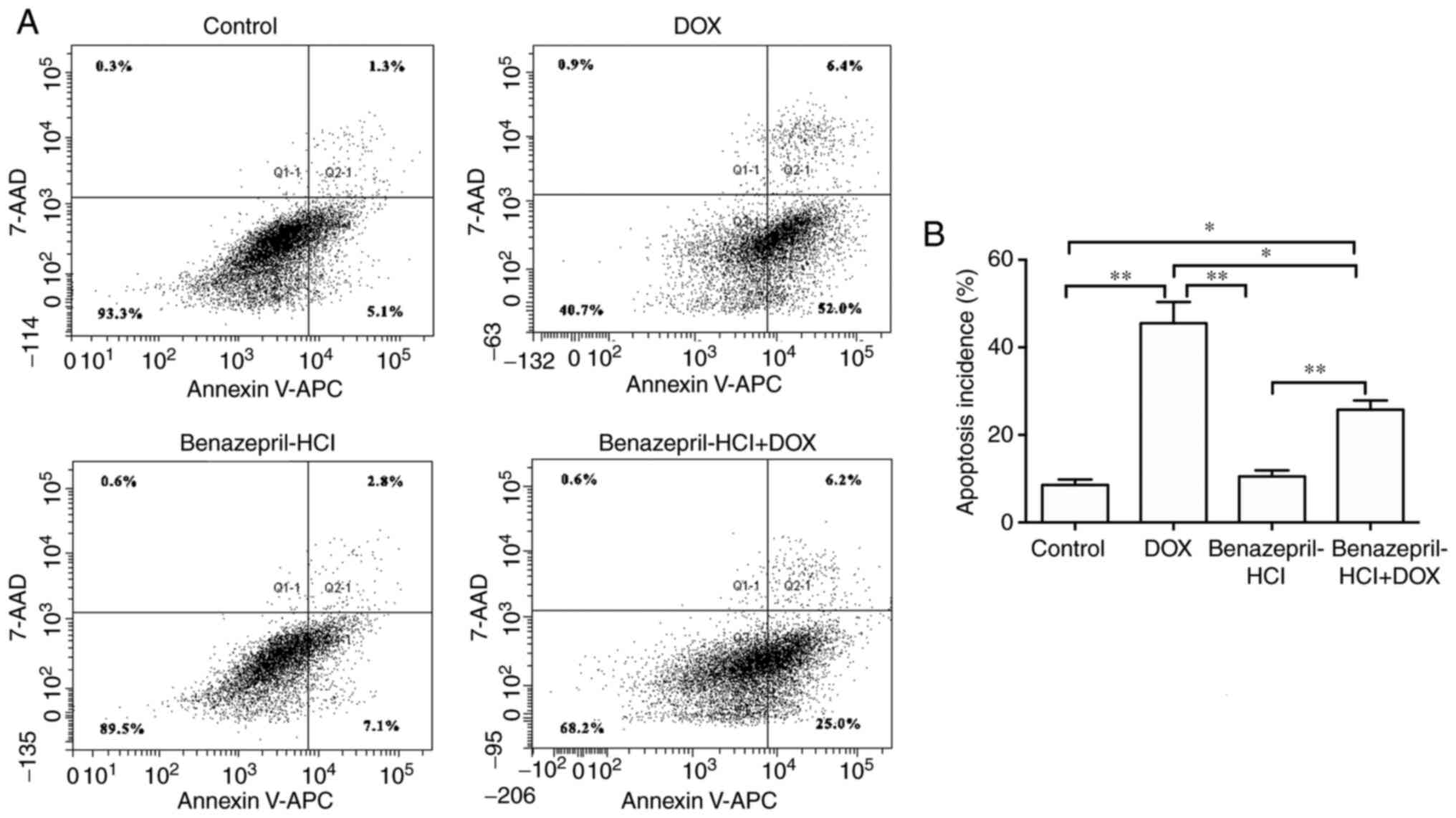
Benazepril-HCl protects H9c2 cells
against DOX-induced apoptosis
As presented in Fig.
3, DOX treatment (2 µM; 24 h) resulted in a significant
increase in the apoptotic rate compared with that in the control
group (45.5±4.82 compared with 8.6±1.21; P<0.01). However,
benazepril-HCL pretreatment significantly decreased DOX-induced
apoptosis to 25.8±2.1 (P<0.05) compared with that in the DOX
treatment group. These results suggested that benazepril-HCl
reduced the DOX-induced apoptosis in H9c2 cells.
