Introduction
Acute respiratory distress syndrome (ARDS) is the
extreme manifestation of acute lung injury (ALI) (1). In 2016, estimates of the incidence of
ARDS in high- and middle-income countries varied between 10.1 and
86.2 per 100,000 individuals per year in the general population
(2). As a pulmonary disease that
occurs in response to stimuli targeting the alveolar-capillary
membrane, ALI/ARDS can lead to increased vascular permeability and
subsequently, pulmonary oedema (3).
Lipopolysaccharide (LPS) is considered to be a major active factor
during the inflammatory response (4,5).
LPS-induced inflammation has been reported to play a critical role
in the pathogenesis of ALI (6).
Consequently, further molecular research on the potential
mechanisms of LPS-induced inflammatory factor release in ALI/ARDS
and the development of new therapeutic targets for ARDS are
urgently required.
There is increasing evidence that long non-coding
(lnc) RNAs have been associated with regulating pulmonary diseases,
including ALI/ARDS (7-9).
Dai et al (7) showed that
MALAT1 overexpression increased the concentration of LPS-induced
inflammatory factors in mouse alveolar cells; therefore,
accelerating the progression of LPS-induced ALI. Wang et al
(8) revealed that THRIL mRNA
expression level was positively correlated with IL-1β and TNF-α
concentration in the tissues of patients with ARDS. Zhou et
al (9) found that NEAT1
expression was markedly elevated in a mouse model of ALI and NEAT1
knockdown inhibited the release of inflammatory factors from mouse
alveolar epithelial cells. Notably, HOTAIR has been
associated with regulating the LPS-induced inflammatory response in
different aspects, such as LPS-induced inflammatory injury in mouse
chondrogenic cells (10), and in
the LPS-induced inflammatory response in mice macrophages (11) and in cardiomyocytes from mice with
sepsis (12). However, the detailed
mechanism of HOTAIR in the LPS-induced inflammatory response
in ALI/ARDS remains unknown.
It is widely known that microRNAs (miRNA/miR) have
been associated with LPS-induced ALI and the release of
inflammatory mediators (13-15).
Li et al (13) revealed that
the overexpression of miR-150 decreased the concentration of
IL-6, IL-1β and tumor necrosis factor (TNF)-α in mice with
LPS-induced ALI. Cheng et al (14) demonstrated that miR-424
upregulation inhibited the release of inflammatory factors from
LPS-induced alveolar epithelial cells. He et al (15) found that lung injury and the
inflammatory response induced by LPS were attenuated by
miR-146b overexpression; therefore, ameliorating LPS-induced
ALI. Notably, miR-30a-5p has been shown to have a protective
role in LPS-induced ALI in children (16). In addition, lncRNAs can act as
competing endogenous RNAs or sponges of miRNAs. HOTAIR has
been reported to regulate numerous miRNAs in several types of
cancer, such as miR-138-5p in ovarian cancer (17), miR-1277-5p in gastric cancer
(18), miR-449b-5p (19) or miR-601 (20) in breast cancer and miR-203 in
lung cancer (21). However, the
mechanisms whereby lncRNA HOTAIR regulates miR-30a-5p
in ARDS/ALI have not yet been reported.
In the present study the effects of lncRNA
HOTAIR knockdown on the concentrations of inflammatory
factors in MLE-12 cells, the regulatory mechanism of how
HOTAIR regulates the miR-30a-5p/PDE7A axis in
LPS-induced MLE-12 cells and the effect of HOTAIR knockdown
on LPS-induced ARDS in vivo were investigated. The results
are aimed to provide a promising therapeutic target for ARDS.
Materials and methods
Cell culture, grouping and
transfection
The murine alveolar epithelial cell line, MLE-12,
was purchased from Baiye Biotech, Ltd. The MLE-12 cells were
cultured at 37˚C in a humidified incubator with 5% CO2,
in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.), supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
and 0.1% penicillin/streptomycin.
A short hairpin (sh)RNA-non-targeting negative
control (sh-NC) and shRNA targeting HOTAIR
(sh-HOTAIR) were purchased from Sangon Biotech, Co., Ltd. A
PDE7A-overexpression plasmid (pcDNA-PDE7A) and its NC (pcDNA-NC),
miR-30a-5p mimics/miR-NC and miR-30a-5p
inhibitor/inhibitor NC were all purchased from Guangzhou RiboBio
Co., Ltd. The aforementioned molecules (all at 20 nM) were
transfected into the MLE-12 cells using Lipofectamine®
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C for 48 h.
Subsequently, the transfected cells were induced with LPS (1 µg/ml;
Sigma-Aldrich; Merck KGaA). MLE-12 cells without any treatments
were served as the controls. The next day, the MLE-12 cells were
harvested for the following experiments.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the mouse cells or lung
tissues using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA (500 ng) was reverse transcribed into
cDNA at 42˚C for 45 min using a First-Strand cDNA Synthesis kit
(APeXBIO Technology LLC) and qPCR was performed with SYBR Green
FAST MasterMix kit (Qiagen GmbH), according to the manufacturer's
instructions. The following thermocycling conditions were used for
qPCR: Initial denaturation at 94˚C for 10 min, followed by 40
cycles at 94˚C for 10 sec, 60˚C for 20 sec and 72˚C for 1 min.
GADPH and U6 were used as the internal references.
The respective sequences of primers were as follows: HOTAIR
forward, 5'-GGTAGAAAAAGCAACCACGAAGC-3' and reverse,
5'-ACATAAACCTCTGTCTGTGAGTGCC-3'; miR-30a-5p forward,
5'-AACGAGACGACGACAGAC-3' and reverse, 5'-TGTAAACATCCTCGACTGGAAG-3';
PDE7A forward, 5'-GGAAATAGTCTAGTAAGCTTAACC-3' and reverse,
5'-GGCAGATGTGAGAATAAGCCTG-3'; GAPDH forward,
5'-TCCGCCCCTTCCGCTGATG-3' and reverse, 5'-CACGGAAGGCCATGCCAGTGA-3';
U6 forward, 5'-CTCGCTTCGGCAGCACA-3' and reverse
5'-AACGCTTCAGAATTTGCGT-3'. Gene expression was quantified using the
2-ΔΔCq method (22).
MTT assay
The viability of the MLE-12 cells was determined
using a MTT assay. Transfected and LPS-treated MLE-12 cells were
seeded into a 96-well plate, at 2x105 cells per well and
incubated at 37˚C. After incubation for 24 h, 20 µl MTT (Shanghai
GeneChem, Co., Ltd.) was added to each well. After incubation for 2
h at 37˚C, cell viability (optical density at 570 nm) was analyzed
using a Multiskan Spectrum microplate reader (Thermo Fisher
Scientific, Inc.).
ELISA
The concentration of the inflammatory factors [TNF-α
(cat. no. 70-EK182HS-96), IL-1β (cat. no. 70-EK101BHS-96) and IL-6
(cat. no. 70-EK106/2-24)] in the MLE-12 cells or mouse
bronchoalveolar lavage fluid (BALF) were measured using specific
ELISA kits (Multisciences Biotech Co., Ltd.), according to the
manufacturer's protocol. A Multiskan Spectrum microplate reader
(Thermo Fisher Scientific, Inc.) was used to determine the
absorbance at 450 nm.
