Introduction
Cardiovascular disease (CVD) represents the most
common cause of death worldwide (1). Endothelial dysfunction is an initial
and aggravating factor of CVD; thus, an endothelial protection
strategy could prevent and decrease the occurrence of CVD (2). Hypoxia is a common pathological
condition that leads to vascular endothelial injury associated with
a variety of CVDs, due to its ability to induce endothelial injury
and dysfunction (3). Therefore,
attenuation of hypoxia-induced vascular endothelial dysfunction is
a promising strategy for the prevention and treatment of CVDs.
Endothelial progenitor cells (EPCs) are a group of
stem cells that can specifically home to areas of angiogenic
organization and differentiate into endothelial cells (4). EPCs as the central type of cell
involved in the physiological and pathological processes of
angiogenesis after birth (5). As
precursors to vascular endothelial cells, EPCs are strongly
implicated in influencing the stromal cells in the bone marrow (BM)
(6) and in the occurrence of
cardiovascular disease, cerebrovascular disease and tumor
angiogenesis.
Chemokine receptor-4 (CXCR4) belongs to the
G-protein coupled receptor (GPCR) family (7). The interactions between chemokines and
chemokine receptors can induce the chemotactic migration of target
cells, rearrangement of cell cytoskeleton, and intensive adhesive
capacity between target cells and endothelial cells, which are
extensively involved in biological functions such as cell growth,
differentiation and apoptosis (8).
Presently, it is known that one of the most important natural
ligands of CXCR4 is stromal cell-derived factor-1 (SDF-1/CXCL12)
(7). The combination of CXCR4 and
SDF-1 can activate downstream signaling pathways including
Ca2+ influx, phosphatidylinositol 3-kinase (PI3K), Akt,
mitogen-activated protein (MAP) kinase, nuclear factor-κB (NF-κB),
proline-rich tyrosine kinase 2 and the CT10 regulator of kinase
(Crk) pathway (9). The significant
effects of SDF-1/CXCR4 interaction have been highlighted in the
regulation of cellular functions including cell adhesion, cell
proliferation, cell migration, cell survival and angiogenesis
(10,11). However, the functions and mechanisms
of CXCR4 in BM-derived EPCs (BM-EPCs) have not previously been
investigated.
The aim of the present study was to investigate the
effects of CXCR4 overexpression on the proliferation, apoptosis and
migration of hypoxia-treated BM-EPCs, and to explore the
involvement of the downstream PI3K signaling pathway in these
behaviors. BM-EPCs were pre-treated with hypoxia, in order to
identify the regulatory mechanism of CXCR4 overexpression in EPCs
under hypoxic conditions.
Materials and methods
Experimental animals
A total of 20 female Sprague Dawley (SD) rats (aged
4-6 weeks, weighing 200-240 g) were provided by Shrek (Shanghai,
China). Animals were allowed free access to food and water,
maintained in a 12-h light/dark cycle at room temperature (21-25˚C)
in 50-60% relative humidity. Routine cleaning and disinfection were
performed daily. The duration of the experiment was 10 months.
Animal health was monitored every 3 months. The animals used in the
present study were cared for in accordance with the Guide for the
Care and Use of Laboratory Animals published by the National
Institutes of Health, and methods were approved by the Animal
Research Committee of Hunan Provincial People's Hospital [Changsha,
China; approval no. (2019)-041].
Isolation and culture of EPCs
SD rats were anesthetized by intraperitoneal
injection of 3% pentobarbital sodium (30 mg/kg body weight) and
then euthanized by means of cervical dislocation. Respiration and
heartbeat were stopped to confirm the death of the animal, and the
bodies were subsequently soaked in 75% ethanol solution for 15 min.
The femur and tibia were then separated under aseptic conditions,
with the bone surface of the femur and tibia carefully removed and
sheared accordingly. The BM cavity was repeatedly washed with
low-glucose Dulbecco's modified Eagle's medium (L-DMEM; Beyotime
Institute of Biotechnology), after which the flushing fluid was
collected. The collected flushing fluid was incubated with the
equivalent volume of lymphocyte separation medium (Wisent, Inc.).
The solution was centrifuged at 2,000 x g for 15 min at room
temperature to collect the cells, which were seeded into a culture
dish with 200 ng/ml fibrinolysin at a concentration of
1x106/cm2. The cells were then incubated in
L-DMEM with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C with 5% CO2.
Phenotype identification of
BM-EPCs
The BM-EPCs were characterized by immunofluorescent
staining and flow cytometry analysis. For immunofluorescent
staining, the BM-EPCs were harvested and washed with phosphate
buffered saline (PBS) and incubated with 200 µl Acetylated DiI
lipoprotein from human plasma (Dil-Ac-LDL; 10 µg/ml; Molecular
Probes; Thermo Fisher Scientific, Inc.) for 4 h at 37˚C with 5%
CO2. Samples were then fixed with 4% paraformaldehyde
for 30 min at 37˚C. Cells were stained with 200 µl fluorescein
isothiocyanate labeled ulex europaeus lectin-1 (FITC-UEA-1; 10
µg/ml; Sigma Aldrich; Merck KGaA) for 1 h at 37˚C with 5%
CO2, followed by observation under a fluorescence
microscope (magnification, x120; Olympus Corporation).
For flow cytometry analysis, the BM-EPCs were lysed
in PBS (Invitrogen; Thermo Fisher Scientific, Inc.) with 0.5%
paraformaldehyde (Sigma Aldrich; Merck KGaA) and 0.5% saponin
(Sigma Aldrich; Merck KGaA) for 5 min at 4˚C. The cells were
labeled with rabbit anti-mouse FITC-CD14 (BD Biosciences), rabbit
anti-mouse PE-CD34 (Bio Legend, Inc.), and rabbit anti-mouse
PerCP/Cy5.5-KDR (BioLegend, Inc.) for 1 h at room temperature. The
same fluorescein-labeled isotype IgG was used as a control to
define the negative population for each stain. After washing, cells
were analyzed with the FACS™ Canto II flow cytometer.
