Introduction
According to the Global Cancer Observatory, 400,000
deaths from esophageal cancer occurred in 2012 and it ranks 6th
among all cancer types worldwide (1-3).
In addition, among the top 10 malignant tumor types in China,
esophageal cancer ranks 4th in males and 8th in females (4). The major pathological subtypes of
esophageal cancer are squamous cell carcinoma, adenocarcinoma and
small cell carcinoma, and squamous cell carcinoma is the major type
in China, accounting for >90% of cases, while adenocarcinoma is
the major type in European countries and in both North and South
America (5). The risk factors of
the different pathological types of esophageal squamous cell
carcinoma (ESCC) are also different and include sex, ethnicity,
smoking, alcohol consumption, diet, nutritional status and
hereditary factors (6).
The aryl hydrocarbon receptor (AHR) is in the basic
helix-loop-helix/Per-ARNT-SIM (bHLH/PAS) subgroup in the bHLH
transcription factor superfamily. The gene is ~60 Kbp long and has
22 exons. An unusual exon/intron junction sequence was detected in
the 11th intron of the gene, which begins at the 5' end of the GC
region. The exon/intron organization of the mouse AhR nuclear
translocator (mArnt) gene is different from the other members in
the same bHLH/PAS family, as it does not contain a TATA box and has
several transcriptional initiation sites (7). The promoter region of the mArnt gene
is rich in GC and contains numerous hypothesized regulatory DNA
sequences, such as two GC-boxes, a cyclic adenosine monophosphate
response element, an E-box, an activator protein-1 locus and a
CAAT-box. The AHR is the only member of the family, which is known
to be activated by a ligand (8).
The AHR is a ligand-activated transcription factor, located in the
cytoplasm and combines with the heat shock protein (HSP)90, the
AHR-interacting protein and the HSP90-interacting protein
P23(9). After the ligands are
combined, the AHR is transported into the nucleus and dimerizes
with the aryl receptor nuclear translocator. Subsequently, it
combines with xenobiotic response elements to regulate and
participate in the downstream signaling pathway (9,10).
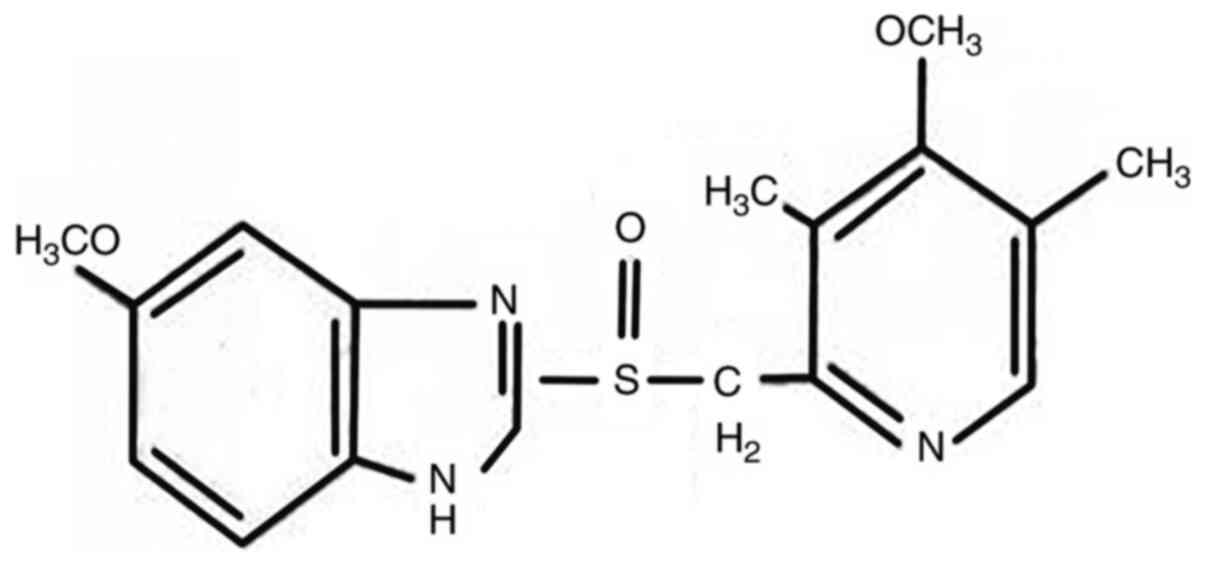
Omeprazole (OME; Fig.
1), a well-known acid suppressant, is used to inhibit the
proton pump in gastric wall cells (H+/K+
enzyme). In addition, animal experiments have demonstrated that OME
had a protective effect on gastric mucosa damage, and was able to
increase gastric mucosa blood flow due to an association with a
variety of different factors, such as glycoproteins, nitric oxide
and TNF-α (11). Furthermore, OME
has an anti-Helicobacter pylori (H. pylori) effect
(12). Due to its mode of action,
OME is widely used in the treatment of peptic ulcers, reflux
esophagitis, upper gastrointestinal hemorrhage, H. pylori
infection, Zole-Aids syndrome and gastric ulcer caused by
non-steroidal anti-inflammatory drugs (13). Following increases in AHR research,
OME has also been indicated to be a selective regulator of AHR
(14). Therefore, the present study
aimed to investigate whether OME regulates the expression level of
AHR in ESCC cells to affect the proliferation, cell cycle,
migration and invasion, to provide a novel treatment for ESCC.
Materials and methods
Cell culture
The human ESCC cell lines TE1 and KYSE150 were
purchased from the Chinese Academy of Sciences. The cells were
cultured in RPMI-1640 medium (Beijing Solarbio Science &
Technology Co., Ltd.) containing 10% FBS (Cellmax Nutrients B.V.)
at 37˚C in a humidified incubator with 5% CO2.
Western blot analysis
OME was first dissolved in DMSO at a high
concentration to produce a stock solution and then dissolved in the
corresponding medium during later use (the concentration of DMSO in
the final solution did not exceed 0.1%). TE1 and KYSE150 cells were
treated with 0, 100 and 300 µM OME for 48 h. The cells were then
lysed with RIPA buffer (Beyotime Institute of Biotechnology) for 15
min and the protein was extracted. The concentration of total
protein was measured using the bicinchoninic acid assay (Thermo
Fisher Scientific, Inc.). Protein aliquots (30 µg) were loaded with
SDS buffer (Beyotime Institute of Biotechnology) and boiled at 99˚C
for 15 min. Equal amounts of protein samples per lane were
separated using SDS-PAGE on a 10% gel. Following electrophoresis,
the cells were transferred to a PVDF membrane (EMD Millipore). Fast
blocking solution (Beyotime Institute of Biotechnology) was used
for blocking for 15 min at room temperature and the membrane was
incubated with the following primary antibodies on a shaking table
overnight at 4˚C: Anti-GAPDH (1:1,000; cat. no. ab181602; Abcam),
anti-AHR (1:500; cat. no. ab182642; Abcam), anti-proliferating cell
nuclear antigen (PCNA; 1:2,000; cat. no. 2586; Cell Signaling
Technology, Inc.) and anti-MMP9 (1:1,000; cat. no. 3582; Cell
Signaling Technology, Inc.). Subsequently, samples were incubated
with the corresponding secondary antibodies: Goat anti-Rabbit
secondary antibody (1:10,000; cat. no. 31460; Thermo Fisher
Scientific, Inc.) and Goat anti-mouse secondary antibody (1:10,000;
cat. no. 31430; Thermo Fisher Scientific, Inc.) for 2 h at room
temperature, and blots were visualized using enhanced
chemiluminescence (Beyotime Institute of Biotechnology) solution.