Target prediction
The miRNA targets of HOTAIR were predicted
using StarBase database (http://starbase.sysu.edu.cn/). In addition, the mRNA
targets of miR-30a-5p were predicted using TargetScan
database (http://www.targetscan.org).
Dual luciferase reporter (DLR)
assay
The 3'-untranslated region sequences containing the
HOTAIR binding site were cloned into the pGL3 vector
(Promega Corporation) to establish the wild-type (WT) vector. The
Phusion Site-Directed Mutagenesis kit (Thermo Fisher Scientific,
Inc.) was used to construct the mutant vector. The WT or mutant
vectors and the miR-146a mimics or miR-NC were
co-transfected into the MLE-12 cells using Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C for 48 h. A
DLR Assay System (Promega Corporation) was used to determine
relative luciferase activity. The activity of firefly luciferase
was normalized to the activity of Renilla luciferase.
Western blot analysis
RIPA buffer (Beyotime Institute of Biotechnology),
containing protease inhibitors was used to extract total protein
from the MLE-12 cells. Protein concentration was then determined
using a BCA Protein Assay kit (Abcam). A total of 50 µg
protein/lane was separated using 10% SDS-PAGE and the resolved
proteins were transferred onto PVDF membranes. The membranes were
blocked with 5% bovine serum albumin (Thermo Fisher Scientific,
Inc.) for 2 h at room temperature. Following which, the membranes
were incubated overnight at 4˚C with the primary antibodies against
PDE7A (1:1,000; cat. no. ab14616; Abcam) and tubulin (1:1,000; cat.
no. ab6160; Abcam). Subsequently, the membranes were washed 3 times
with TBS-Tween-20 (0.05%), then incubated with an HRP-conjugated
anti-rabbit IgG secondary antibody (1:5,000; cat. no. ab6721;
Abcam) and an HRP-conjugated anti-rat IgG secondary antibody
(1:5,000; cat. no ab6734; Abcam), respectively, at room temperature
for 1 h. Tubulin was used as the internal reference. An enhanced
chemiluminescence detection kit (Thermo Fisher Scientific, Inc.)
was used to detect the bands, which were then quantified using
Gel-Pro Analyzer software (v4.0; Media Cybernetics, Inc.).
Construction of an ARDS mouse
model
A total of 20 female BALB/c nude mice (4-5 weeks;
weighing 20±2 g) were purchased from Cavens Lab. All mice were
housed under controlled conditions (25˚C; 50% humidity; 12 h
light/dark cycle) and had free access to food and water. The mouse
model of ARDS was constructed by intratracheal instillation of 2
mg/kg LPS. The mice were randomly divided into sham, ARDS, ARDS +
sh-NC and ARDS + sh-HOTAIR groups (n=5). After LPS treatment
for 48 h, the mice in the ARDS + sh-HOTAIR group were
treated with sh-HOTAIR lentivirus (2x107
transducing units/ml by intravenous injection), while those in the
ARDS + sh-NC group were intravenously injected with an equal
quantity of sh-NC lentivirus. Simultaneously, the mice in the sham
group were intravenously injected with an equal volume of saline.
The next day, the mice were anesthetized with sodium pentobarbital
(50 mg/kg) and euthanized via cervical dislocation. Subsequently,
the BALF samples were collected and centrifuged, and the resulting
supernatants were used to measure the concentration of TNF-α, IL-6
and IL-1β.
Histopathological examination using
H&E staining
Fresh mouse lung samples were resected, then fixed
with 4% paraformaldehyde at 37˚C for 1 day. The samples were then
embedded in paraffin and cut into 6-µm thick sections. The sections
were immediately stained with H&E for 15 min at room
temperature, then observed using a light microscope (magnification,
x200). The injury score was calculated based on a previous study
(23).
Lung function assay
After sacrifice of the mice, arterial blood samples
(5 ml) were collected from mouse carotid arteries. An automatic
blood gas analyzer (Radiometer Medical ApS) was used to determine
the partial pressure of oxygen (PaO2) and carbon dioxide
(PaCO2). The wet weight of the fresh lung tissues was
then determined. The tissues were dried at 60˚C for 72 h and the
dry weight was then determined. The wet/dry weight ratio of the
lung samples was used as a measure of tissue oedema.
Statistical analysis
SPSS software (v20.0; IBM, Corp.) was used to
perform statistical analyses. The data are presented as the mean ±
standard deviation. A Student's t-test (unpaired) was used to
assess the differences between two groups, while one-way ANOVA was
used to evaluate the differences among multiple groups, followed by
a Tukey's multiple comparisons test. P<0.05 was considered to
indicate a statistically significant difference. All experiments
were performed in independently 3 times in triplicate.
Results
lncRNA HOTAIR knockdown decreases the
concentration of the inflammatory factors in LPS-induced MLE-12
cells
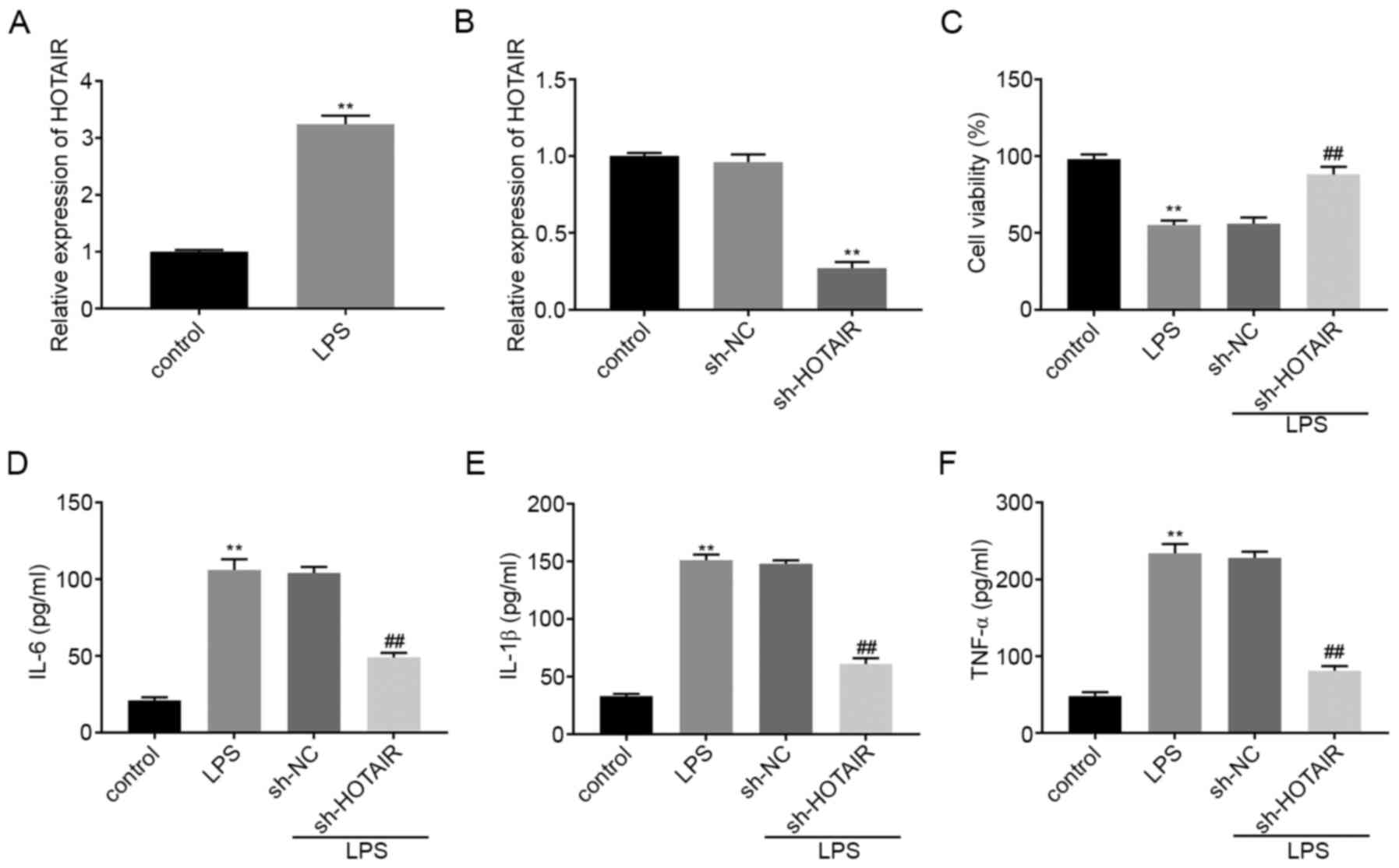
HOTAIR mRNA expression level was upregulated
in the MLE-12 cells in the LPS group compared with that in the
control group (P<0.01; Fig. 1A).