CD14, CD34 and KDR were examined using the FACSDiva software (v.6;
BD Biosciences) for fluorescence-activated cell sorting, based on
the manufacturer's instructions.
Adenovirus transfection
The BM-EPCs at passage number 2 were used for
transduction. Viral vector, packaging plasmid, adenovirus related
vectors (rAAV6 vector and pHelper1.0 vector) and 293T cells were
purchased from Cyagen Biosciences Inc. 293T cells were transfected
by the calcium phosphate method. The recombinant plasmids
pAAV2.1-CMV-GFP (0.5 µg/ml) or pAAV2.1-CMV-CXCR4 (5 µg/ml) and the
packaging plasmid pRC6 (2 µg/ml) and pHelper (10 µg/ml) were added
to CaCl2, then mixed with 2X HBS to make Calcium-DNA.
293T cells were then co-transfected with Calcium-DNA at 37˚C with
5% CO2. After 48 h, the viral supernatant was harvested
with centrifuged at 2,000 x g for 20 min at 4˚C. The concentration
of recombinant adeno-associated virus was determined by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
BM-EPCs (1x105/well) were seeded in 24-well cell culture
plates and cultured overnight. Then, BM-EPCs were subsequently
transfected with rAAV6-GFP and rAAV6-CXCR4 at the indicated
multiplicity of infection (moi). After incubation at 37˚C for 48 h,
the transfection efficiency of the lentivirus vectors in
transduced-EPCs was identified using fluorescence microscope. The
level of CXCR4 expression was detected by western blotting, RT-qPCR
and flow cytometry.
Cell treatment
After the BM-EPCs were transfected with
rAAV6-GFP and rAAV6-CXCR4, they were cultured
at 37˚C with 5% CO2 in a humidified atmosphere with
Endothelial Cell Growth Medium-2 (Lonza Group, Inc.), supplemented
with 10% FBS, 100 U/ml penicillin and streptomycin sulfate (Thermo
Fisher Scientific, Inc.), and SDF-1 at 20 µg/l for conventional
culture. Initially, the BM-EPCs in the normoxia group were cultured
under normoxic conditions (5% CO2, 95% air atmosphere).
BM-EPCs in the hypoxia group were cultured under anaerobic
conditions (5% CO2, 95% N2) free of FBS for 4
h, and then cultured under normoxic conditions with PBS for 6 h.
The BM-EPCs were subsequently divided into four groups: Normoxia +
Sham (Empty vector) group, Normoxia + CXCR4 group, Hypoxia + Sham
group, and Hypoxia + CXCR4 group. In additional experiments, the
BM-EPCs were divided into these four groups: Hypoxia + Sham group,
Hypoxia + CXCR4 group, Hypoxia + CXCR4 + PI3K inhibitor (LY294002,
20 µmol/l for 1 h) group, and Hypoxia + CXCR4 + CXCR4 inhibitor
(AMD3100) group.
Cell counting kit-8 (CCK-8) assay
The treated BM-EPCs (2x103 cells/well)
were seeded into 96-well plates at 37˚C. CCK-8 reagent (cat. no.
CK04; Dojindo Molecular Technologies, Inc.) was added at the 24,
48, 72 and 96 h time points. After an additional 3 h, the
absorbance value was determined by a microplate reader (Multiscan
MK3; Thermo Fisher Scientific, Inc.) at 450 nm. The mean optical
density (OD) of the three wells in each group was used to reflect
the percentage of cell proliferation.
Transwell assay
The treated BM-EPCs (5x104 cells/well)
were added to the top of chambers (BD Biosciences) with serum-free
medium. The complete medium was then added to the lower chambers.
After 24 h, the non-migrated cells were removed, while the migrated
cells were fixed using 4% paraformaldehyde for 20 min, and stained
with 0.1% crystal violet solution for 5-10 min at 37˚C (Sigma
Aldrich; Merck KGaA). Lastly, images were captured under a light
microscope (magnification, x200). The experiment was performed
three times.
Annexin V-FITC/PI double staining
assay
The treated BM-EPCs (1x106 cells/ml) were
stained with Annexin V-FITC/PI (Multsciences) under dark conditions
for 15 min at room temperature. Cell apoptosis was detected using a
FACS Calibur flow cytometry (BD Biosciences) with CellQuest
software (version FACS101; BD Biosciences). The experiment was
performed three times.
RT-qPCR assay
The total RNA of the treated BM-EPCs was extracted
using TRIzol reagent (cat. no. 9109; Takara Biotechnology Co.
Ltd.). Then, 3 µg RNA was reverse transcribed to cDNA using a cDNA
synthesis kit (cat. no. 6130; Takara Biotechnology Ltd.) following
the manufacturer's instructions. The expression levels of CXCR4
were detected using SYBR Green PCR Master Mix kit (Takara
Biotechnology Co. Ltd.) on an ABI-Prism 7300 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). β-actin was used as an
endogenous control. Results were calculated using the
2-ΔΔCq method (12). The
following primers were used: β-actin (product length, 150 bp)
forward, 5'-GGAGATTACTGCCCTGGCTCCTA-3'; β-actin reverse,
5'-GACTCATCGTACTCCTGCTTGCTG-3'; CXCR4 (product length 113 bp)
forward, 5'-CCATCCTCTACGCCTTCC-3'; CXCR4 reverse,
5'-TCCACCCCGTTTCCCTT-3'. The qPCR reaction mixture consisted of the
following: 1 µl cDNA, 0.5 µl forward primers and 0.5 µl reverse
primers, 10 µl SYBR Green PCR Master mix and 8 µl ddH2O.
The PCR reaction was as follow: (94˚C, 2 min; 94˚C, 20 sec; 58˚C,
20 sec; 72˚C, 20 sec; 40 cycles in total) followed by a dissolution
curve.