The grayscale values of the resulting bands were measured using
ImageJ software (version, 1.46r; National Institutes of Health).
Each experiment was performed in triplicate.
Cell proliferation assay
The TE1 and KYSE150 cells were cultured, collected
and then counted. Subsequently, the cells were seeded in a 96-well
plate (~2,000 cells/well; 200 µl/well). After the cells had
attached, they were treated with 0, 100 and 300 µM OME for at 24,
48, 72 or 96 h. Cell Counting Kit-8 solution (MedChemExpress) was
added for 2 h and cell proliferation was measured at 560 nm using a
microplate reader (Thermo Fisher Scientific, Inc.). At the same
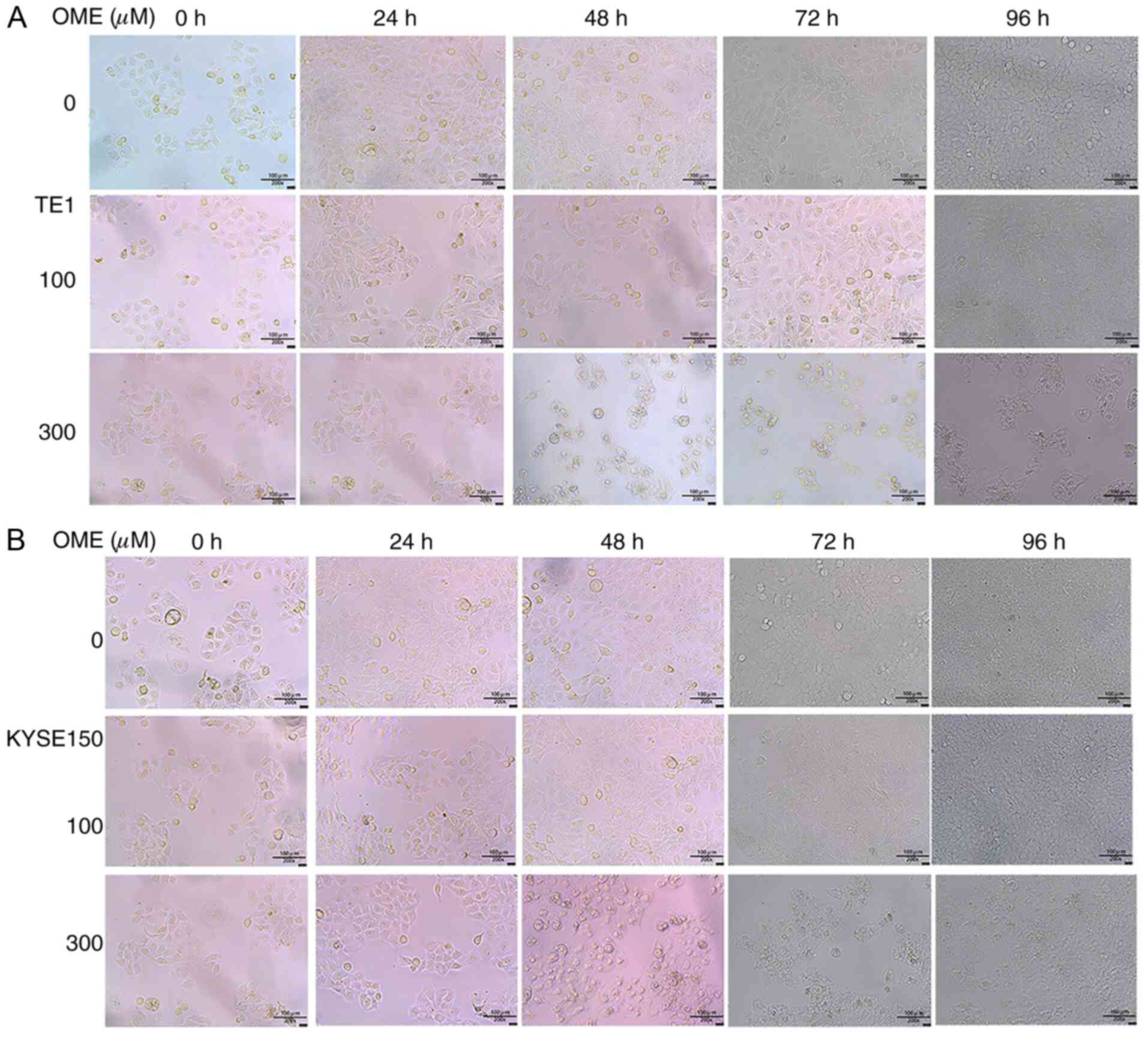
time, the photomicrographs of the ESCCs were captured at 0, 24, 48,
72 and 96 h with an inverted microscope (magnification, x200). Each
experiment was performed in triplicate.
Wound-healing assay
The TE1 and KYSE150 cells were cultured and seeded
in a six-well plate. After the cells grew to 90% confluence
(~1x106 cells per well), they were scratched with a
200-µl pipette tip and the serum-free medium was replaced with
serum-free OME at 0, 100 and 300 µM. Images of the wounds were
captured at 0, 24 and 48 h with an inverted microscope
(magnification, x50). Wound areas were measured using ImageJ
(version 1.46r; National Institutes of Health). The migration rate
was then calculated as follows: (The difference of the wound area
between 24, 48 and 0 h)/(wound area at 0 h). Each experiment was
performed in triplicate.
Transwell assay
The TE1 and KYSE150 cells were collected and then
seeded in a six-well plate. After the cells had attached, they were
treated with OME at 0, 100 and 300 µM for 48 h. The cells were then
collected and ~5,000 cells/well in 200 µl/well were seeded into the
upper chamber of a Transwell insert (Corning, Inc.) that had been
pre-coated with Matrigel™ (1:8; BD Biosciences) for 30 min at 37˚C
for the invasion assay. RPMI-1640 medium containing 20% FBS (500
µl/well) was added to the lower chamber. The cells were fixed with
methanol for 10 min and stained with crystal violet for 20 min at
room temperature. After 48 h of incubation, cells on the upper
surface were scraped off and cells on the lower surface were viewed
under a light microscope (Leica Microsystems; magnification, x100).
Images were captured using ImageJ software.
Cell cycle analysis
The TE1 and KYSE150 cells were collected and seeded
in a six-well plate. Subsequently, the cells were treated with
serum-free medium for 24 h and then with OME at 0, 100 and 300 µM.
After 48 h, the cells were collected and fixed overnight in a flow
tube (BD Biosciences) at 4˚C. A total of 490 µl PBS was added to
the cells and RNase A (Sigma Aldrich; Merck KGaA) was added.