Effects of benazepril-HCl on
DOX-induced oxidative stress in H9c2 cells
DOX-induced ROS production within the mitochondrial
respiratory chain is considered a primary contributor to H9c2 cell
death (26). To assess whether the
DOX-induced oxidative stress may be mitigated by pretreatment with
benazepril-HCl, cellular ROS levels were measured in cells treated
with benazepril-HCl, DOX or benazepril-HCl + DOX stained with 10 µM
DCFH-DA. As presented in Fig. 4A,
the DCF fluorescence intensity was markedly increased by exposure
to DOX in the absence of benazepril-HCl compared with that in the
control group, indicating that DOX induced oxidative stress in H9c2
cells. However, pretreatment with benazepril-HCl significantly
reduced DOX-induced intracellular ROS concentrations (Fig. 4B).
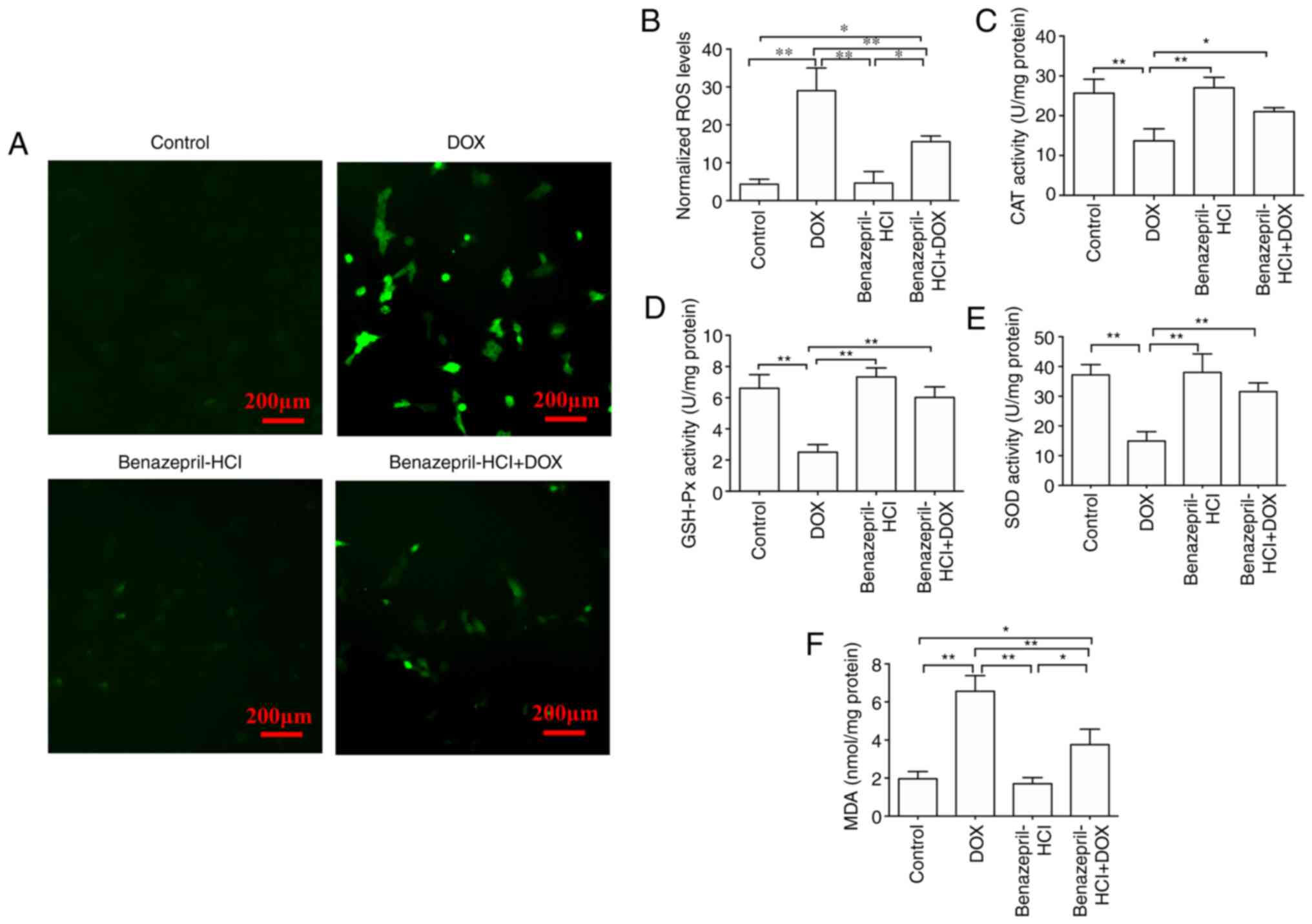 | Figure 4Effects of benazepril-HCl on
DOX-induced oxidative stress in H9c2 cells. (A) DOX-induced ROS
generation was inhibited in the presence of benazepril-HCl (scale
bar, 200 mm). (B) Quantified comparison of the ROS levels. Effects
of benazepril-HCl on (C) CAT, (D) GSH-Px and (E) SOD activities,
and (F) MDA concentrations in H9c2 cells. *P<0.05,
**P<0.01. N=3 per group. DOX, doxorubicin; ROS,
reactive oxygen species; CAT, catalase; GSH-Px, glutathione
peroxidase; SOD, superoxide dismutase; MDA, malondialdehyde. |
CAT, GSH-Px, SOD and MDA are commonly used as key
biomarkers for oxidative stress and their changes are widely used
as indicators of oxidative injury (27). The levels of these markers were
measured to assess whether the DOX-induced metabolic imbalances in
myocardial cells may be improved by pretreatment with
benazepril-HCl. In comparison with the controls, CAT, SOD and
GSH-Px activity levels were significantly reduced in DOX-treated
H9c2 cells, whereas the MDA level was significantly increased (all