Western blotting
Total protein from the treated BM-EPCs was extracted
using a cell lysis buffer for Western and IP with proteinase
inhibitors (Beyotime Institute of Biotechnology, Inc.). The
concentration of total protein was determined using a BCA kit (cat.
no. 23227; Thermo Fisher Scientific, Inc.). Then, 40 µg protein was
separated using 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
fluoride membranes (EMD Millipore). The membranes were blocked with
5% milk for 2 h at room temperature, then blotted with primary
antibodies at 4˚C overnight, followed by blotting with specific
HRP-conjugated AffiniPure goat anti-rabbit secondary antibodies
(cat. no. BA1054; Boster Biological Technology) at 37˚C for 1 h
after TBST (TBS with 0.05% Tween-20) washing. Finally, protein
bands were visualized using enhanced chemiluminescence reagents
(Pierce; Thermo Fisher Scientific, Inc.). on a Gel Imaging System
(cat. no. 1708370; Edwards Life Sciences). The primary antibodies
used were: Anti-GAPDH (1:10,000; RC-5G5, Kangcheng), anti-CXCR4
(1:1,000; cat. no. PA1237; Boster Biological Technology), anti-PI3K
(1:1,000; cat. no. A01091-1; Boster Biological Technology) and
anti-Akt (1:1,000; cat. no. A00024;Boster Biological
Technology).
Statistical analysis
All data are presented as mean ± standard deviation
(SD). Statistical analysis was performed using SPSS software
(version 13.0; SPSS, Inc.). One-way analysis of variance (ANOVA)
was used for data with n <50. The least significant difference
method was employed to compare samples when the variance was
homogeneous. The Dunnett T3 test was used to compare samples when
variance was not homogeneous. P<0.05 was considered to indicate
a statistically significant difference.
Results
Phenotype identification of
BM-EPCs
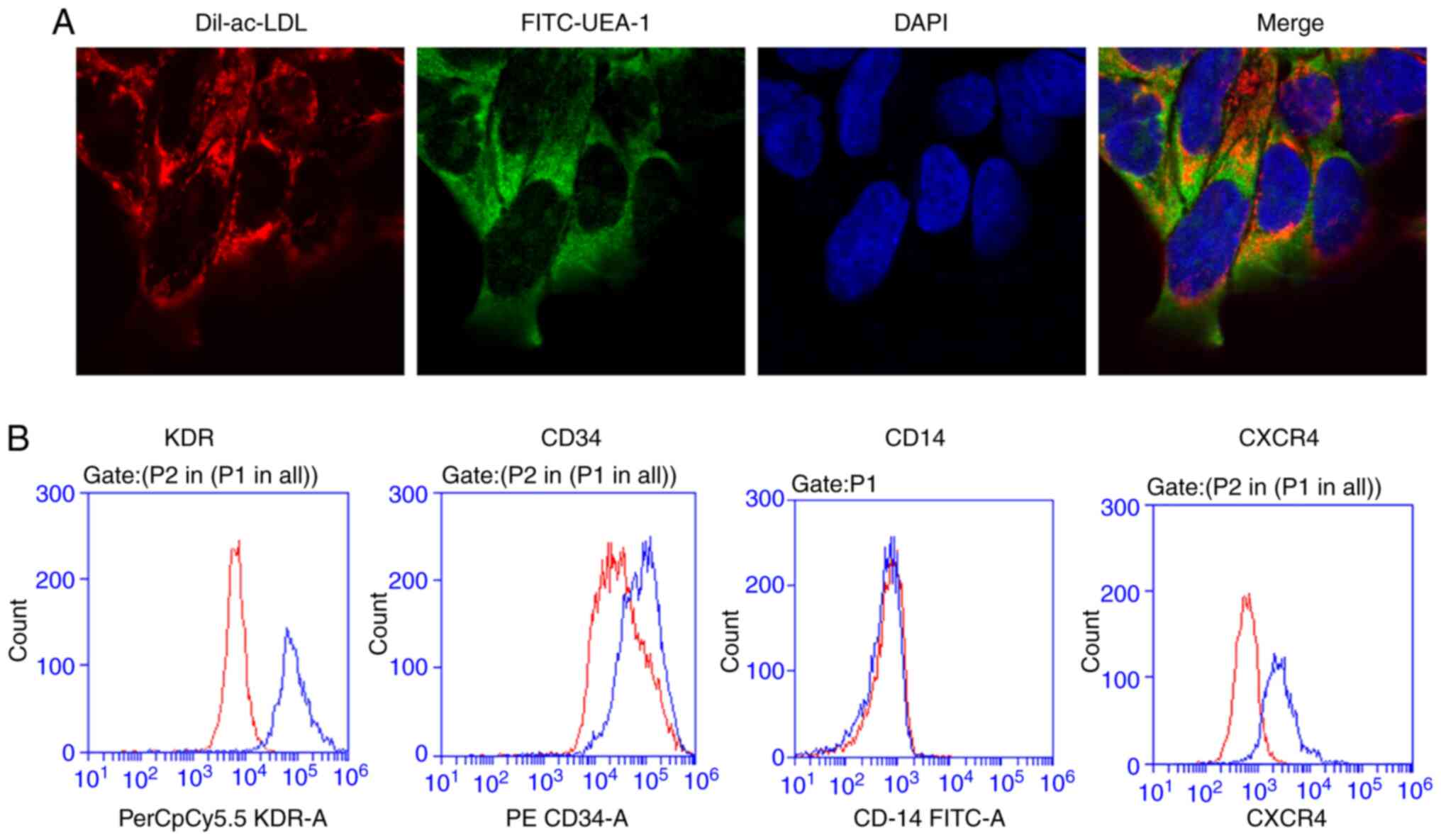
The phenotype of BM-EPCs was identified through the
application of immunofluorescent staining with Dil-ac-LDL and
FITC-UEA-1. As depicted in Fig. 1A,
the cell nuclei of BM-EPCs were blue after staining with DAPI.
Endocytic vesicles were fluorescent red around the nucleus within
the cytoplasm because of Dil-ac-LDL. The cell membrane was green
after staining with FITC-UEA-1. The aforementioned results indicate
that the expression of the EPC-specific surface marker CD14 was
negative, while that of CD34 and KDR was positive (Fig. 1B).