Following incubation in a 37˚C water bath for ~30 min, the cells
was stained with 10 µl propidium iodide (PI; 50 µg/ml, BD
Biosciences) for 30 min on ice in the dark. The cell cycle was then
measured using flow cytometry. The cell cycle distribution was
detected by a BD LSRFortessa instrument (BD Biosciences) and the
results were analyzed with ModFit software (version, LT 4.1; BD
Biosciences).
Statistical analysis
All of the data were analyzed using SPSS software
(version 20; IBM Corp.) and GraphPad Prism 5.04 (GraphPad Software,
Inc.). One-way ANOVA and two-way ANOVA with Bonferroni's post-hoc
test was used for comparing means of multiple samples. Experimental
data are presented as the mean ± standard deviation. The survival
analysis was performed using the Kaplan-Meier method. P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibitory effect of OME on AHR
expression
AHR, a ligand-activated transcription factor, has a
variety of endogenous and exogenous ligands (15). In previous studies investigating
other types of cancer, OME was indicated to regulate the expression
level of AHR in a non-ligand manner (16). In addition, numerous studies have
demonstrated the promoting effect of AHR on tumor development
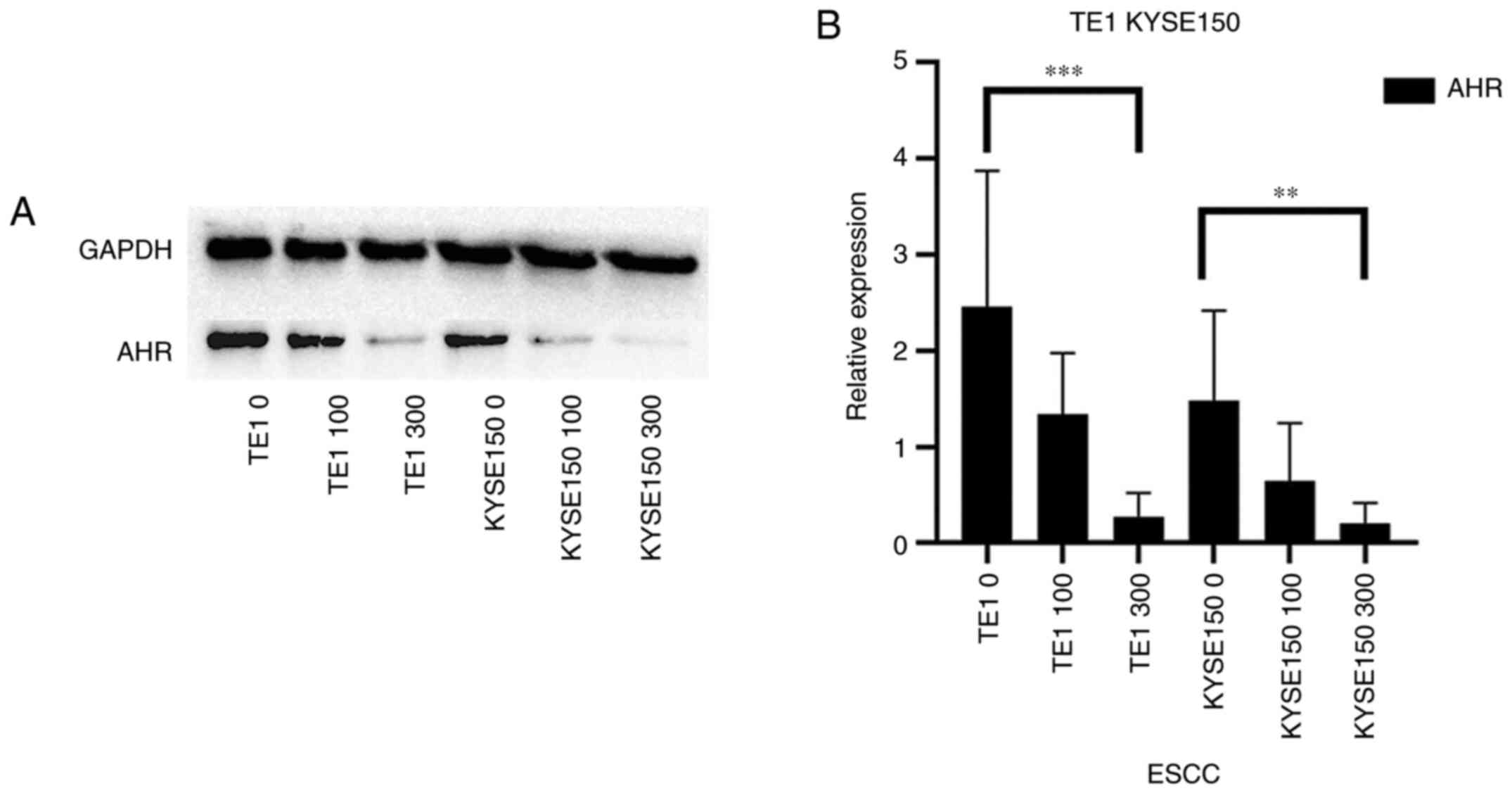
(17). In the present study, the
inhibitory effect of OME on the protein expression level of AHR was
determined using western blot analysis (Fig. 2A), which was also indicated to be
significantly decreased with increasing doses of OME (Fig. 2B).
OME inhibits the proliferation of TE1
and KYSE150 cells
Numerous studies suggested that the ligands of AHR
regulate the expression levels of AHR and thus the development of
tumors (18,19). In addition, OME was reported to be a
selective regulator of AHR and to lower the expression level of AHR
in previous research (16).
Therefore, western blot analysis and cell proliferation assays were
used in the present study to verify whether OME is able to reduce
the proliferation of the ESCCs via AHR.