P<0.01). Compared with those in the DOX-treated group, following
pretreatment with benazepril-HCl, cardiac CAT, SOD and GSH-Px
activity levels were significantly enhanced and the MDA levels were
significantly decreased (P<0.01; Fig. 4C-F). These results suggested that
benazepril-HCl prevented DOX-induced oxidative stress in cardiac
cells.
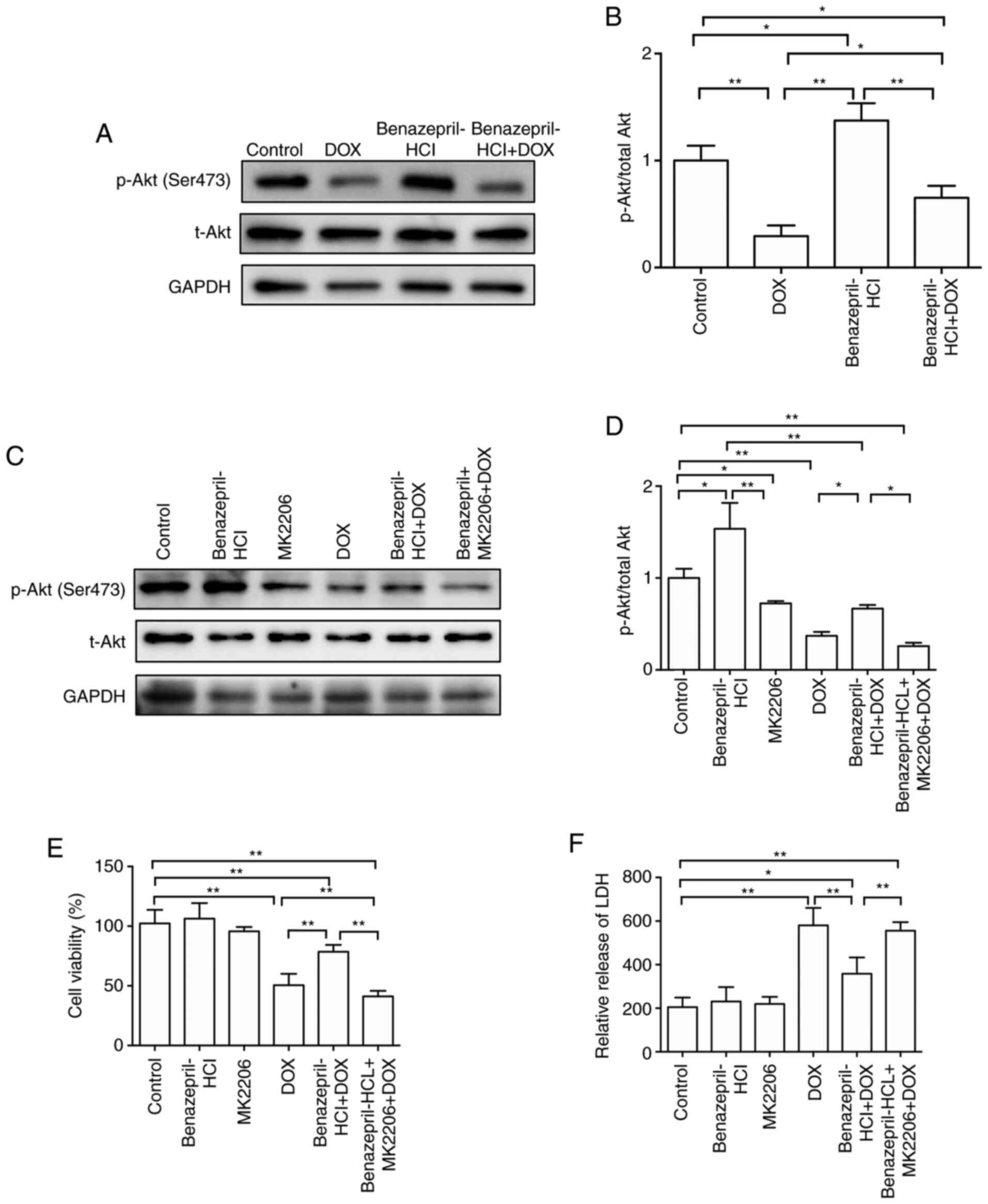
Benazepril-HCl alleviates DOX-induced
cardiomyocyte injury via PI3K/Akt signaling
The expression levels of proteins associated with
the PI3K/Akt signaling pathway were determined by western blotting.
As presented in Fig. 5A and
B, the Akt phosphorylation levels
were significantly reduced by DOX compared with those in the
control group (P<0.01). However, benazepril-HCl + DOX
significantly recovered the phosphorylation levels of Akt compared
with those in the DOX-treated group (P<0.05; Fig. 5A and B). Furthermore, MK2206, an Akt inhibitor,
was used to reveal whether the PI3K/Akt pathway mediated the
protective effect of benazepril-HCl on DOX-induced cardiomyocyte
injury. MK2206 supplementation (benazepril-HCl + MK2206 + DOX
group) inhibited Akt phosphorylation (P<0.05, Fig. 5C and D) and reduced cell viability (P<0.01,
Fig. 5E) significantly, compared
with those in the Benazepril + DOX treated group. In addition, LDH
release was significantly increased after cells were incubated with
MK2206 (benazepril-HCl + MK2206 + DOX group) compared with that in
the benazepril-HCl + DOX group (P<0.05; Fig. 5F). These results suggested that the
protective effects of benazepril-HCl on DOX-induced cardiotoxicity
may be due to a reduction in oxidative stress and the activation of
the PI3k/Akt signaling.
Discussion
The present study aimed to investigate the
protective roles and underlying mechanisms of benazepril-HCl on
DOX-induced cardiotoxicity. The results indicated that pretreatment
with benazepril-HCl of H9c2 cells alleviated DOX-induced oxidative