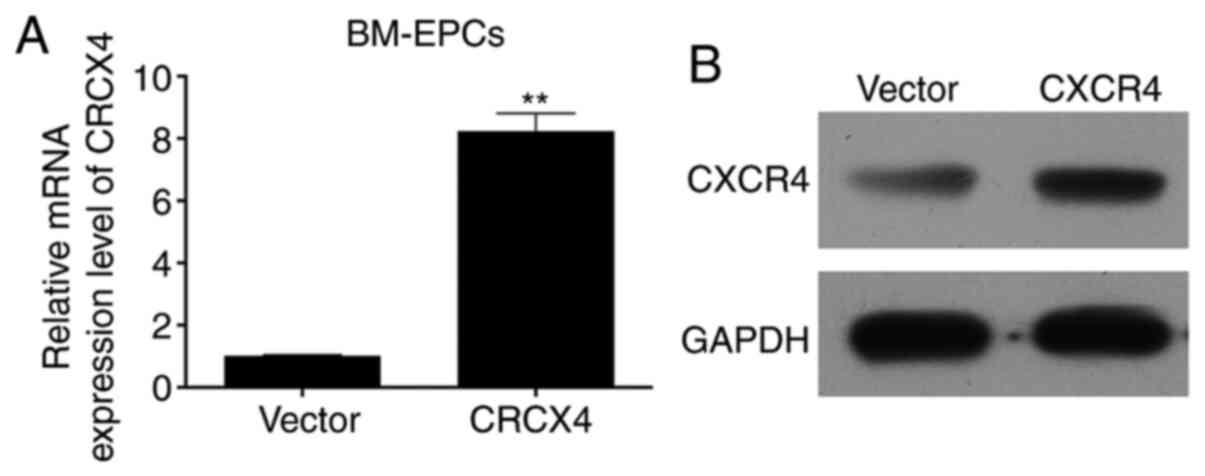
CXCR4 is overexpressed in BM-EPCs
In order to investigate the mechanism as well as the
functions associated with CXCR4 in BM-EPCs, adenoviruses were
constructed (rAAV6-GFP and rAAV6-CXCR4), and the empty vector or
adenovirus-CXCR4 were transfected into BM-EPCs. CXCR4 was stably
overexpressed in BM-EPCs, as measured through flow cytometry assays
(Fig. 1B). The mRNA and protein
expression of CXCR4 was determined by RT-qPCR and western blot
assays, respectively. The RT-qPCR results showed that CXCR4 mRNA
was highly expressed in the CXCR4 group compared with the vector
group (P<0.01; Fig. 2A). Western
blotting results revealed that CXCR4 protein was also highly
expressed in the CXCR4 group compared with the vector group
(Fig. 2B). The aforementioned
results confirmed efficient transfection, and subsequent high
expression of adenovirus-CXCR4 in BM-EPCs.
CXCR4 promotes proliferation and
migration, while inhibiting apoptosis of hypoxia-induced
BM-EPCs
In order to demonstrate the functional mechanism of
CXCR4 in BM-EPCs under hypoxic conditions, BM-EPCs were treated
with Hypoxia + CXCR4, Normoxia + CXCR4, Hypoxia + Sham (Empty
vector), or Normoxia + Sham. Initially, western blotting revealed
that the expression level of CXCR4, PI3K and Akt were all
significantly elevated in the Normoxia + CXCR4 group compared with
the Normoxia + Sham group (P<0.01). The expression level of
CXCR4, PI3K and Akt were also significantly increased in the
Hypoxia + CXCR4 group vs. the Hypoxia + Sham group (P<0.01;
Fig. 3A). Thus, it was concluded
that CXCR4 upregulated the expression level of PI3K and Akt in
BM-EPCs under hypoxic conditions. Moreover, the present results
indicated that cell proliferation ability was significantly
strengthened in the Normoxia + CXCR4 group compared with the
Normoxia + Sham group (P<0.01), while a dramatic increase was
observed in the Hypoxia + CXCR4 group compared with the Hypoxia +
Sham group (P<0.05). In addition, cell proliferation ability was
significantly inhibited in the Hypoxia + Sham group compared with
the Normoxia + Sham group, and was also markedly suppressed in the
Hypoxia + CXCR4 group compared with the Normoxia + CXCR4 group
(P<0.01; Fig. 3B).