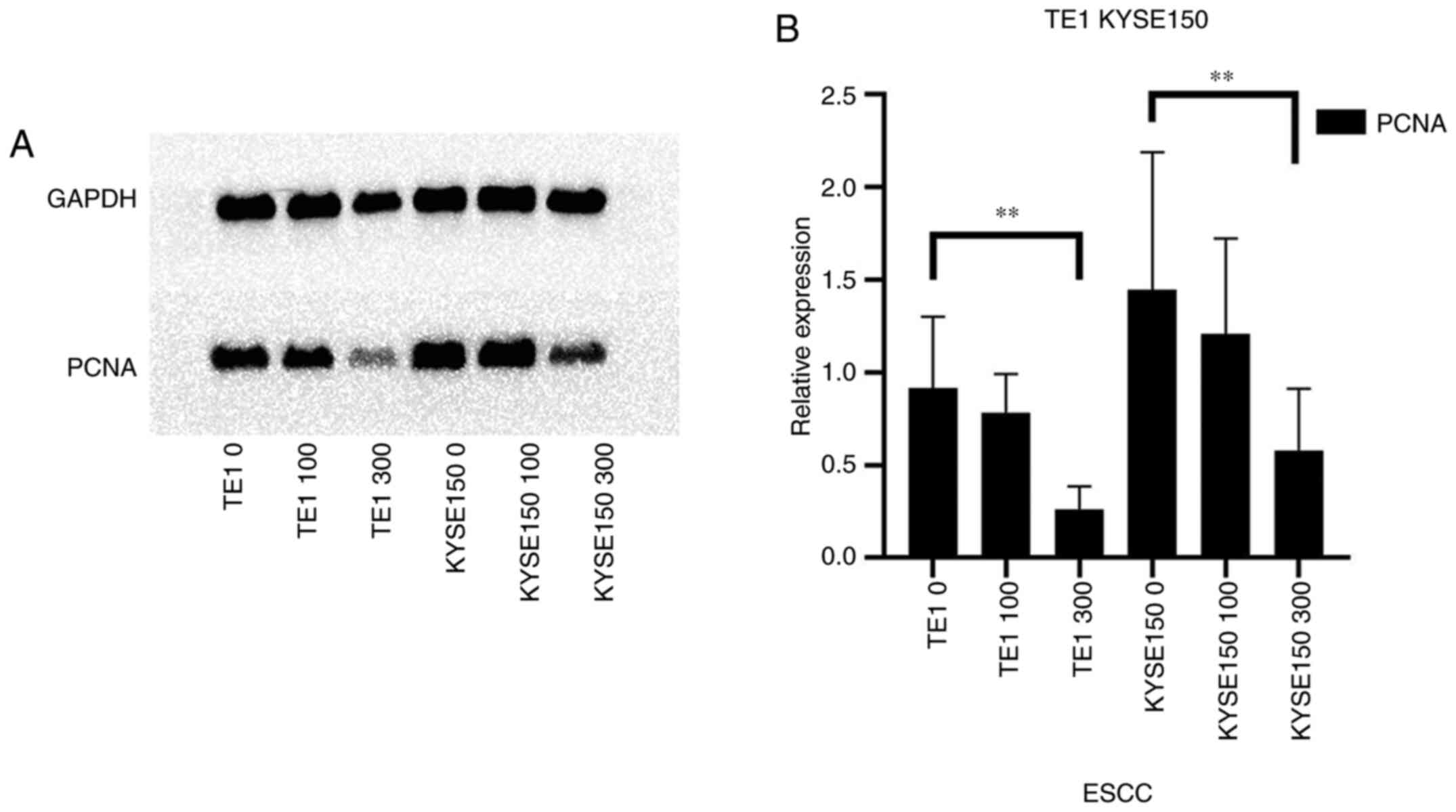
PCNA occurs in normal and tumor cells. The protein
expression level of PCNA was decreased in a dose-dependent manner
in the TE1 and KYSE150 cell lines treated with 0, 100 and 300 µM
OME for 48 h (Fig. 3A). As
presented in Fig. 3B, the protein
expression level of PCNA decreased with the increase in the OME
concentration; however, the effect was small when the cells were
treated with 100 µM OME, which may be associated with the
sensitivity of the different cell lines to the drug. The decrease
in PCNA protein expression levels was significant when the cells
were treated with 300 µM OME.
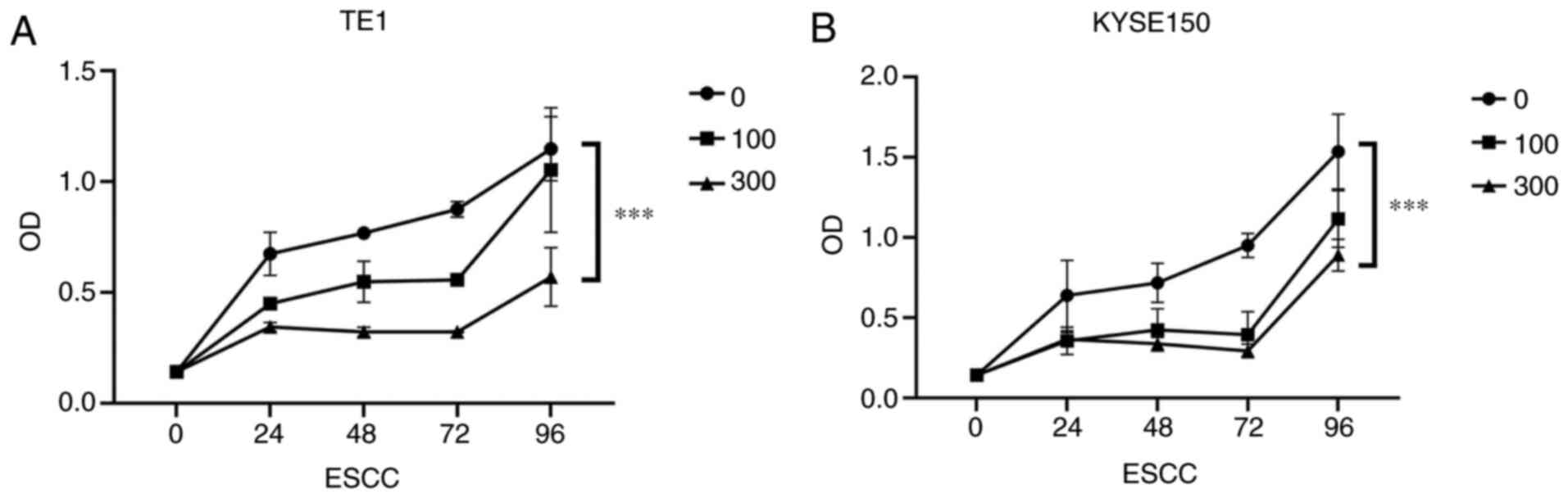
In addition, to further confirm the effect of OME on
the proliferation of the ESCCs, a CCK-8 assay was performed. It was
revealed that the proliferation of the ESCCs was inhibited
following OME treatment and the effect was significant and
dose-dependent (Fig. 4A and
B). Photomicrographs of the ESCCs
in the different groups are presented in Fig. 5.