stress, reduced the cardiomyocyte apoptosis and attenuated
DOX-induced cardiotoxicity, suggesting that the use of
benazepril-HCl may be a potential therapeutic approach to assist in
the prevention of DOX-induced cardiotoxicity.
Anthracyclines, including DOX, are highly effective
anticancer therapeutics; however, DOX-induced cardiac toxicity is a
notable side effect of its long-term clinical use for anticancer
treatment (28). Therefore,
reducing DOX-induced cardiomyopathy is needed for the clinical
success of this anticancer chemotherapy, which has been a challenge
for pharmacologists and oncologists. Accumulating evidence has
demonstrated the potential protective effects of ACEIs on the heart
(29-32).
In particular, the protective function of benazepril-HCl against
heart failure has been confirmed in an isoproterenol-induced
chronic heart failure model via amelioration of hypertrophy and
dysfunction of heart ventricles (33,34).
Additionally, alleviation of myocardial injury induced by ischemia
and ventricular arrhythmia by benazepril-HCl has also been
demonstrated (35). Furthermore,
benazepril-HCl inhibits ventricular remodeling, myocardial
hypertrophy and cardiac fibrosis in spontaneously hypertensive rats
(36). Its anti-inflammatory
activity prevents myocardial injury during septic shock, and its
protective effect against aldosterone-induced myocardial injury has
been identified to be similar to aldosterone inhibitors such as
spironolactone and eplerenone (37).
The results of the present study revealed that the
administration of benazepril-HCl at a dose of 1 µM improved the
viability of DOX-treated H9c2 cells. DOX treatment has been
previously reported to increase the release of LDH from myocardial
tissues (36). LDH has been
demonstrated to be important for the diagnosis of DOX-induced
myocardial cardiotoxicity (37).