RT-qPCR was then used to determine the transfection efficiency of
sh-HOTAIR and sh-NC in the MLE-12 cells and the results
illustrated that HOTAIR expression was significantly decreased in
the sh-HOTAIR group compared with that in the sh-NC group
(P<0.01; Fig. 1B). A MTT assay
revealed that, compared with that in the control cells, treatment
with LPS significantly decreased cell viability. Cell viability in
the sh-HOTAIR + LPS group was also significantly increased compared
with that in the sh-NC + LPS group (P<0.01; Fig. 1C). ELISA results showed that the
concentration of IL-6, IL-1β and TNF-α were all upregulated in the
MLE-12 cells in the LPS group compared with that in the control
group. However, transfection with sh-HOTAIR significantly
decreased the concentration of the inflammatory factors in the
MLE-12 cells treated with LPS compared with that in cells
transfected with sh-NC (P<0.01; Fig.
1D-F).
lncRNA HOTAIR targets miR-30a-5p
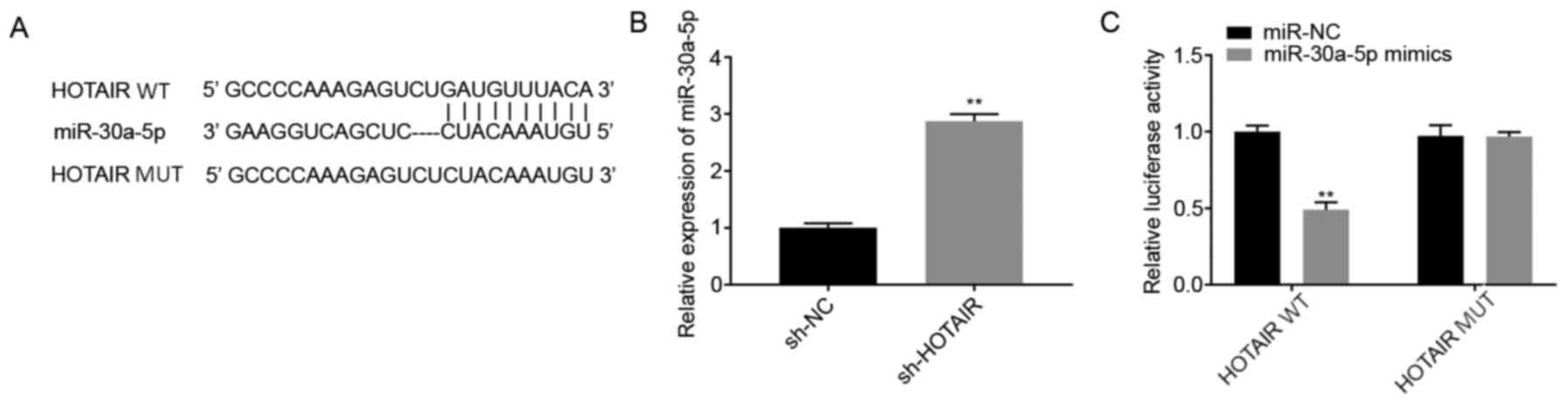
The potential binding sites between HOTAIR
and miR-30a-5p were predicted using StarBase database
(Fig. 2A). Among the 46 miRNA
targets predicted, miR-30a-5p was selected for further analysis as
it has been associated with ALI (16) and its unknown regulatory association
with HOTAIR. The mRNA expression levels of miR-30a-5p
following transfection of the MLE-12 cells with sh-HOTAIR or
sh-NC were also determined. The results showed that
miR-30a-5p expression level was increased in the
sh-HOTAIR group compared with that in the sh-NC group
(P<0.01; Fig. 2B). Subsequently,
a DLR assay was performed to further verify the interaction between
HOTAIR and miR-30a-5p. The results showed that
luciferase activity was lower in the HOTAIR WT +
miR-30a-5p mimics group compared with that in the
HOTAIR WT + miR-NC group (P<0.01; Fig. 2C).