OME induces cell cycle arrest in
G1 phase
To identify how OME affects the cell cycle in the
ESCCs, flow cytometry was performed and the number of cells in
G1/G0, S and G2 phase was
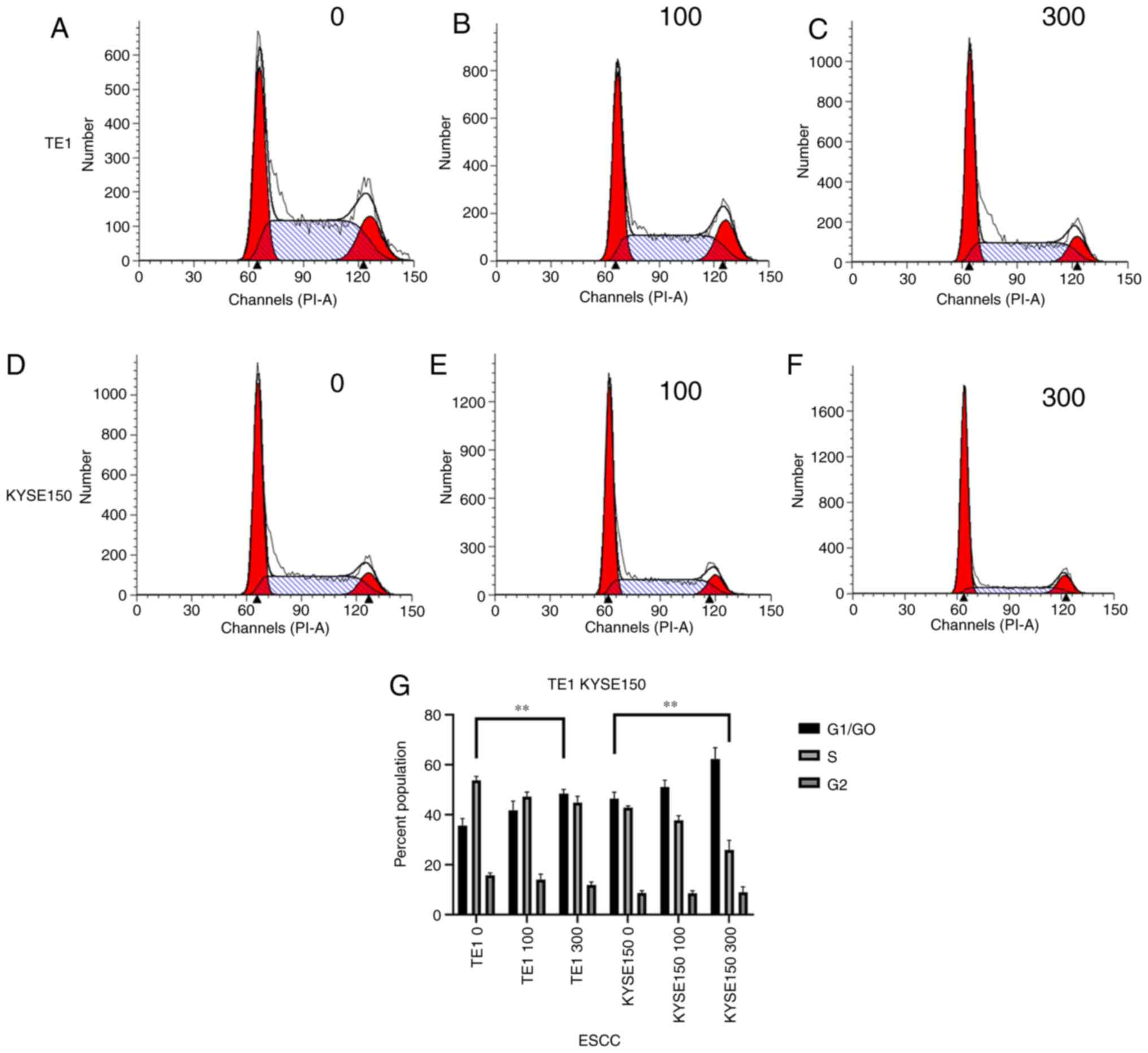
measured. As presented in Fig. 6,
after the TE1 cell line was treated with 0, 100 and 300 µM OME, the
percentages of cells in G1/G0 phase were
32.74, 38.29 and 46.80%, those in S phase were 52.34, 45.29 and
42.10% and those in G2/M phase were 14.92, 16.42 and
11.10%, respectively. Following treatment of the KYSE150 cell line
with 0, 100 and 300 µM OME, the percentages of cells in
G1/G0 phase were 48.23, 51.47 and 66.96%,
those in S phase were 42.14, 39.15 and 21.75%, and those in
G2/M phase were 9.63, 9.38 and 11.29%, respectively.
These data indicated that OME induced G1-phase arrest in
the ESCCs in a dose-dependent manner, as increasing concentrations
of omeprazole enhanced the proportion of cells that were blocked in
G1/G0 phase.
OME reduces cell migration and
invasion
Western blot analysis, wound healing and Transwell
assays were used to confirm whether OME is able to affect cell
migration and invasion.
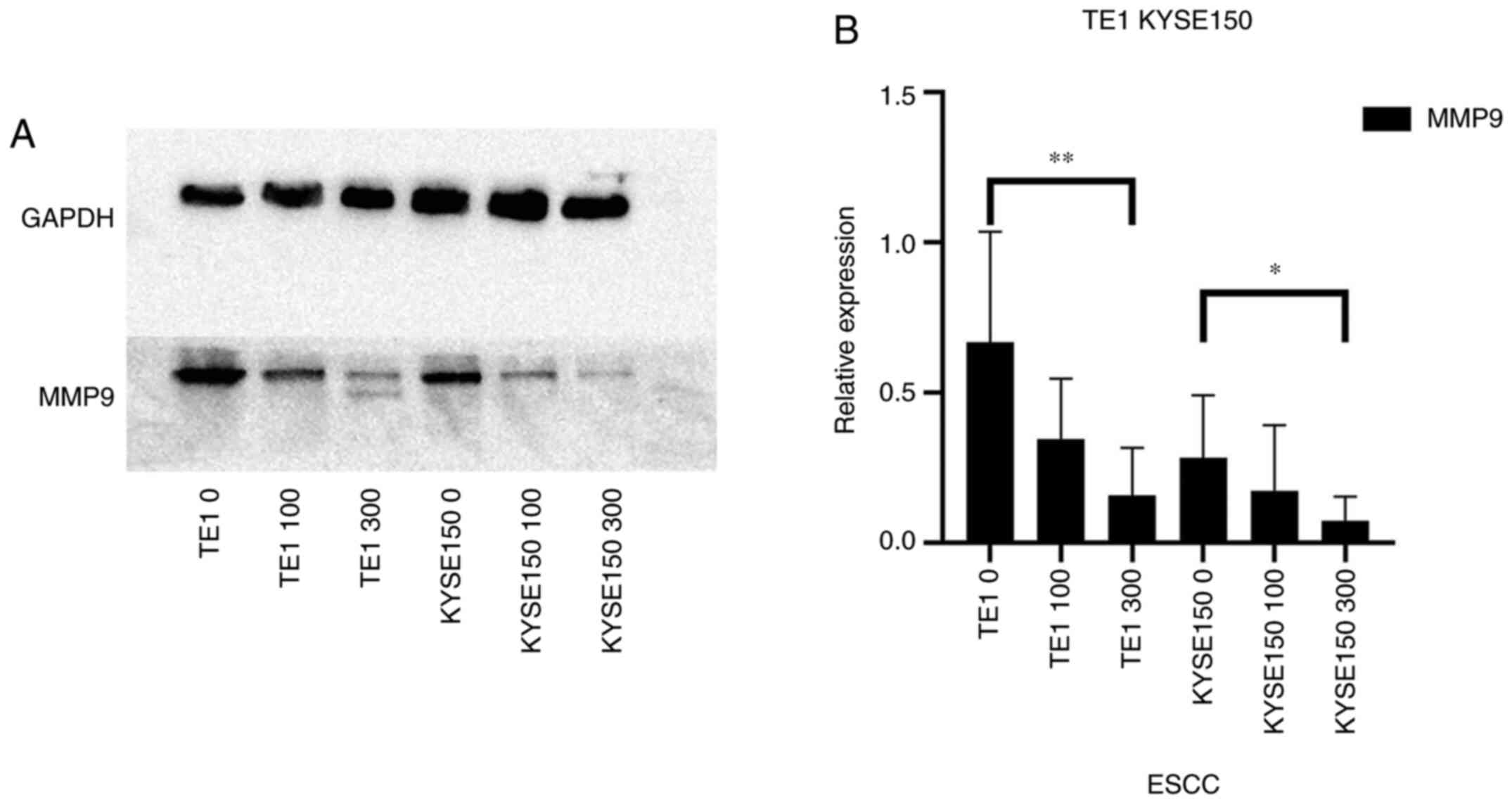
Proteins of the MMP family are involved in
extracellular matrix breakdown in normal physiological processes
(such as embryonic development, reproduction and tissue remodeling)
and in disease processes (such as arthritis and metastasis)
(20). Thus, analyzing the protein
expression levels of the MMP family may be used to investigate the
invasive capacity of cells. The ESCCs were treated with different
concentrations of OME and the protein expression levels of MMP9
were then analyzed (Fig. 7). Of
note, OME reduced the protein expression level of MMP9, suggesting
that OME may inhibit cell invasion.