The present study revealed an increased level of LDH in the culture
medium of DOX-treated H9c2 cells, whereas the LDH release was
significantly inhibited in H9c2 cells treated by a combination of
benazepril-HCL and DOX. These results indicated the protective role
of benazepril-HCl against DOX-induced cardiotoxicity in H9c2
cells.
Despite the lack of understanding of the primary
mechanism for DOX-induced cardiotoxicity, apoptosis of
cardiomyocytes may serve a notable part in the side effects of DOX
(38). The results of the present
study confirmed a high level of apoptosis in DOX-treated H9c2
cells, whereas the apoptotic rate was significantly reduced in
cells pretreated with benazepril-HCl compared with that in the
cells treated with DOX alone. Therefore, the inhibition of
cardiomyocyte apoptosis by pretreatment with benazepril-HCl may
contribute to the alleviation of DOX-induced cardiotoxicity.
A high level of free radicals is an important
contributor to DOX-induced cardiotoxicity (39). After DOX enters cardiomyocytes, it
is reduced by mitochondrial enzymes into its semiquinone form,
which produces ROS, including superoxide anion, hydrogen peroxide
and hydroxyl radicals (37,40). In comparison with other organs in
mammalian species, the heart contains a lower concentration of
enzymes, such as SOD, CAT and GSH-Px, for attenuating ROS toxicity
and protecting against oxidative stress (8). Therefore, the heart is a primary organ
associated with DOX-induced toxicity. In addition to the generation
of damaging free radicals, endogenous antioxidant levels are also
decreased by DOX, which may result in cardiomyocyte apoptosis
(41). The results of the present
study demonstrated that DOX decreased the expression levels of SOD,
CAT and GSH-Px and increased cardiomyocyte apoptosis, which were
consistent with previous studies (41,42).
Due to of the occurence of oxidative stress after
DOX treatment, antioxidant administration in combination with DOX
may attenuate DOX-induced cardiotoxicity. Benazepril-HCl exhibits
antioxidant properties that have been confirmed by its
neuroprotective effects on intracerebral hemorrhage, excitotoxicity
and diabetes-associated cognitive deficits, as well as its effects
against melanoma, renal ischemia-reperfusion injury and pulmonary
hypertension (43-45).
The present study demonstrated that benazepril-HCl pretreatment
reduced the levels of SOD, CAT and GSH-Px in DOX-treated H9c2 cells
and decreased the DOX-induced ROS concentration in H9c2 cells.
These results suggested that benazepril-HCl pretreatment may
protect H9c2 cells from DOX-induced cardiotoxicity by decreasing
oxidative stress.
Several previous studies have provided evidence that
DOX-induced cardiac dysfunction is associated with reduced
p-Akt/Akt levels (46-48).
In addition, certain cadioprotective agents, such as neuregulin-1β
and insulin-like growth factor-1, protect against DOX-induced
cardiotoxicity by regulating the Akt-dependent signaling pathway
(49,50). The results of the present study
revealed that pretreatment with benazepril-HCl significantly
increased the phosphorylation of Akt. These results suggested that
the protective effects of benazepril-HCl on DOX-induced
cardiotoxicity may be due to a reduction in oxidative stress and
the activation of the PI3k/Akt signaling. Furthermore, pretreatment
with benazepril-HCl significantly enhanced the phosphorylation of
Akt. Akt phosphorylation and LDH release in H9c2 cells pretreated
with benazepril-HCl were significantly inhibited by an Akt
inhibitor MK2206. These results suggested that the protective
effects of benazepril-HCl on DOX-induced cardiotoxicity may be due
to a reduction in oxidative stress and the activation of the
PI3k/Akt signaling.
The present study focused on the protction of
benazepril against DOX-induced cardiotoxicity in H9c2 cells but not
in animals. Although H9c2 cell line cells are special clonal
cardiomyoblasts that have a number of cardiomyocyte
characteristics, increasing evidence suggests that there may be
differences between H9c2 cell experiments in vitro and
animal models in vivo (51,52).
Thus, further studies in an animal model are needed to confirm the
pharmacological antagonism of benazepril against DOX-induced
cardiomyopathy.