Overexpression of miR-30a-5p decreases
the concentration of the inflammatory factors in the LPS-induced
MLE-12 cells
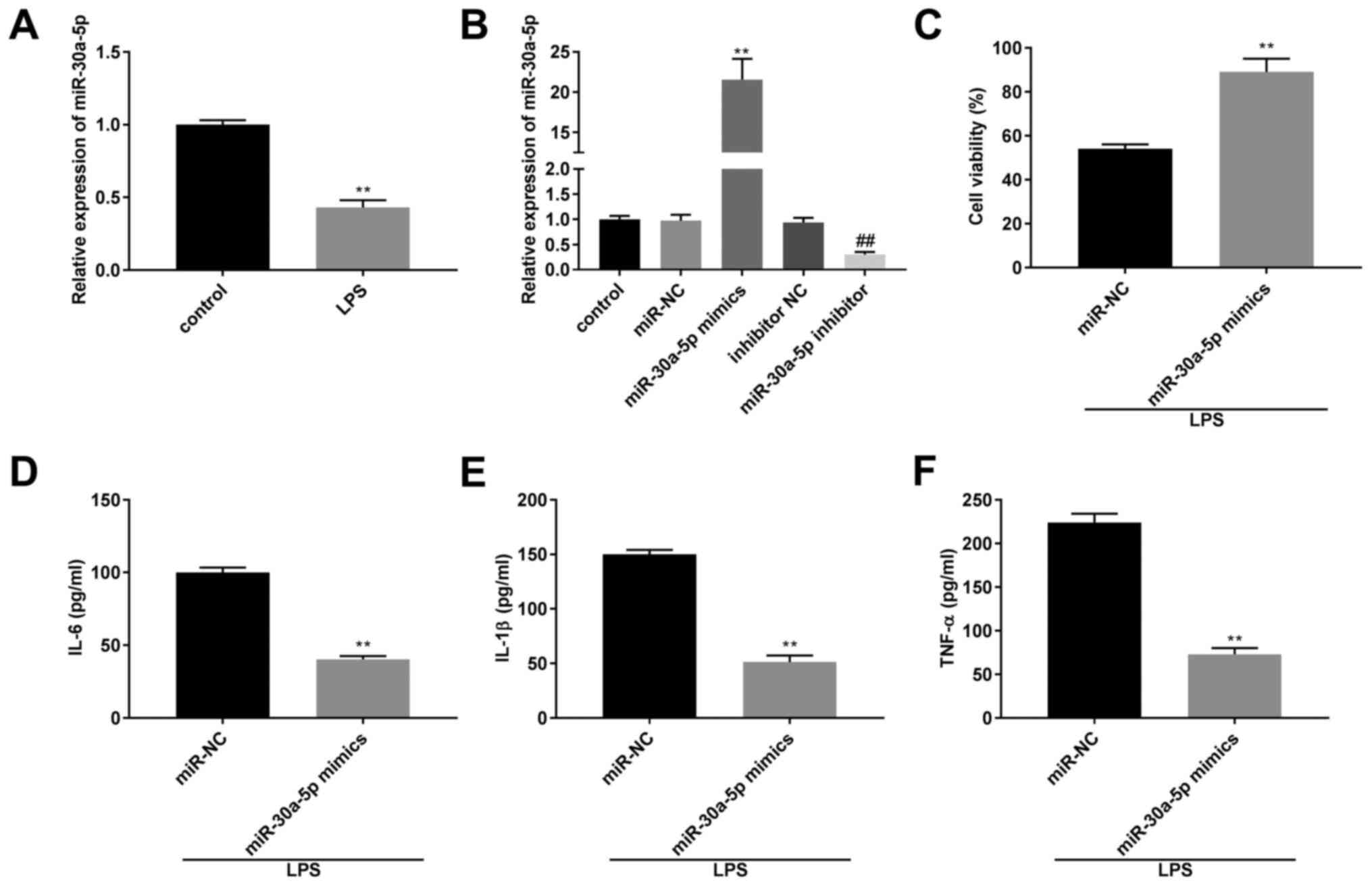
The mRNA expression level of miR-30a-5p was
significantly lower in the LPS group compared with that in the
control group (P<0.01; Fig. 3A).
The transfection efficiency of the miR-30a-50p mimics,
inhibitor and their respective NCs in the MLE-12 cells was
determined using RT-qPCR. The results revealed that
miR-30a-5p mRNA expression levels were downregulated by the
miR-30a-5p inhibitor and upregulated by miR-30a-5p
mimics (P<0.01; Fig. 3B). After
treatment with LPS, the results from a MTT assay and ELISA
demonstrated that compared with that in the miR-NC group, cell
viability and the concentration of IL-6, IL-1β and TNF-α was
increased and decreased, respectively, in the miR-30a-5p
mimics group (P<0.01; Fig.
3C-F).
miR-30a-5p targets PDE7A
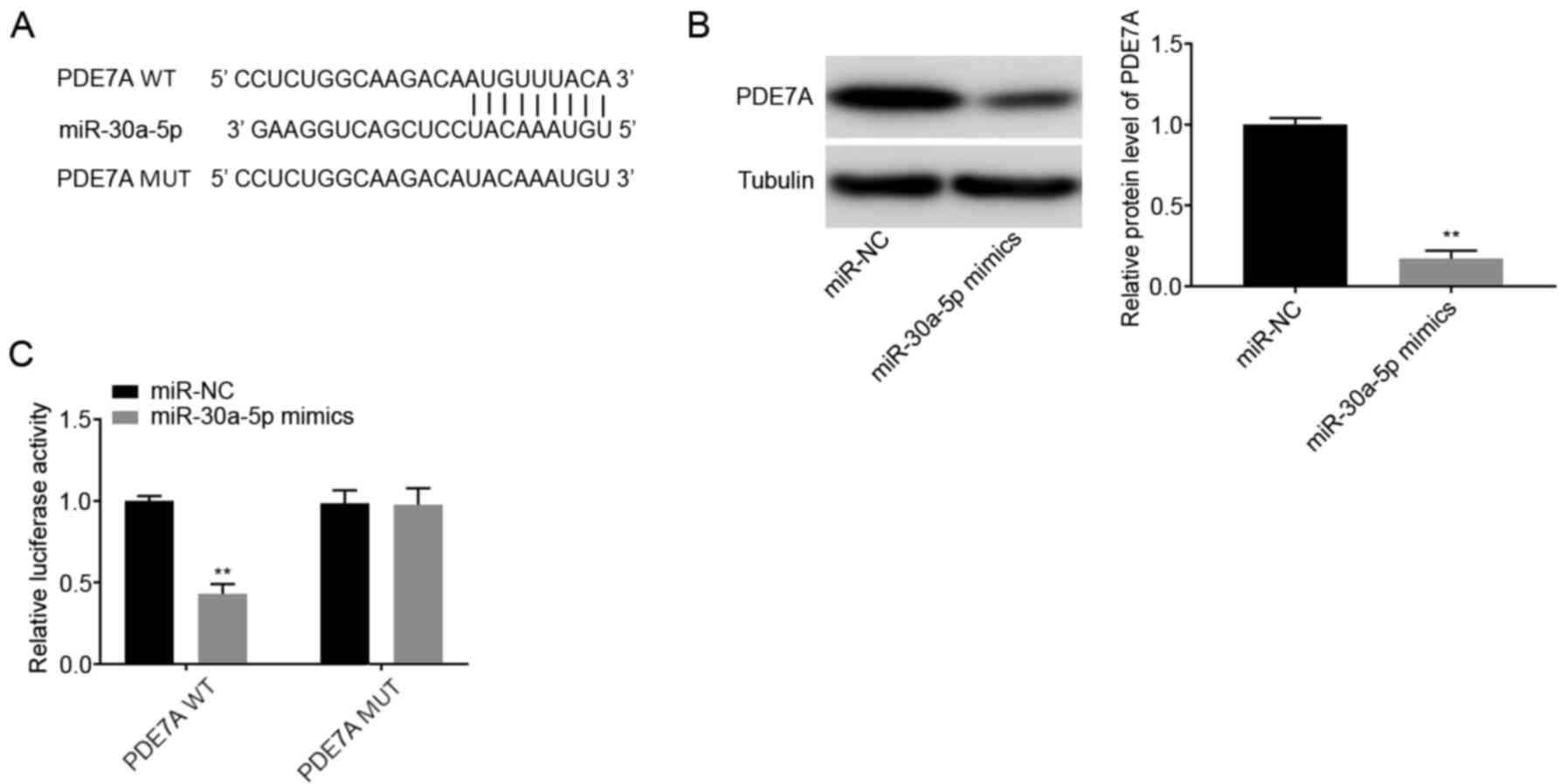
Using TargetScan database, the potential binding
site between miR-30a-5p and PDE7A was predicted
(Fig. 4A). A total of 1,570 targets
were predicted. PDE7A was selected for further analysis as it has
been associated with pulmonary inflammatory diseases, such as
chronic obstructive pulmonary disease and asthma (24). Western blot analysis showed that the
protein expression levels of PDE7A were decreased in the
miR-30a-5p mimics group compared with that in the miR-NC
group (P<0.01; Fig. 4B). DLR
assays showed that luciferase activity was lower in the
PDE7A WT + miR-30a-5p mimics group compared with that
in the PDE7A WT + miR-NC group (P<0.01; Fig. 4C).