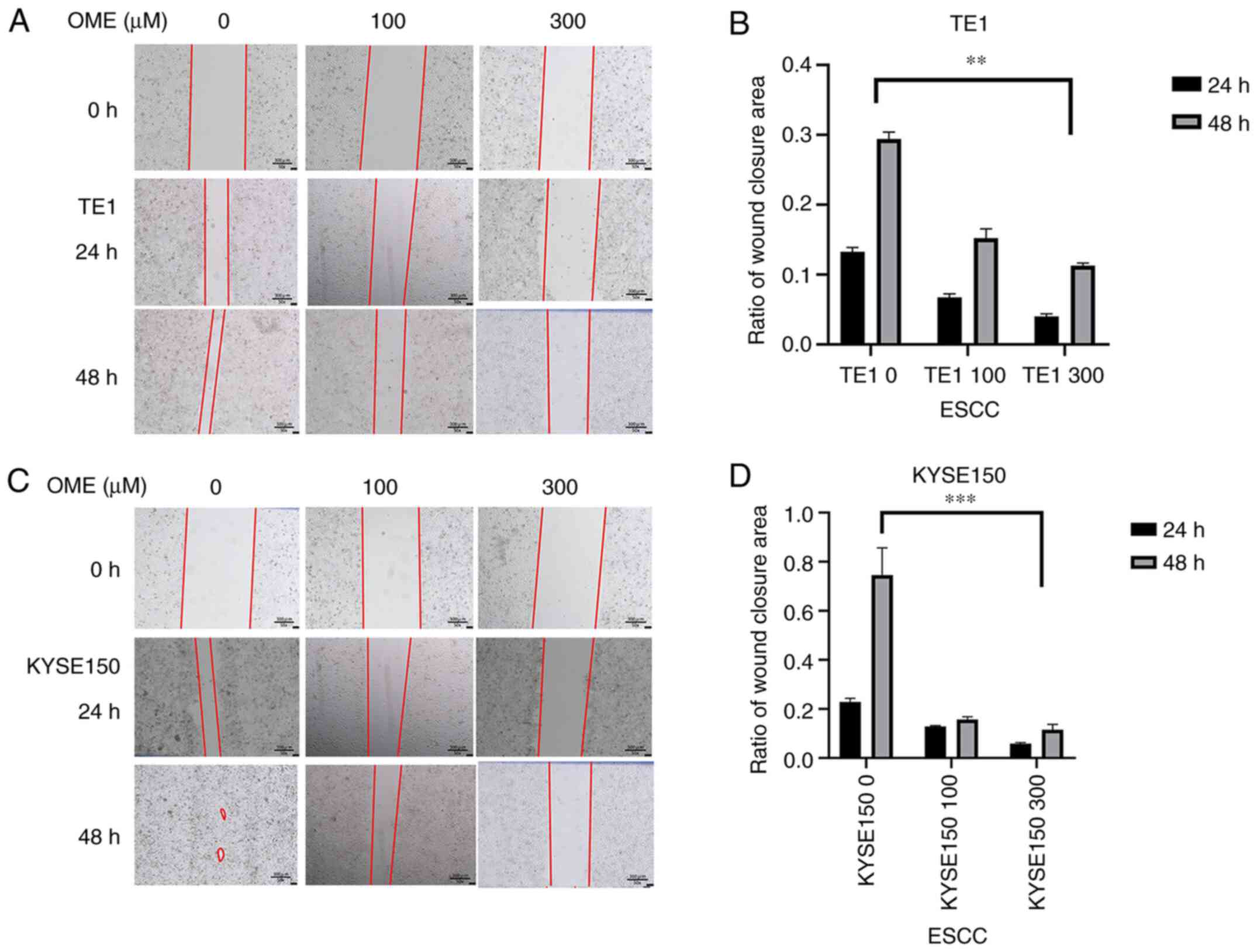
The wound-healing assay indicated that the migratory
ability of the TE1 and KYSE150 cell lines treated with OME was
reduced and the high concentration of OME inhibited migration to
the greatest extent (Fig. 8). In
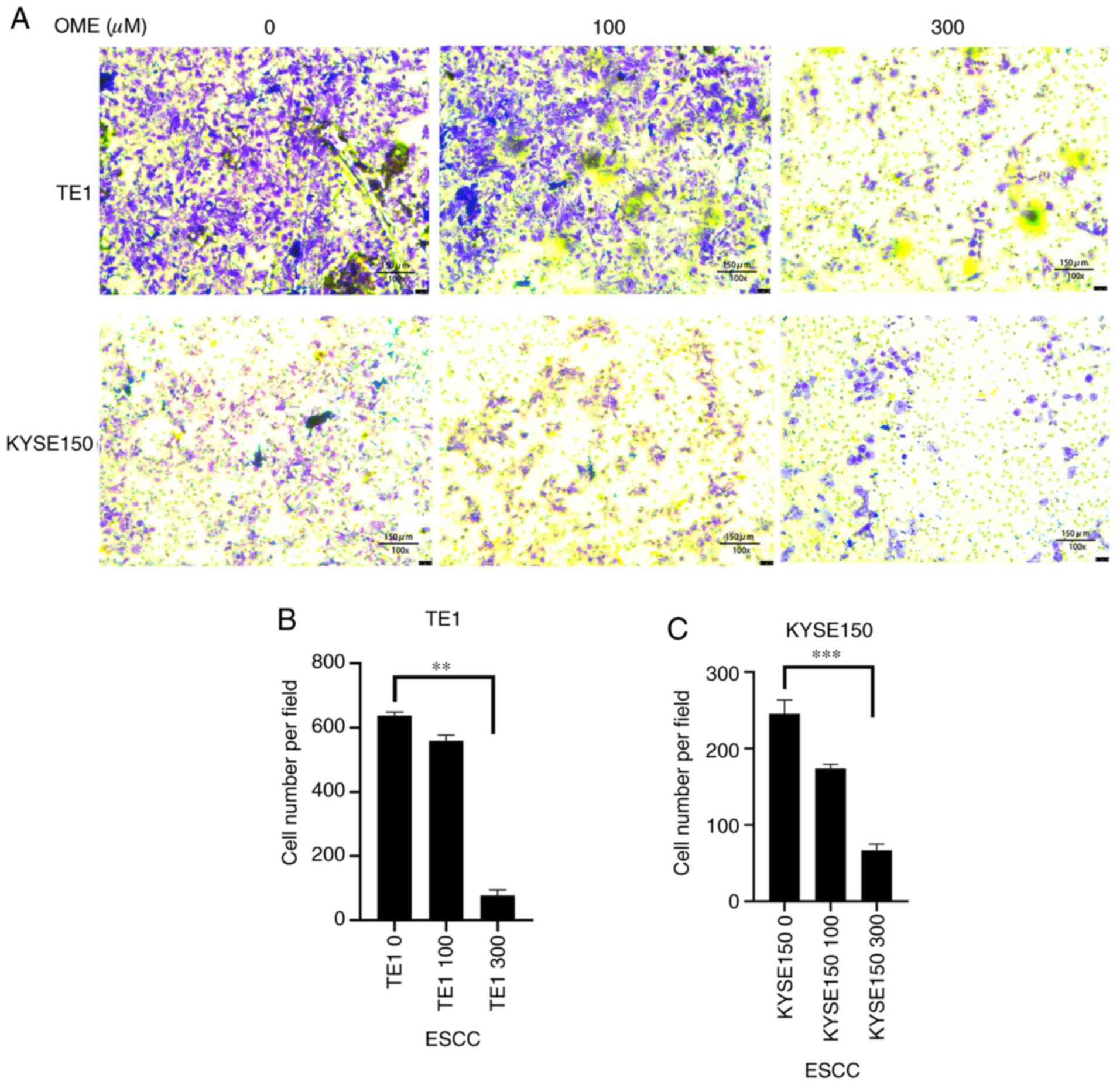
addition, as expected, the results of the Transwell assay revealed
that the invasive ability of the TE1 and KYSE150 cells treated with
OME was also reduced and this effect was observed to be
dose-dependent (Fig. 9).