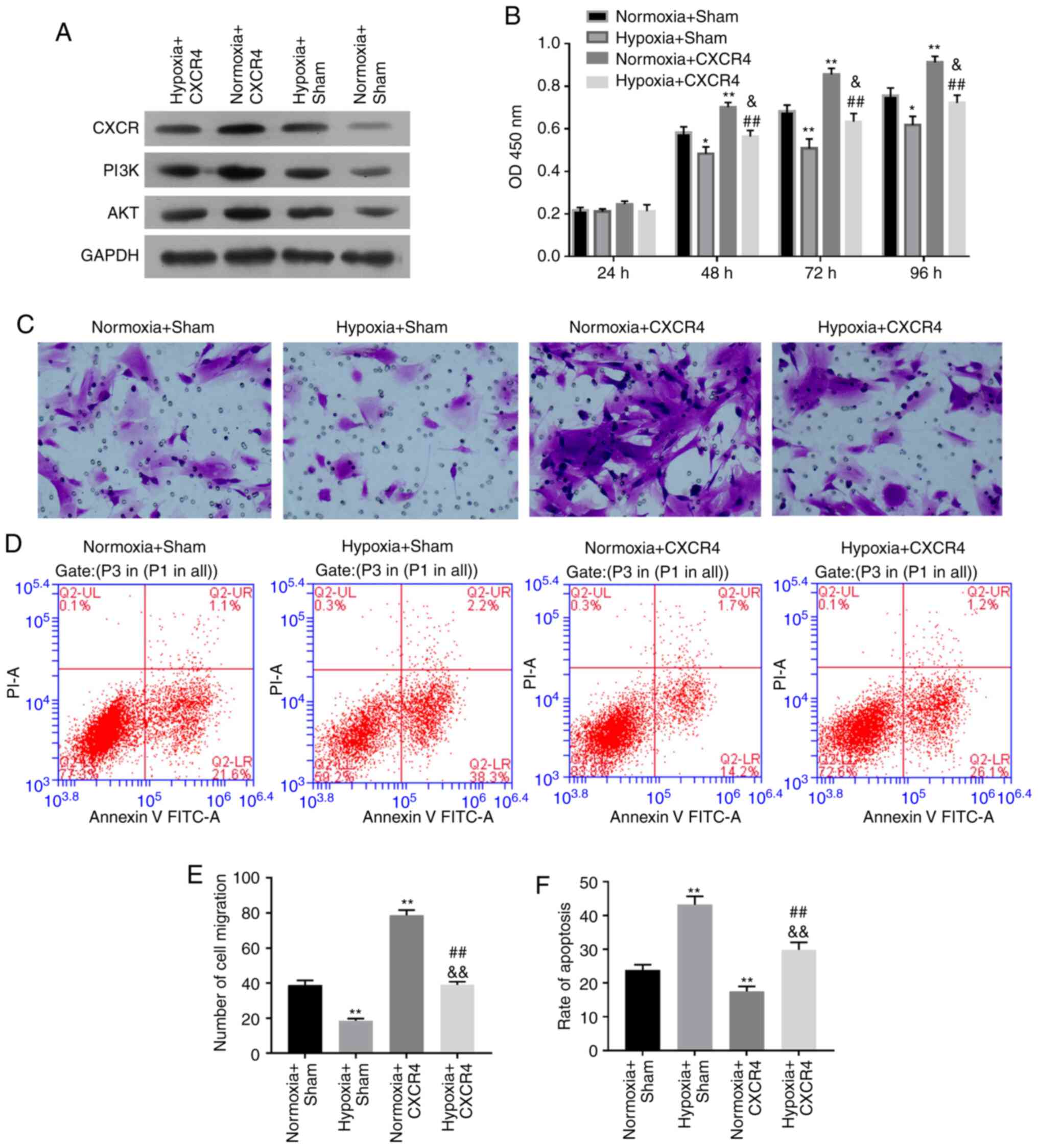 | Figure 3CXCR4 promotes proliferation and
migration, and inhibits apoptosis of hypoxia-induced BM-EPCs.
BM-EPCs were divided into four groups: Hypoxia + CXCR4, Normoxia +
CXCR4, Hypoxia + Sham, and Normoxia + Sham groups. (A) The protein
expression levels of CXCR4, PI3K and AKT were measured by western
blotting. GAPDH was regarded as the loading control. (B) Cell
proliferation ability detected by Cell Counting Kit-8 assay. (C)
Migration capacity was determined by Transwell assay
(magnification, x200). (D) Apoptosis capacity was detected by flow
cytometry. (E) Quantification of CXCR4-promoted migration of
hypoxia-induced BM-EPCs. (F) Quantification of CXCR4-inhibited
apoptosis in hypoxia-induced BM-EPCs. Experiments were repeated 3
times; data are expressed as mean ± SD. *P<0.05 and
**P<0.01 vs. Normoxia + Sham;
&P<0.05 and &&P<0.01 vs.
Hypoxia + Sham; ##P<0.01 vs. Normoxia + CXCR4.
BM-EPCs, bone marrow-derived endothelial progenitor cells; PI3K,
phosphatidylinositol 3-kinase; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase; CXCR4, chemokine receptor-4. |
The aforementioned results suggest that hypoxia
inhibits cell proliferation of BM-EPCs, while CXCR4 promotes the
proliferation ability of BM-EPCs with or without hypoxia treatment.
Transwell assays further indicated that hypoxia resulted in the
inhibition of BM-EPC cell migration, in addition to demonstrating
that CXCR4 significantly enhanced the migration ability of BM-EPCs
with or without hypoxia treatment (P<0.01; Fig. 3C and E). Furthermore, hypoxia markedly
accelerated cell apoptosis of BM-EPCs, and it was found that CXCR4
markedly suppressed the apoptosis ability of BM-EPCs with or
without hypoxia treatment (P<0.01; Fig. 3D and F).