In conclusion, in the present study, H9c2 cells were
protected from DOX-induced cardiotoxicity by pretreatment with
benazepril-HCl. This protection may be mediated by decreasing
oxidative stress, reducing apoptosis and inducing alterations in
the myocardial architecture. The present study supported the
clinical benefits of co-administration of benazepril-HCl with DOX
to reduce the risk of DOX-induced cardiotoxicity. However, further
research is required to increase the understanding of the
mechanisms of inhibition of oxidative stress and to confirm whether
the administration of benazepril-HCl in combination with or before
DOX treatment may interfere with the antitumor activity of DOX.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by The National Natural Science
Foundation of China (grant nos. 81870221, 81670249, 31271226 and
31071001).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LZ conducted the experiments, collected the data and
drafted the manuscript. XW obtained materials and conducted
analysis. YZ acquired and analyzed data, provided general
administrative support and confirmed the authenticity of all the
raw data. GZ performed cell apoptosis examination and interpreted
the data. YD and XC performed and analyzed the western blotting. WJ
provided ideas, interpreted data, acquired funding and revised the
manuscript. SW obtained materials, conducted analaysis, was a major
contributor in writing the manuscript, and confirmed the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rosen MR, Myerburg RJ, Francis DP, Cole GD
and Marbán E: Translating stem cell research to cardiac disease
therapies: Pitfalls and prospects for improvement. J Am Coll
Cardiol. 64:922–937. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Khouri MG, Klein MR, Velazquez EJ and
Jones LW: Current and emerging modalities for detection of
cardiotoxicity in cardio-oncology. Future Cardiol. 11:471–484.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Angsutararux P, Luanpitpong S and
Issaragrisil S: Chemotherapy-induced cardiotoxicity: Overview of
the roles of oxidative stress. Oxid Med Cell Longev.
2015(795602)2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ma ZG, Kong CY, Wu HM, Song P, Zhang X,
Yuan YP, Deng W and Tang QZ: Toll-like receptor 5 deficiency
diminishes doxorubicin-induced acute cardiotoxicity in mice.
Theranostics. 10:11013–11025. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cappetta D, De Angelis A, Sapio L,
Prezioso L, Illiano M, Quaini F, Rossi F, Berrino L, Naviglio S and
Urbanek K: Oxidative stress and cellular response to doxorubicin: A
common factor in the complex milieu of anthracycline
cardiotoxicity. Oxid Med Cell Longev. 2017(1521020)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Nithipongvanitch R, Ittarat W, Cole MP,
Tangpong J, Clair DK and Oberley TD: Mitochondrial and nuclear p53
localization in cardiomyocytes: Redox modulation by doxorubicin
(Adriamycin)? Antioxid Redox Signal. 9:1001–1008. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Phaniendra A, Jestadi DB and Periyasamy L:
Free radicals: Properties, sources, targets, and their implication
in various diseases. Indian J Clin Biochem. 30:11–26.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Murabito A, Hirsch E and Ghigo A:
Mechanisms of anthracycline-induced cardiotoxicity: Is
mitochondrial dysfunction the answer? Front Cardiovasc Med.
7(35)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wallace KB, Sardão VA and Oliveira PJ:
Mitochondrial determinants of doxorubicin-induced cardiomyopathy.
Circ Res. 126:926–941. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fu X, Kong L, Tang M, Zhang J, Zhou X, Li
G, Wang H and Fu F: Protective effect of ocotillol against
doxorubicin-induced acute and chronic cardiac injury. Mol Med Rep.
9:360–364. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang H, Yu P, Gou H, Zhang J, Zhu M, Wang
ZH, Tian JW, Jiang YT and Fu FH: Cardioprotective effects of 20
(S)-ginsenoside Rh2 against doxorubicin-induced cardiotoxicity in
vitro and in vivo. Evid Based Complement Alternat Med.
2012(506214)2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nazmi AS, Ahmad SJ, Pillai KK, Akhtar M,
Ahmad A and Najmi AK: Protective effects of Bombyx mori,
quercetin and benazepril against doxorubicin induced cardiotoxicity
and nephrotoxicity. J Saudi Chem Soc. 20 (Suppl 1):S573–S578.
2016.
|
|
13
|
Ganatra S, Nohria A, Shah S, Groarke JD,
Sharma A, Venesy D, Patten R, Gunturu K, Zarwan C, Neilan TG, et
al: Upfront dexrazoxane for the reduction of anthracycline-induced
cardiotoxicity in adults with preexisting cardiomyopathy and
cancer: A consecutive case series. Cardiooncology.