Discussion
Esophageal cancer is one of the most common types of
cancer in the Western world, with high aggressiveness and a low
5-year survival rate. Despite advances in diagnosis and treatment,
the overall 5-year survival rate for patients with esophageal
cancer is only 15-20% in the US (21). There are two major subtypes of
esophageal cancer: Squamous cell carcinoma and adenocarcinoma, and
both account for >95% of cases of esophageal cancer. Clinical
studies have indicated that the combination of OME and aspirin
reduced the mortality rate in patients with esophageal
adenocarcinoma (22). Therefore,
the present study focused on ESCC and OME.
AHR, as a ligand-activated transcription factor, has
been investigated in numerous types of tumor, such as colorectal
(23), breast (24,25),
lung (26-28),
stomach (29), skin (30), prostate (31) and pancreatic cancers (32,33).
High protein expression levels were confirmed in the present study
and promoted the process of tumor development. AHR has a variety of
endogenous and exogenous ligands, and in previous studies, OME, a
widely known acid inhibitor, has also been determined to be a
selective regulator of AHR. OME is a proton pump inhibitor, which
is widely used in the treatment of various diseases, including
digestive tract ulcers and reflux esophagitis. In addition, OME was
indicated to regulate the expression level of AHR in a non-ligand
manner, thereby affecting the occurrence and development of tumors
(14,16,29).
However, the underlying mechanism has remained elusive, and to the
best of our knowledge, no previous studies have investigated the
effect of the interaction between OME and AHR on tumorigenesis and
progression of ESCC.
From the results of the present study, several
conclusions may be drawn. First, western blot analysis revealed
that the TE1 and KYSE150 cell lines treated with 0, 100 and 300 µM
OME for 48 h exhibited decreased AHR protein expression levels and
this effect was dose-dependent. Combined with the results of
previous studies by our group (34,35),
it may be preliminarily suggested that OME affects the
proliferation, migration, invasion and cell cycle of ESCCs.
Furthermore, the protein expression level of PCNA was also
decreased when the TE1 and KYSE150 cell lines were treated with 0,
100 and 300 µM OME. In addition, the results of the cell
proliferation assay suggested that the proliferation of the cells
treated with OME was significantly inhibited and the inhibitory
effect was more notable when the concentration of OME was
increased. Cell cycle analysis then revealed that TE1 and KYSE150
cells treated with 0, 100 and 300 µM OME for 48 h exhibited gradual
increases in the G1/G0 phase population and
decreases in the percentages of cells in the S phases. This
suggested that OME induced G1/G0 phase arrest
in ESCCs. Furthermore, from the wound-healing and Transwell assays,
it was concluded that OME inhibited the migration and invasion of
the ESCCs, which was also associated with the concentration of OME.
As a limitation, the present study did not determine the underlying
mechanism by which OME affected the expression level of AHR.
However, an apoptosis experiment was performed, which had an
invalid result. Furthermore, there are numerous agonists and
inhibitors of AHR, but no further research was performed as they
lie beyond the scope of the present study. However, the agonists
and inhibitors of AHR may act as the potential focus of future
investigations. Although the effect of OME on the growth of tumors
has not been investigated in vivo, OME, via the AHR pathway,
was demonstrated to be able to inhibit the proliferation, migration
and invasion of ESCC cells and cause cell cycle arrest at the
G1/G0 phase, thus exerting inhibitory effects
on ESCC.
In summary, OME selectively regulated and inhibited
AHR expression, thereby exerting inhibitory effects on tumor cell
proliferation, migration and invasion, along with the induction of
G1/G0-phase arrest. The mechanism by which
OME regulated AHR was not determined in the present study; however,
the results proved the effect of OME on ESCC via the AHR pathway,
which may be a potential treatment for ESCC in the future.
Acknowledgements
The authors gratefully acknowledge the contribution
of Dr Rui-Qun Qi (Department of Dermatology, The First Hospital of
China Medical University, Shenyang, China) for providing all
aforementioned instruments.
Funding
Funding: The present research was supported by grants from the
Natural Science Foundation of China (grant no. 81201890) and the
Research Foundation of the Education Bureau of Liaoning Province,
China (grant no. LK201614).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SGZ and YB designed the study. YB, PZ and KZ
performed the research and recorded and analyzed the results. SGZ
and YB wrote the manuscript. PZ and KZ carefully edited the
manuscript and provided guidance and advice. BY and SGZ confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ilic M, Kocic S, Radovanovic D, Macuzic IZ
and Ilic I: Trend in esophageal cancer mortality in Serbia,
1991-2015 (a population-based study): An age-period-cohort analysis
and a joinpoint regression analysis. J BUON. 24:1233–1239.
2019.PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Malhotra G, Yanala U, Ravipati A, Follet
M, Vijayakumar M and Are C: Global trends in esophageal cancer. J
Surg Oncol. 115:564–579. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lin Y, Totsuka Y, He Y, Kikuchi S, Qiao Y,
Ueda J, Wei W, Inoue M and Tanaka H: Epidemiology of esophageal
cancer in Japan and China. J Epidemiol. 23:233–242. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Smyth E, Lagergren J, Fitzgerald R,
Lordick F, Shah M, Lagergren P and Cunningham D: Oesophageal
cancer. Nat Rev Dis primers. 3(17048)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Arnal MJ, Arenas ÁA and Arbeloa ÁL:
Esophageal cancer: Risk factors, screening and endoscopic treatment
in Western and Eastern countries. World J Gastroenterol.