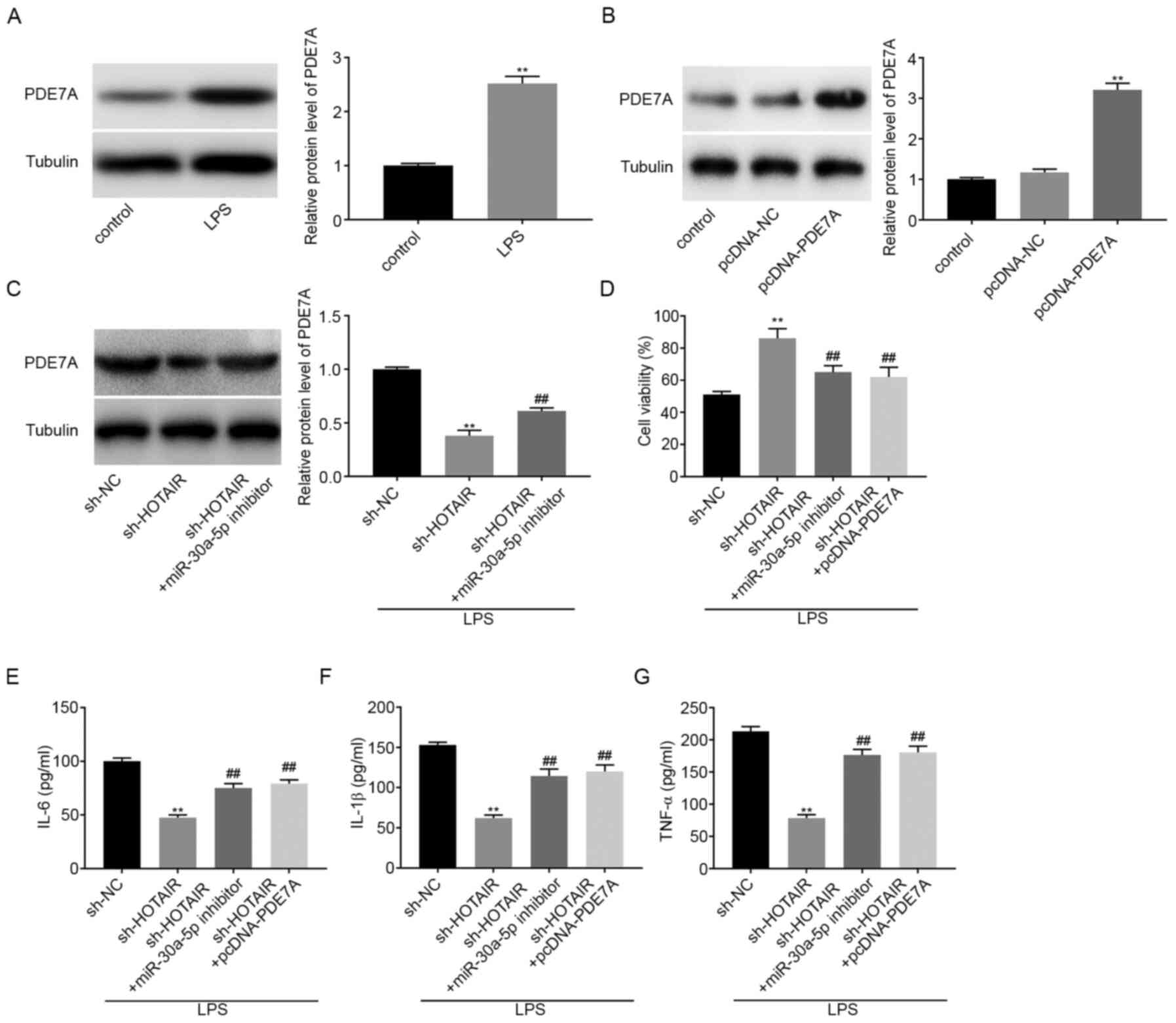
lncRNA HOTAIR knockdown decreases
inflammatory factor concentration in LPS-induced MLE-12 cells by
regulating the miR-30a-5p/PDE7A axis
Western blot analysis was used to determine the
protein expression levels of PDE7A in LPS-induced MLE-12 cells. The
results showed that LPS treatment significantly increased PDE7A
protein expression levels (P<0.01; Fig. 5A). pcDNA-PDE7A or pcDNA-NC were then
transfected into the MLE-12 cells and the results from western blot
analysis demonstrated that PDE7A overexpression significantly
increased the protein expression levels of PDE7A (P<0.01;
Fig. 5B). Subsequently, sh-NC,
sh-HOTAIR, or sh-HOTAIR + miR-30a-5p inhibitor
was transfected into the MLE-12 cells, then stimulated with LPS.
The protein expression level of PDE7A was subsequently analyzed and
the results indicated that HOTAIR knockdown decreased the
protein expression level of PDE7A, whereas downregulation of
miR-30-5p reversed the inhibitory effect of HOTAIR
knockdown on PDE7A protein expression levels (P<0.01; Fig. 5C). The results from the MTT assay
showed that cell viability increased in the sh-HOTAIR group
compared with that in the sh-NC group, whereas cell viability in
both the sh-HOTAIR + miR-30a-5p inhibitor group and
the sh HOTAIR + pcDNA-PDE7A group was partly reduced
compared with that in the sh-HOTAIR group (P<0.01;
Fig. 5D). ELISA results showed that
the concentrations of IL-6, IL-1β, and TNF-α were all decreased by
HOTAIR knockdown. However, downregulation of
miR-30a-5p and upregulation of PDE7A both reversed the
suppressive effect of HOTAIR knockdown on the concentration
of the inflammatory factors (P<0.01; Fig. 5E-G).
lncRNA HOTAIR knockdown attenuates
LPS-induced ARDS and the ARDS-related inflammatory response in
vivo
The mRNA expression levels of HOTAIR,
miR-30a-5p, and PDE7A in mouse lung tissues were
determined using RT-qPCR. The results showed that, compared with
that in the sham group, HOTAIR and PDE7A mRNA
expression levels were elevated in the ARDS group, whereas
miR-30a-5p mRNA expression levels were lower in the ARDS
group (P<0.05; Fig. 6A-C).
Following an intravenous injection with sh-HOTAIR or sh-NC
lentiviruses, the mRNA expression levels of HOTAIR and
PDE7A were downregulated in the lung tissues of mice in the
ARDS + sh-HOTAIR group compared with that in the ARDS group.