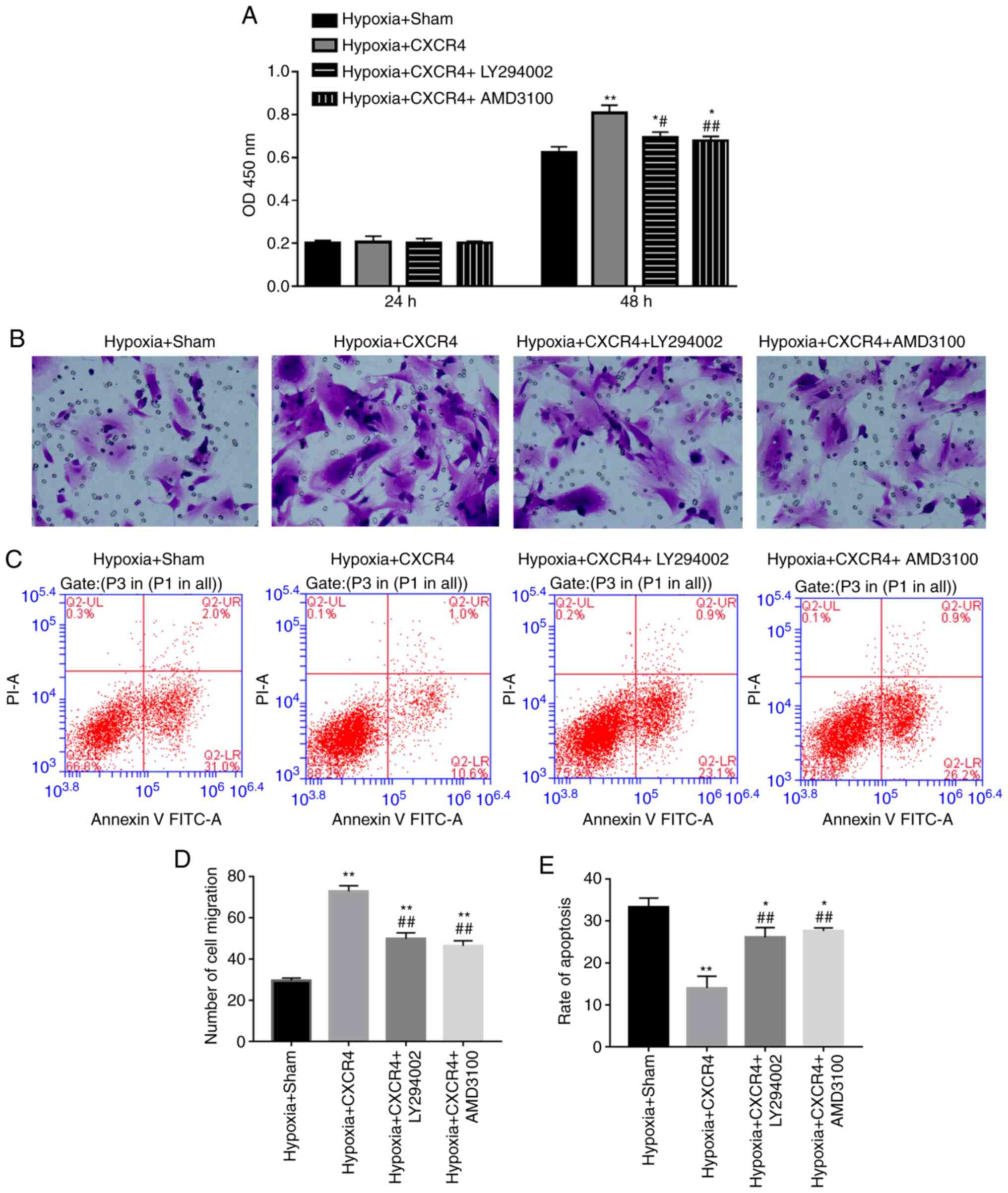
PI3K inhibitor and AMD3100 suppresses
proliferation and migration, and accelerates apoptosis of
CXCR4-mediated BM-EPCs under the hypoxic condition
Based on the observation that CXCR4 could upregulate
the expression levels of PI3K and Akt in BM-EPCs, it was
subsequently hypothesized that CXCR4 is implicated in the
biological process of BM-EPCs by regulating the PI3K/Akt signaling
pathway. In order to test the hypothesis, BM-EPCs were treated with
Hypoxia + Sham, Hypoxia + CXCR4, Hypoxia + CXCR4 + PI3K inhibitor
(LY294002), or Hypoxia + CXCR4 + CXCR4 inhibitor (AMD3100). The
CCK-8 assay indicated that cell proliferation ability was
significantly increased in the Hypoxia + CXCR4 group compared with
the Hypoxia + Sham group (P<0.01). A significant decrease in
cell proliferation ability was identified in the Hypoxia + CXCR4 +
LY294002 group compared with the Hypoxia + CXCR4 group (P<0.05),
and similar results were observed in the Hypoxia + CXCR4 + AMD3100
group compared with the Hypoxia + CXCR4 group (P<0.01; Fig. 4A).
The aforementioned findings suggest that CXCR4
functioning on the EPC surface promoted BM-EPC proliferation under
hypoxic conditions, and that the PI3K inhibitor acts to attenuate
the stimulation mediated by CXCR4 in BM-EPCs under hypoxic
conditions. Transwell assays further verified that CXCR4
significantly accelerated the migration of BM-EPCs under hypoxic
conditions, while illustrating that PI3K inhibitor and CXCR4
inhibitor weakened the acceleration mediated by CXCR4 in BM-EPCs
under hypoxic conditions (P<0.01; Fig. 4B and D). More importantly, CXCR4 significantly
repressed the apoptosis of BM-EPCs under hypoxic conditions, but
PI3K inhibitor or CXCR4 inhibitor blocked the inhibition mediated
by CXCR4 in BM-EPCs under hypoxic conditions (P<0.01; Fig. 4C and E). These findings have significant
implications on the understanding of how CXCR4 promotes the
proliferation and migration of BM-EPCs, and suppresses their
apoptosis through the PI3K signaling pathway.
Discussion
The pathogenesis of the majority of CVDs involves
vascular endothelial damage (13),
and hypoxia represents a chief factor implicated in vascular
endothelial damage (14).
Therefore, strategies to prevent vascular endothelial cell injury
could potentially decrease the occurrence and severity of CVDs. The
present study focused on the role of CXCR4 in protecting BM-derived
EPCs in hypoxia, and found that the overexpression of CXCR4
promoted the proliferation and migration abilities of
hypoxia-induced BM-EPCs and simultaneously inhibited the apoptosis
ability of hypoxia-induced BM-EPCs through the PI3K/Akt signaling
pathway.
Firstly, mononuclear cells were successfully
isolated from the bone marrow of rats under a hypoxic environment
and EPCs were characterized by immunofluorescence assay with
DIL-ac-LDL and FITC-UEA-1 labeling, resulting in cells that were
CD14-negative, CD34- and KDR-positive, suggesting that EPCs have
similar characteristics to endothelial cells. EPCs are defined as
precursors of endothelial cells with positive expression of CD34,
CD133 and VEGFR2(15). EPCs are
also BM mononuclear cells with partial endothelial function and
differentiation ability, which can bind to ulex europaeus
agglutinin-1 (UEA-1) and acetylated low-density lipoprotein (AcLDL)
(16). EPCs derived from BM possess
the ability to proliferate, migrate and differentiate into mature
endothelial cells, as well as participate in angiogenesis, bind to
blood vessels, and stimulate the proliferation of neighboring
endothelial cells (17). Studies
have highlighted the key role of EPCs in CVDs (18). Therefore, CXCR4 may be a potential
therapeutic target for vascular damage and regeneration.