21:7933–7943. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schulte K, Green E, Wilz A, Platten M and
Daumke O: Structural basis for aryl hydrocarbon receptor-mediated
gene activation. Structure. 25:1025–1033. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang F, Gao J, Mimura J, Kobayashi A,
Sogawa K and Fujii-Kuriyama Y: Structure and expression of the
mouse AhR nuclear translocator (mArnt) gene. J Biol Chem.
273:24867–24873. 1998.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nebert D: Aryl hydrocarbon receptor (AHR):
‘Pioneer member’ of the basic-helix/loop/helix per-Arnt-sim
(bHLH/PAS) family of ‘sensors’ of foreign and endogenous signals.
Prog Lipid Res. 67:38–57. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shiizaki K, Ohsako S, Kawanishi M and Yagi
T: Omeprazole alleviates benzo[a]pyrene cytotoxicity by inhibition
of CYP1A1 activity in human and mouse hepatoma cells. Basic Clin
Pharmacol Toxicol. 103:468–475. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gao W, Li HY, Wang LX, Hao LJ, Gao JL,
Zheng RJ, Cai CJ and Si YL: Protective effect of omeprazole on
gastric mucosal of cirrhotic portal hypertension rats. Asian Pac J
Trop Med. 7:402–406. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Weil J, Bell GD, Powell K, Morden A,
Harrison G, Gant PW, Jones PH and Trowell JE: Omeprazole and
helicobacter pylori: Temporary suppression rather than true
eradication. Aliment Pharmacol Ther. 5:309–313. 1991.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Blum R: Lansoprazole and omeprazole in the
treatment of acid peptic disorders. Am J Health Syst Pharm.
53:1401–1415. 1996.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jin UH, Lee SO and Safe S: Aryl
hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR
modulators in MDA-MB-468 and BT474 breast cancer cells. J Pharmacol
Exp Ther. 343:333–341. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nguyen LP and Bradfield CA: The search for
endogenous activators of the aryl hydrocarbon receptor. Chem Res
Toxicol. 21:102–116. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jin UH, Kim SB and Safe S: Omeprazole
inhibits pancreatic cancer cell invasion through a nongenomic aryl
hydrocarbon receptor pathway. Chem Res Toxicol. 28:907–918.
2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Murray IA, Patterson AD and Perdew GH:
Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat
Rev Cancer. 14:801–814. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Patrizi B and Siciliani de Cumis M: TCDD
toxicity mediated by epigenetic mechanisms. Int J Mol Sci.
19(4101)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Opitz C, Litzenburger U, Sahm F, Ott M,
Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller
M, et al: An endogenous tumour-promoting ligand of the human aryl
hydrocarbon receptor. Nature. 478:197–203. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Huang H: Matrix metalloproteinase-9
(MMP-9) as a cancer biomarker and MMP-9 biosensors: Recent
advances. Sensors (Basel). 18(3249)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jankowski JAZ, de Caestecker J, Love SB,
Reilly G, Watson P, Sanders S, Ang Y, Morris D, Bhandari P, Brooks
C, et al: Esomeprazole and aspirin in Barrett's oesophagus
(AspECT): A randomised factorial trial. Lancet. 392:400–408.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shiizaki K, Kido K and Mizuta Y: Insight
into the relationship between aryl-hydrocarbon receptor and
β-catenin in human colon cancer cells. PLoS One.
14(e0224613)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tomblin JK, Arthur S, Primerano DA,
Chaudhry AR, Fan J, Denvir J and Salisbury TB: Aryl hydrocarbon
receptor (AHR) regulation of L-type amino acid transporter 1
(LAT-1) expression in MCF-7 and MDA-MB-231 breast cancer cells.
Biochem Pharmacol. 106:94–103. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Donovan M, Selmin O and Romagnolo D: Aryl
hydrocarbon receptor diet and breast cancer risk. Yale J Biol Med.
91:105–127. 2018.PubMed/NCBI
|
|
26
|
Terashima J, Jimma Y, Jimma K, Hakata S,
Yachi M, Habano W and Ozawa S: The regulation mechanism of AhR
activated by benzo[a]pyrene for CYP expression are different
between 2D and 3D culture of human lung cancer cells. Drug Metab
Pharmacokinet. 33:211–214. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gao H, Ye G, Lin Y, Chi Y and Dong S:
Benzo[a]pyrene at human blood equivalent level induces human lung
epithelial cell invasion and migration via aryl hydrocarbon
receptor signaling. J Appl Toxicol. 40:1087–1098. 2020.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Dong S, Zhu P and Zhang S: Expression of
collagen type 1 alpha 1 indicates lymph node metastasis and poor
outcomes in squamous cell carcinomas of the lung. PeerJ.
8(e10089)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yoshinari K, Ueda R, Kusano K, Yoshimura
T, Nagata K and Yamazoe Y: Omeprazole transactivates human CYP1A1
and CYP1A2 expression through the common regulatory region
containing multiple xenobiotic-responsive elements. Biochem
Pharmacol. 76:139–145. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Vogeley C, Esser C, Tüting T, Krutmann J
and Haarmann-Stemmann T: Role of the aryl hydrocarbon receptor in
environmentally induced skin aging and skin carcinogenesis. Int J
Mol Sci. 20(6005)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chen Z, Cai A, Zheng H, Huang H, Sun R,
Cui X, Ye W, Yao Q, Chen R and Kou L: Carbidopa suppresses prostate
cancer via aryl hydrocarbon receptor-mediated ubiquitination and
degradation of androgen receptor. Oncogenesis. 9(49)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang L, Tang W, Yang S, He P, Wang J,
Gaedcke J, Ströbel P, Azizian A, Ried T, Gaida MM, et al: NO
/RUNX3/kynurenine metabolic signaling enhances disease
aggressiveness in pancreatic cancer. Int J Cancer. 146:3160–3169.
2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Masoudi S, Nemati AH, Fazli HR, Beygi S,
Moradzadeh M, Pourshams A and Mohamadkhani A: An increased level of
aryl hydrocarbon receptor in patients with pancreatic cancer.
Middle East J Dig Dis. 11:38–44. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhu P, Yu H, Zhou K, Bai Y, Qi R and Zhang
S: 3,3'-Diindolylmethane modulates aryl hydrocarbon receptor of
esophageal squamous cell carcinoma to reverse
epithelial-mesenchymal transition through repressing
RhoA/ROCK1-mediated COX2/PGE2 pathway. J Exp Clin Cancer Res.
39(113)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhu P, Zhou K, Lu S, Bai Y, Qi R and Zhang
S: Modulation of aryl hydrocarbon receptor inhibits esophageal
squamous cell carcinoma progression by repressing COX2/PGE2/STAT3
axis. J Cell Commun Signal. 14:175–192. 2020.PubMed/NCBI View Article : Google Scholar
|