However, miR-30a-5p mRNA expression levels were upregulated
in the ARDS + sh-HOTAIR group compared with that in the ARDS
group. As shown in Fig. 6D, the
histopathological changes and injury scores of lung tissues were
analyzed using H&E staining. Inflammatory cell infiltration and
interstitial oedema were more severe in the ARDS group compared
with that in the sham group. HOTAIR knockdown markedly
ameliorated these effects. The injury scores were higher in the
ARDS group compared with that in the sham group. HOTAIR
knockdown significantly decreased the injury scores of the lung
tissues (P<0.01; Fig. 6D). Lung
function assays demonstrated that, compared with that in the sham
group, PaCO2 levels and the lung wet/dry weight ratios
were increased, whereas PaO2 levels were decreased in
the ARDS group (P<0.01; Fig.
6E-G). HOTAIR knockdown decreased PaCO2
levels and lung wet/dry weight ratios, and increased
PaO2 levels. ELISA results showed that IL-6, IL-1β and
TNF-α concentration in the mouse BALF samples were all elevated in
the ARDS group compared with that in the sham group (P<0.01;
Fig. 6H-J). However, HOTAIR
knockdown significantly reversed the promoting effect of ARDS on
inflammatory factor levels.
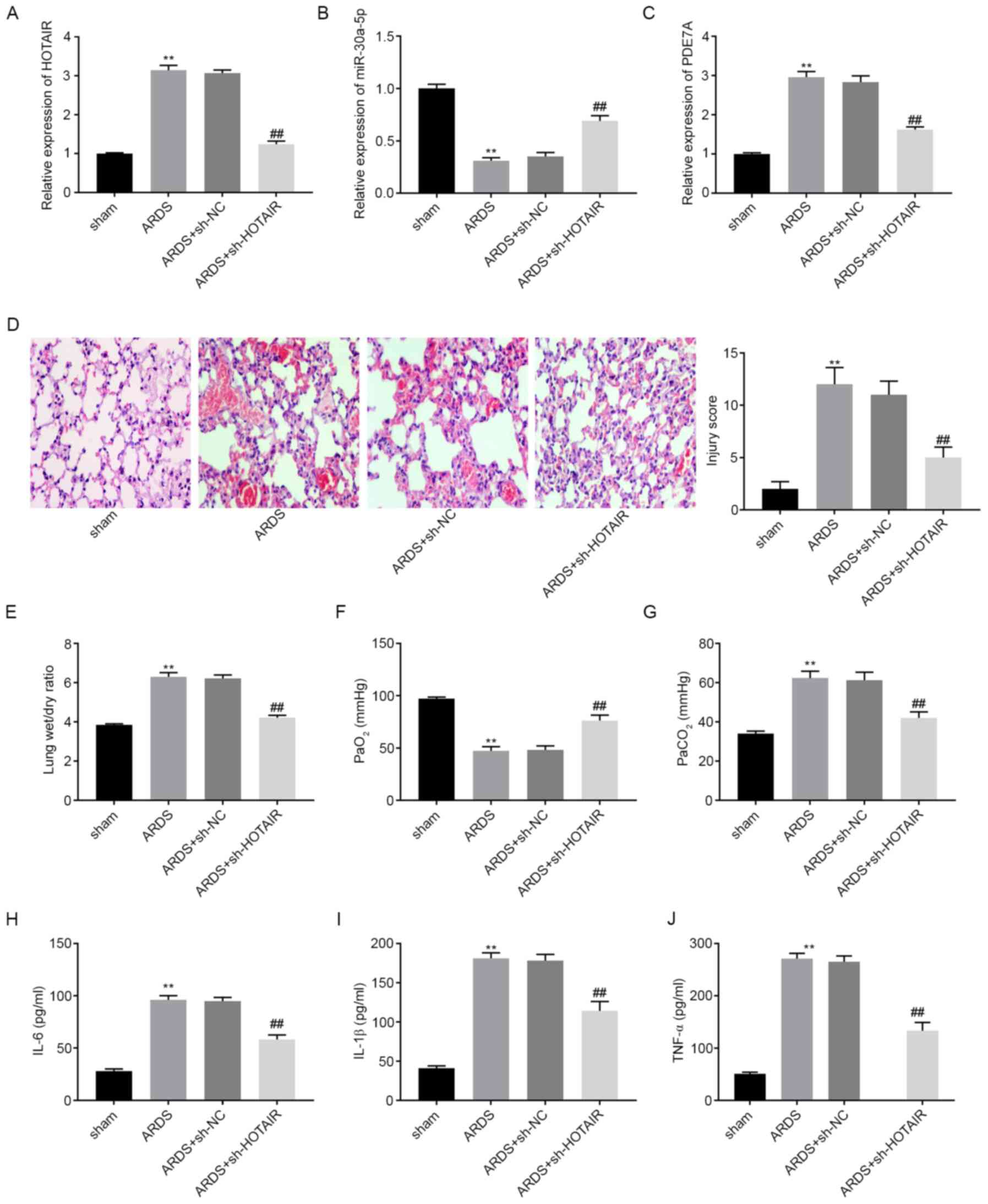 | Figure 6lncRNA HOTAIR knockdown attenuates
LPS-induced ARDS and ARDS-related inflammatory response in
vivo. The mRNA expression level of (A) HOTAIR, (B) miR-30a-5p
and (C) PDE7A in the lung tissues of mice was detected using
reverse transcription-quantitative PCR. (D) Histopathological
change and injury score were detected using H&E staining.
Magnification, x200. (E) Lung wet/dry weight ratio, (F)
PaO2 and (G) PaCO2 was measured using an
automatic blood gas analyzer. The concentration of (H) IL-6, (I)
IL-1β and (J) TNF-α in BALF of mice was measured using ELISA.
**P<0.01 vs. sham. ##P<0.01 vs. ARDS.
Pa, partial pressure; TNF, tumor necrosis factor; miR, microRNA;
NC, negative control; sh, short hairpin; ARDS, acute respiratory
distress syndrome. |
Discussion
As a severe form of ALI, the cost to treat ARDS is
high, which severely affects the health and quality of life in
patients with ARDS (25). Several
lncRNAs have been associated with the progression of ALI and ARDS
(7-9).
Dai et al (7) found high
protein expression levels of MALAT1 in the lung tissues from an
LPS-induced rat ALI model. Wang et al (8) showed that THRIL expression was
upregulated in lung tissues from patients with ARDS. Zhou et
al (9) demonstrated that NEAT1
was highly expressed in the lung tissues of mice with LPS-induced
ALI and in LPS-treated mouse alveolar epithelial cells. Consistent
with the aforementioned results, the present study showed that
HOTAIR mRNA expression levels were increased in the lung
tissues from mice with LPS-induced ARDS and in LPS-treated MLE-12
cells. Therefore, the results from the present study suggested that
HOTAIR may act as a pathogenic factor in ARDS.
Previous studies have reported that lncRNAs act as
crucial regulators in hypoxic lung diseases and inflammatory
epithelial injury (26,27), by affecting processes, such as
inflammation and interstitial oedema (7,28,29).