5(1)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sacco G, Bigioni M, Evangelista S, Goso C,
Manzini S and Maggi CA: Cardioprotective effects of zofenopril, a
new angiotensin-converting enzyme inhibitor, on doxorubicin-induced
cardiotoxicity in the rat. Eur J Pharmacol. 414:71–78.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Guglin M, Munster P, Fink A and Krischer
J: Lisinopril or Coreg CR in reducing cardiotoxicity in women with
breast cancer receiving trastuzumab: A rationale and design of a
randomized clinical trial. Am Heart J. 188:87–92. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Spallarossa P, Guerrini M, Arboscello E
and Sicbaldi V: Enalapril and carvedilol for preventing
chemotherapy-induced left ventricular systolic dysfunction. J Am
Coll Cardiol. 62:2451–2452. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Choi HS, Park ES, Kang HJ, Shin HY, Noh
CI, Yun YS, Ahn HS and Choi JY: Dexrazoxane for preventing
anthracycline cardiotoxicity in children with solid tumors. J
Korean Med Sci. 25:1336–1342. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zablocki D and Sadoshima J: Angiotensin II
and oxidative stress in the failing heart. Antioxid Redox Signal.
19:1095–1109. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
King JN, Font A, Rousselot JF, Ash RA,
Bonfanti U, Brovida C, Crowe ID, Lanore D, Pechereau D, Seewald W
and Strehlau G: Effects of benazepril on survival of dogs with
chronic kidney disease: A multicenter, randomized, blinded,
placebo-controlled clinical trial. J Vet Intern Med. 31:1113–1122.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chan KK, Buch A, Glazer RD, John VA and
Barr WH: Site-differential gastrointestinal absorption of
benazepril hydrochloride in healthy volunteers. Pharm Res.
11:432–437. 1994.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Muñoz-Durango N, Fuentes CA, Castillo AE,
González-Gómez LM, Vecchiola A, Fardella CE and Kalergis A: Role of
the renin-angiotensin-aldosterone system beyond blood pressure
regulation: Molecular and cellular mechanisms involved in end-organ
damage during arterial hypertension. Int J Mol Sci.
17(797)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Parving HH: Diabetic nephropathy.
Prevention and treatment. Kidney Int. 60:2041–2055. 2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yim HE and Yoo KH: Renin-angiotensin
system-considerations for hypertension and kidney. Electrolyte
Blood Press. 6:42–50. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang Z, Wang J, Xie R, Xie R, Liu R and Lu
Y: Mitochondria-derived reactive oxygen species play an important
role in doxorubicin-induced platelet apoptosis. Int J Mol Sci.
16:11087–11100. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kuyumcu F and Aycan A: Evaluation of
oxidative stress levels and antioxidant enzyme activities in burst
fractures. Med Sci Monit. 24:225–234. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ma ZG, Yuan YP, Xu SC, Wei WY, Xu CR,
Zhang X, Wu QQ, Liao HH, Ni J and Tang QZ: CTRP3 attenuates cardiac
dysfunction, inflammation, oxidative stress and cell death in
diabetic cardiomyopathy in rats. Diabetologia. 60:1126–1137.
2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kumari H, Huang WH and Chan MWY: Review on
the role of epigenetic modifications in doxorubicin-induced
cardiotoxicity. Front Cardiovasc Med. 7(56)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Munger MA: Use of angiotensin receptor
blockers in cardiovascular protection: Current evidence and future
directions. P T. 36:22–40. 2011.PubMed/NCBI
|
|
31
|
Haybar H, Shahrabi S, Deris Zayeri Z and
Pezeshki S: Strategies to increase cardioprotection through
cardioprotective chemokines in chemotherapy-induced cardiotoxicity.
Int J Cardiol. 269:276–282. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yakubova A, Thorrez L, Svetlichnyy D,
Zwarts L, Vulsteke V, Laenen G, Oosterlinck W, Moreau Y, Dehaspe L,
Van Houdt J, et al: ACE-inhibition induces a cardioprotective
transcriptional response in the metabolic syndrome heart. Sci Rep.
8(16169)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gujral DM, Lloyd G and Bhattacharyya S:
Effect of prophylactic betablocker or ACE inhibitor on cardiac
dysfunction and heart failure during anthracycline chemotherapy ±
trastuzumab. Breast. 37:64–71. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Steele JL, Henik RA and Stepien RL:
Effects of angiotensin-converting enzyme inhibition on plasma
aldosterone concentration, plasma renin activity, and blood
pressure in spontaneously hypertensive cats with chronic renal
disease. Vet Ther. 3:157–166. 2002.PubMed/NCBI
|
|
35
|
Wan W, Jiang X, Li X, Zhang C and Yi X:
Silencing of angiotensin-converting enzyme by RNA interference
prevents H9c2 cardiomyocytes from apoptosis induced by
anoxia/reoxygenation through regulation of the intracellular
renin-angiotensin system. Int J Mol Med. 32:1380–1386.