Secondly, CXCR4 was found to protect BM-derived EPCs
under hypoxia. CXCR4, as a receptor for SDF-1, represents a crucial
factor in the regulatory process of EPC homing (19). The blockade of CXCR4 signaling was
previously shown to alleviate the decreased homing of EPCs to
injured arteries (20), indicating
that CXCR4 signaling may be a potential molecular target for gene
therapy associated with the dysfunction of EPCs. The SDF-1/CXCR4
signaling pathway plays a key role in cell proliferation, tumor
metastasis, angiogenesis, and the reendothelialization capacity of
EPCs (21,22). The SDF-1α/CXCR4 axis depletes EPC
apoptosis under serum deprivation conditions through the PI3K/Akt
signaling pathway (23), but the
functions and mechanisms of CXCR4 in ischemic vascular diseases
remain unknown. Adenoviruses were constructed (rAAV6-GFP and
rAAV6-CXCR4), and successfully transfected into BM-EPCs. It was
found that overexpression of CXCR4 promoted the proliferation and
migration abilities of hypoxia-induced BM-EPCs. Simultaneously, the
overexpression of CXCR4 inhibited the apoptosis ability of
hypoxia-induced BM-EPCs. Therefore, CXCR4 may serve as a potential
therapeutic target for the regeneration of impaired blood
vessels.
Thirdly, CXCR4 upregulates the expression level of
the PI3K and Akt signaling pathway in BM EPCs under hypoxic
conditions; PI3K inhibitor (ly294002) inhibits proliferation and
migration under hypoxia, and promotes the apoptosis of BM-EPC
mediated by CXCR4. The PI3K signaling pathway is activated by
various cellular stimuli or toxic insults, and induces the
phosphorylation of key downstream effectors such as Akt. The
activation of PI3K leads to an increase in
phosphatidylinositol-(3,4,5)-triphosphate (PIP3) (24), which can bind to the pleckstrin
homology on PDK1. Then, PDK1 can phosphorylate Akt at the
activation loop (25). Akt
phosphorylates several antiapoptotic proteins, cell-cycle-related
proteins and transcription factors (24,25).
Akt can regulate intracellular signals, affect cell responses to
extrinsic stimuli, and participate in cell proliferation and
survival (26). Extensive research
has shown that PI3K/Akt are involved in an array of growth factor
signal transduction pathways, and exert multiple biological
functions (27). The present study
found that CXCR4 upregulates PI3K and Akt expression in BM-EPCs
under hypoxic conditions. In addition, PI3K inhibitor (LY294002)
inhibits proliferation and migration, while promoting the apoptosis
of CXCR4-mediated BM-EPCs under hypoxic conditions.
CXCR4 has been found to be expressed in >23
different types of cancer (28).
SDF-1/CXCR4 is involved in breast cancer, glioma and neuroblastoma,
which promotes endothelial progenitor cells homing to form
neovascularization (29-31).
In the present study, CXCR4-overexpressing EPCs showed enhanced
cellular proliferation and decreased apoptosis under hypoxia, as
well as normoxia. The intrinsic increase in PI3K/Akt is one of the
major causes of transformation. Therefore, the potential risk of
tumors and hemangiomas caused by CXCR4-overexpressing EPCs should
be carefully considered.
A limitation of the present study was that there
were no set gradient values for SDF-1, LY294002 and AMD3100; thus,
the most effective relative concentration for the interaction
between SDF-1 and CXCR4, and the relative concentration of CXCR4
inhibition by LY294002 and AMD3100 were unknown.
In conclusion, the present study suggests that CXCR4
accelerates proliferation and migration, and suppresses apoptosis
of hypoxia-induced BM-EPCs through the PI3K signaling pathway in
rats. These results open new avenues of research to better
understand the role of BM-EPCs in vascular repair.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Outstanding Youth
Project of the Education Bureau of Hunan Province (grant no.
18B036), the Natural Science Foundation of Hunan Province (grant
no. 2020JJ4406), the Type B vertical project of the Health and
Family Planning Commission (grant no. 20190489), the Hunan Natural
Science Foundation joint project of the Science and Technology
Commission (grant no. 2020JJ8082), and the Scientific Research
Project of the Hunan Provincial Health Commission (grant no.
B2019066).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZYL, ZFZ, BLZ and QXY conceived and designed the
study; YC, JJR, JL, YT, CLW, XP, QCZ, LZ, LBX, FL and ZHY acquired
the experimental data; HWP, JH, JZ, JW, YZ, JQP and ZFZ analyzed
and interpreted the data; and ZYL, QXY and BLZ drafted the article
or revised it critically for intellectual content. ZFZ and BLZ
agree to be accountable for all aspects of the work, ensuring that
questions related to the accuracy or integrity of any part of the
work are appropriately investigated and resolved. ZYL, ZFZ, BLZ and
QXY were responsible for confirming the authenticity of all the raw
data, All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
This study was conducted with the approval of the
Ethics Committee of the Hunan Provincial People's Hospital
(Changsha, China) [approval no. (2019)-041]. The animals used in
this study were cared for in accordance with the Guide for the
Care and Use of Laboratory Animals published by the National
Institutes of Health. All efforts were made to minimize the
suffering of the animals included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
GBD 2016 Causes of Death Collaborators.
Global, regional, and national age-sex specific mortality for 264
causes of death, 1980-2016: A systematic analysis for the global
burden of disease study 2016. Lancet. 390:1151–1210.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Townsend N, Wilson L, Bhatnagar P,
Wickramasinghe K, Rayner M and Nichols M: Cardiovascular disease in
Europe: Epidemiological update 2016. Eur Heart J. 37:3232–3245.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ramakrishnan S, Anand V and Roy S:
Vascular endothelial growth factor signaling in hypoxia and
inflammation. J Neuroimmune Pharmacol. 9:142–60. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jaipersad AS, Lip GY, Silverman S and
Shantsila E: The role of monocytes in angiogenesis and
atherosclerosis. J Am Coll Cardiol. 63:1–11. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sharifpanah F, Behr S, Wartenberg M and
Sauer H: Mechanical strain stimulates vasculogenesis and expression
of angiogenesis guidance molecules of embryonic stem cells through
elevation of intracellular calcium, reactive oxygen species and
nitric oxide generation. Biochim Biophys Acta. 1863:3096–3105.