Dai et al (7) have shown
that MALAT1 inhibition led to a marked decrease in the
concentration of IL-6, IL-1β and TNF-α in rat lung tissues. In
addition, histopathological examination showed that MALAT1
inhibition distinctly attenuated lung tissue injury. Jiang et
al (29) showed that the
PaO2/FiO2 ratio was decreased and the lung
wet/dry weight ratio was increased in ARDS. Kcnq1ot1 knockdown in
mice was shown to reverse the suppressive effect of LPS on the
PaO2/FiO2 ratio and the positive effect of
LPS on the lung wet/dry weight ratio (29). Consistent with the aforementioned
results, in the present study, it was found that injury scores,
lung wet/dry weight ratios, PaCO2 levels and the
concentration of IL-6, IL-1β and TNF-α were increased, and
PaO2 levels were decreased in a mouse model of ARDS.
HOTAIR knockdown using a sh HOTAIR lentivirus
injection reversed the positive effect of LPS on injury scores,
lung wet/dry weight ratios, PaCO2 levels, and the
concentration of IL-6, IL-1β and TNF-α, and the negative effect of
LPS on PaO2 levels. These results suggested that
HOTAIR knockdown attenuated the LPS-induced inflammatory
response and LPS-induced ALI. To further verify this hypothesis,
further experiments were performed using the MLE-12 cells and it
was found that sh-HOTAIR reversed the negative effect of LPS
on cell viability and the positive effect of LPS on the
concentration of IL-6, IL-1β and TNF-α. The results from the
present study are consistent with those of other studies (10-12),
suggesting that HOTAIR knockdown attenuated the LPS-induced
inflammatory response and LPS-induced ALI in vivo and in
vitro.
Increasing evidence has indicated that miRNA
expression levels were downregulated in LPS-induced rat models of
ALI or LPS-treated cells. For example, miR-497a-5p (30) and miR-381-3p (29) expression levels were downregulated
in an LPS-induced mouse model of ALI, while miR-424
(14) expression levels were
downregulated in alveolar epithelial cells, and miR-146b
(15) expression levels were
downregulated in murine lung alveolar macrophages. Similarly, in
the present study it was found that miR-30a-5p expression
levels were downregulated in both an LPS-induced mouse model of
ARDS and in LPS-treated MLE-12 cells. In addition, previous studies
have shown that miRNAs have a role in modulating pulmonary
inflammation (13,14,30).
Cheng et al (14) showed
that upregulation of miR-424 suppressed the secretion of
IL-6 and IL-8 from LPS-treated alveolar epithelial cells. Guo et
al (30) showed that
miR-497a-5p overexpression decreased the concentration of
IL-6, IL-1β and TNF-α in LPS-treated RAW264.7 cells. Li et
al (13) reported that high
expression levels of miR-150 inhibited the secretion of
IL-6, IL-1β and TNF-α from LPS-treated human pulmonary epithelial
cells. In the present study, it was found that miR-30a-5p
upregulation suppressed the secretion of IL-6, IL-1β and TNF-α from
LPS-treated MLE-12 cells. These results suggested that
miR-30a-5p may be a suppressor of the inflammatory response.
Consistent with these results, miR-30a-5p inhibition has
been shown to promote the release of IL-6, IL-1β and TNF-α from
PC-12 cells (31). In addition,
miR-30a-5p was shown to be the target of HOTAIR in
the present study. Downregulation of miR-30a-5p reversed the
positive effect of sh-HOTAIR on MLE-12 cell viability and
the inhibitory effect of sh-HOTAIR on inflammatory factor
release from MLE-12 cells. This suggested that HOTAIR
knockdown alleviated the LPS-induced inflammatory response by
modulating miR-30a-5p.
Enzymes within the phosphodiesterase (PDE) family
are found in most proinflammatory and immune cells (24). As a member of the PDE family, PDE7A
has been associated with the inflammatory response (24,32-34).
Goto et al (32) reported
that PDE7A downregulation ameliorated concanavalin A-induced
hepatitis in mice by suppressing the concentration of IL-4 and
TNF-α. Kadoshima-Yamaoka et al (33) showed that ASB16165, a PDE7A
inhibitor, reduced cutaneous TNF-α concentration in
12-o-tetradecanoylphorbol-13-acetate-induced skin inflammation in
mice. Yamamoto et al (34)
demonstrated that YM-393059, an inhibitor of PDE7A, inhibited
LPS-induced TNF-α production in mice. In the present study, it was
found that PDE7A protein expression levels were upregulated in
LPS-treated MLE-12 cells and PDE7A overexpression accelerated
inflammatory factor release from the MLE-12 cells. These results
suggested that PDE7A was associated with promoting the secretion of
the inflammatory factors, whereas inhibiting PDE7A may be
beneficial in preventing inflammation in ARDS. In addition, PDE7A
overexpression reversed the positive effect of sh-HOTAIR on
cell viability and the inhibitory effect of sh-HOTAIR on
inflammatory factor release from the MLE-12 cells. This suggested
that HOTAIR knockdown attenuated the LPS-induced
inflammatory response by regulating PDE7A. At the same time,
PDE7A was the target gene of miR-30a-5p. We further
hypothesized that knockdown of HOTAIR ameliorated the
LPS-induced inflammatory response by regulating the
miR-30a-5p/PDE7A axis.
Taken together, the results of the current study
showed that HOTAIR knockdown ameliorated the LPS-induced
inflammatory response and ARDS in vivo and reduced
LPS-induced inflammatory factor production by regulating the
miR-30a-5p/PDE7A axis in vitro. However, there
were also some limitations. First, the detailed mechanism involving
HOTAIR, miR-30a-5p and PDE7A was not confirmed in
vivo. Second, the detailed mechanism of this axis and the
signaling pathways involved remain unclear. Further experiments
will be performed to elucidate these in the future. In conclusion,
these findings may contribute to the development of a new strategy
for treating ARDS.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
XM conceived and designed the present study. HW and
SS performed the experiments, analyzed the data and drafted the
article. XM revised the article critically for important
intellectual content. XM, HW and SS confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were conducted in
agreement with the principles approved by the ethical committee of
Yantai Yuhuangding hospital (Yantai, China; approval no.
2019017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Patel VJ, Roy SB, Mehta HJ, Joo M and
Sadikot RT: Alternative and Natural therapies for acute lung injury
and acute respiratory distress syndrome. Biomed Res Int.
2018(2476824)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: Epidemiology, patterns of care, and mortality for
patients with acute respiratory distress syndrome in intensive care
units in 50 countries. JAMA. 315:788–800. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Villar J, Blanco J and Kacmarek RM:
Current incidence and outcome of the acute respiratory distress
syndrome. Curr Opin Crit Care. 22:1–6. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tsai CL, Lin YC, Wang HM and Chou TC:
Baicalein, an active component of Scutellaria baicalensis, protects
against lipopolysaccharide-induced acute lung injury in rats. J
Ethnopharmacol. 153:197–206. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Opitz B, Van Laak V, Eitel J and Suttorp
N: Innate immune recognition in infectious and noninfectious
diseases of the lung. Am J Respir Crit Care Med. 181:1294–1309.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Han S and Mallampalli RK: The acute
respiratory distress syndrome: From mechanism to translation. J
Immunol. 194:855–860. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dai L, Zhang G, Cheng Z, Wang X, Jia L,
Jing X, Wang H, Zhang R, Liu M, Jiang T, et al: Knockdown of LncRNA
MALAT1 contributes to the suppression of inflammatory responses by
up-regulating miR-146a in LPS-induced acute lung injury. Connect
Tissue Res. 59:581–592. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang Y, Fu X, Yu B and Ai F: Long
non-coding RNA THRIL predicts increased acute respiratory distress
syndrome risk and positively correlates with disease severity,
inflammation, and mortality in sepsis patients. J Clin Lab Anal.