2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li YW, Li YM, Hon Y, Wan QL, He RL, Wang
ZZ and Zhao CH: AT1 receptor modulator attenuates the
hypercholesterolemia-induced impairment of the myocardial ischemic
post-conditioning benefits. Korean Circ J. 47:182–192.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nie P, Meng F, Zhang J, Wei X and Shen C:
Astragaloside IV exerts a myocardial protective effect against
cardiac hypertrophy in rats, partially via activating the Nrf2/HO-1
signaling pathway. Oxid Med Cell Longev.
2019(4625912)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Maron BA and Leopold JA: Aldosterone
receptor antagonists: Effective but often forgotten. Circulation.
121:934–939. 2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Gorini S, De Angelis A, Berrino L, Malara
N, Rosano G and Ferraro E: Chemotherapeutic drugs and mitochondrial
dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxid
Med Cell Longev. 2018(7582730)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jing S, Sun G, Cui X, Meng X, Qin M and
Sun X: Myricitrin protects against doxorubicin-induced
cardiotoxicity by counteracting oxidative stress and inhibiting
mitochondrial apoptosis via ERK/P53 pathway. Evid Based Complement
Alternat Medi. 2016(6093783)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Varga ZV, Ferdinandy P, Liaudet L and
Pacher P: Drug-induced mitochondrial dysfunction and
cardiotoxicity. Am J Physiol Heart Circ Physiol. 309:H1453–H1467.
2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Mantawy EM, El-Bakly WM, Esmat A, Badr AM
and EI-Demerdash E: Chrysin alleviates acute doxorubicin
cardiotoxicity in rats via suppression of oxidative stress,
inflammation and apoptosis. Eur J Pharmacol. 728:107–118.
2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kurutas EB: The importance of antioxidants
which play the role in cellular response against
oxidative/nitrosative stress: Current state. Nutr J.
15(71)2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wu YZ, Zhang L, Wu ZX, Shan TT and Xiong
C: Berberine ameliorates doxorubicin-induced cardiotoxicity via a
SIRT1/p66Shc-mediated pathway. Oxid Med Cell Longev.
2019(2150394)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Malik ZA, Singh M and Sharma PL:
Neuroprotective effect of momordica charantia in global cerebral
ischemia and reperfusion induced neuronal damage in diabetic mice.
J Ethnopharmacol. 133:729–734. 2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Jiang M, Li J, Peng Q, Liu Y, Liu W, Luo
C, Peng J, Li J, Yung KK and Mo Z: Neuroprotective effects of
bilobalide on cerebral ischemia and reperfusion injury are
associated with inhibition of pro-inflammatory mediator production
and down-regulation of JNK1/2 and p38 MAPK activation. J
Neuroinflammation. 11(167)2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Singal PK, Li T, Kumar D, Danelisen I and
Iliskovic N: Adriamycin-induced heart failure: Mechanism and
modulation. Mol Cell Biochem. 207:77–86. 2000.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Taniyama Y and Walsh K: Elevated
myocardial Akt signaling ameliorates doxorubicin-induced congestive
heart failure and promotes heart growth. J Mol Cell Cardiol.
34:1241–1247. 2002.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Timolati F, Ott D, Pentassuglia L, Giraud
MN, Perriard JC, Suter TM and Zuppinger C: Neuregulin-1 beta
attenuates doxorubicin-induced alterations of
excitation-contraction coupling and reduces oxidative stress in
adult rat cardiomyocytes. J Mol Cell Cardiol. 41:845–854.
2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lai HC, Liu TJ, Ting CT, Sharma PM and
Wang PH: Insulin-like growth factor-1 prevents loss of
electrochemical gradient in cardiac muscle mitochondria via
activation of PI3 kinase/Akt pathway. Mol Cell Endocrinol.
205:99–106. 2003.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro Cell Dev Biol
Anim. 47:125–131. 2001.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Branco AF, Pereira SP, Susana G, Gusev O,
Rizvanov AA and Olivira PJ: Gene expression profiling of H9c2
myoblast differentiation towards a cardiac-like phenotype. PLoS
One. 10(e0129303)2015.PubMed/NCBI View Article : Google Scholar
|