2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rafii S, Butler JM and Ding BS: Angiocrine
functions of organ-specific endothelial cells. Nature. 529:316–325.
2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Döring Y, Pawig L, Weber C and Noels H:
The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular
disease. Front Physiol. 5(212)2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schober A and Zernecke A: Chemokines in
vascular remodeling. Thromb Haemost. 97:730–737. 2007.PubMed/NCBI
|
|
9
|
Jacobson O and Weiss ID: CXCR4 chemokine
receptor overview: Biology, pathology and applications in imaging
and therapy. Theranostics. 3:1–2. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yamaguchi J, Kusano KF, Masuo O, Kawamoto
A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner
JM and Asahara T: Stromal cell-derived factor-1 effects on ex vivo
expanded endothelial progenitor cell recruitment for ischemic
neovascularization. Circulation. 107:1322–1328. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Segal MS, Shah R, Afzal A, Perrault CM,
Chang K, Schuler A, Beem E, Shaw LC, Li Calzi S, Harrison JK, et
al: Nitric oxide cytoskeletal-induced alterations reverse the
endothelial progenitor cell migratory defect associated with
diabetes. Diabetes. 55:102–109. 2006.PubMed/NCBI
|
|
12
|
Navidshad B, Liang JB and Jahromi MF:
Correlation coefficients between different methods of expressing
bacterial quantification using real time PCR. Int J Mol Sci.
13:2119–2132. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Matsuzawa Y, Guddeti RR, Kwon TG, Lerman
LO and Lerman A: Treating coronary disease and the impact of
endothelial dysfunction. Prog Cardiovasc Dis. 57:431–442.
2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dai Z, Li M, Wharton J, Zhu MM and Zhao
YY: Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells
and hematopoietic cells induces obliterative vascular remodeling
and severe pulmonary arterial hypertension in mice and humans
through hypoxia-inducible factor-2α. Circulation. 133:2447–2458.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cesari F, Sofi F, Molino Lova R, Vannetti
F, Pasquini G, Cecchi F, Marcucci R, Gori AM and Macchi C: Mugello
Study Working Group. Aging process, adherence to mediterranean diet
and nutritional status in a large cohort of nonagenarians: Effects
on endothelial progenitor cells. Nutr Metab Cardiovasc Dis.
28:84–90. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dai X, Yan X, Zeng J, Chen J, Wang Y, Chen
J, Li Y, Barati MT, Wintergerst KA, Pan K, et al: Elevating CXCR7
improves angiogenic function of EPCs via Akt/GSK-3β/Fyn-mediated
Nrf2 activation in diabetic limb ischemia. Circ Res. 120:e7–e23.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bianconi V, Sahebkar A, Kovanen P,
Bagaglia F, Ricciuti B, Calabrò P, Patti G and Pirro M: Endothelial
and cardiac progenitor cells for cardiovascular repair: A
controversial paradigm in cell therapy. Pharmacol Ther.
181:156–168. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Werner N, Kosiol S, Schiegl T, Ahlers P,
Walenta K, Link A, Böhm M and Nickenig G: Circulating endothelial
progenitor cells and cardiovascular outcomes. N Engl J Med.
353:999–1007. 2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Napoli C, Hayashi T, Cacciatore F,
Casamassimi A, Casini C, Al-Omran M and Ignarro LJ: Endothelial
progenitor cells as therapeutic agents in the microcirculation: An
update. Atherosclerosis. 215:9–22. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wara AK, Manica A, Marchini JF, Sun X,
Icli B, Tesmenitsky Y, Croce K and Feinberg MW: Bone marrow-derived
Kruppel-like factor 10 controls reendothelialization in response to
arterial injury. Arterioscler Thromb Vasc Biol. 33:1552–1560.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ruiz A, Ruiz L, Colón-Caraballo M,
Torres-Collazo BJ, Monteiro JB, Bayona M, Fazleabas AT and Flores
I: Pharmacological blockage of the CXCR4-CXCL12 axis in
endometriosis leads to contrasting effects in proliferation,
migration, and invasion. Biol Reprod. 98:4–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bianchi ME and Mezzapelle R: The chemokine
receptor CXCR4 in cell proliferation and tissue regeneration. Front
Immunol. 11(2109)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zheng H, Dai T, Zhou B, Zhu J, Huang H,
Wang M and Fu G: SDF-1alpha/CXCR4 decreases endothelial progenitor
cells apoptosis under serum deprivation by PI3K/Akt/eNOS pathway.
Atherosclerosis. 201:36–42. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Walton KL, Holt L and Sartor RB:
Lipopolysaccharide activates innate immune responses in murine
intestinal myofibroblasts through multiple signaling pathways. Am J
Physiol Gastrointest Liver Physiol. 296:G601–G611. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang YQ, Zhang JR, Li SD, He YY, Yang YX,
Liu XL and Wan XP: Aberrant methylation of breast and ovarian
cancer susceptibility gene 1 in chemosensitive human ovarian cancer
cells does not involve the phosphatidylinositol 3'-kinase-Akt
pathway. Cancer Sci. 101:1618–1623. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhou W, Guo S, Liu M, Burow ME and Wang G:
Targeting CXCL12/CXCR4 axis in tumor immunotherapy. Curr Med Chem.
26:3026–3041. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S,
Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al: Glioblastoma
stem cells generate vascular pericytes to support vessel function
and tumor growth. Cell. 153:139–152. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Du R, Lu KV, Petritsch C, Liu P, Ganss R,
Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z and Bergers G:
HIF1alpha induces the recruitment of bone marrow-derived vascular
modulatory cells to regulate tumor angiogenesis and invasion.
Cancer Cell. 13:206–220. 2008.PubMed/NCBI View Article : Google Scholar
|