33(e22882)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhou H, Wang X and Zhang B: Depression of
lncRNA NEAT1 Antagonizes LPS-evoked acute injury and inflammatory
response in alveolar epithelial cells via HMGB1-RAGE signaling.
Mediators Inflamm. 2020(8019467)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Duan G, Song S and Niu S: WITHDRAWN: Long
non-coding RNA HOTAIR promotes LPS-induced inflammatory injury by
down-regulation of microRNA-124 in murine chondrogenic ATDC5 cells.
Life Sci: July 20, 2018 (Epub ahead of print).
|
|
11
|
Obaid M, Udden SMN, Deb P, Shihabeddin N,
Zaki MH and Mandal SS: LncRNA HOTAIR regulates
lipopolysaccharide-induced cytokine expression and inflammatory
response in macrophages. Sci Rep. 8(15670)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wu H, Liu J, Li W, Liu G and Li Z:
LncRNA-HOTAIR promotes TNF-α production in cardiomyocytes of
LPS-induced sepsis mice by activating NF-κB pathway. Biochem
Biophys Res Commun. 471:240–246. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li P, Yao Y, Ma Y and Chen Y: MiR-150
attenuates LPS-induced acute lung injury via targeting AKT3. Int
Immunopharmacol. 75(105794)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cheng D, Zhu C, Liang Y, Xing Y and Shi C:
MiR-424 overexpression protects alveolar epithelial cells from
LPS-induced apoptosis and inflammation by targeting FGF2 via the
NF-κB pathway. Life Sci. 242(117213)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
He R, Li Y, Zhou L, Su X, Pan P and Hu C:
miR-146b overexpression ameliorates lipopolysaccharide-induced
acute lung injury in vivo and in vitro. J Cell Biochem.
120:2929–2939. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhou T and Chen YL: The functional
mechanisms of miR-30b-5p in acute lung injury in children. Med Sci
Monit. 25:40–51. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang Y, Ai H, Fan X, Chen S, Wang Y and
Liu L: Knockdown of long non-coding RNA HOTAIR reverses cisplatin
resistance of ovarian cancer cells through inhibiting
miR-138-5p-regulated EZH2 and SIRT1. Biol Res.
53(18)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wei Z, Chen L, Meng L, Han W, Huang L and
Xu A: LncRNA HOTAIR promotes the growth and metastasis of gastric
cancer by sponging miR-1277-5p and upregulating COL5A1. Gastric
Cancer. 23:1018–1032. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang S, Wang B, Xiao H, Dong J, Li Y, Zhu
C, Jin Y, Li H, Cui M and Fan S: LncRNA HOTAIR enhances breast
cancer radioresistance through facilitating HSPA1A expression via
sequestering miR-449b-5p. Thorac Cancer. 11:1801–1816.
2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang Y, Gong G, Xu J, Zhang Y, Wu S and
Wang S: Long noncoding RNA HOTAIR promotes breast cancer
development by targeting ZEB1 via sponging miR-601. Cancer Cell
Int. 20(320)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang C, Xu L, Deng G, Ding Y, Bi K, Jin
H, Shu J, Yang J, Deng H, Wang Z and Wang Y: Exosomal HOTAIR
promotes proliferation, migration and invasion of lung cancer by
sponging miR-203. Sci China Life Sci. 63:1265–1268. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Guo Z, Li Q, Han Y, Liang Y, Xu Z and Ren
T: Prevention of LPS-induced acute lung injury in mice by
progranulin. Mediators Inflamm. 2012(540794)2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Smith SJ, Brookes-Fazakerley S, Donnelly
LE, Barnes PJ, Barnette MS and Giembycz MA: Ubiquitous expression
of phosphodiesterase 7A in human proinflammatory and immune cells.
Am J Physiol Lung Cell Mol Physiol. 284:L279–L289. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Petrucci N and De Feo C: Lung protective
ventilation strategy for the acute respiratory distress syndrome.
Cochrane Database Syst Rev. 2013(CD003844)2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sun H, Chen J, Qian W, Kang J, Wang J,
Jiang L, Qiao L, Chen W and Zhang J: Integrated long non-coding RNA
analyses identify novel regulators of epithelial-mesenchymal
transition in the mouse model of pulmonary fibrosis. J Cell Mol
Med. 20:1234–1246. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liang H, Gu Y, Li T, Zhang Y, Huangfu L,
Hu M, Zhao D, Chen Y, Liu S, Dong Y, et al: Integrated analyses
identify the involvement of microRNA-26a in epithelial-mesenchymal
transition during idiopathic pulmonary fibrosis. Cell Death Dis.
5(e1238)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dunkel B: Acute lung injury and acute
respiratory distress syndrome in foals. Equine Vet J. 5:127–133.
2006.
|
|
29
|
Jiang X, Yu M, Zhu T, Lou L, Chen X, Li Q,
Wei D and Sun R: Kcnq1ot1/miR-381-3p/ETS2 axis regulates
inflammation in mouse models of acute respiratory distress
syndrome. Mol Ther Nucleic Acids. 19:179–189. 2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Guo S, Chen Y, Liu J, Yang J, Yang C,
Zhang T, Jiang K, Wu Z, Shaukat A and Deng G: miR-497a-5p
attenuates lipopolysaccharide-induced inflammatory injury by
targeting IRAK2. J Cell Physiol. 234:22874–22883. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhu S, Zhou Z, Li Z, Shao J, Jiao G, Huang
YE and Lin Y: Suppression of LINC00707 alleviates
lipopolysaccharide-induced inflammation and apoptosis in PC-12
cells by regulated miR-30a-5p/Neurod 1. Biosci Biotechnol Biochem.
83:2049–2056. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Goto M, Tanaka Y, Murakawa M,
Kadoshima-Yamaoka K, Inoue H, Murafuji H, Nagahira A, Kanki S,
Hayashi Y, Nagahira K, et al: Inhibition of phosphodiesterase 7A
ameliorates Concanavalin A-induced hepatitis in mice. Int
Immunopharmacol. 9:1347–1351. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kadoshima-Yamaoka K, Goto M, Murakawa M,
Yoshioka R, Tanaka Y, Inoue H, Murafuji H, Kanki S, Hayashi Y,
Nagahira K, et al: ASB16165, a phosphodiesterase 7A inhibitor,
reduces cutaneous TNF-alpha level and ameliorates skin edema in
phorbol ester 12-O-tetradecanoylphorbol-13-acetate-induced skin
inflammation model in mice. Eur J Pharmacol. 613:163–166.
2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yamamoto S, Sugahara S, Naito R, Ichikawa
A, Ikeda K, Yamada T and Shimizu Y: The effects of a novel
phosphodiesterase 7A and -4 dual inhibitor, YM-393059, on
T-cell-related cytokine production in vitro and in vivo. Eur J
Pharmacol. 541:106–114. 2006.PubMed/NCBI View Article : Google Scholar
|