Introduction
Diabetes mellitus is a group of metabolic diseases
characterized by chronic hyperglycemia from defective insulin
secretion and/or impaired biological effects (1). Diabetic nephropathy (DN) is a major
microvascular complication of diabetes mellitus and the leading
cause of end-stage renal disease, thereby contributing to the high
mortality rates (2).
Several mechanisms contribute to the onset and
development of DN, including hemodynamic factors, oxidative stress
and cytokine signaling (3,4). Recently, increasing evidence has
suggested that inflammation plays a key role in the pathogenesis of
DN, although it is commonly considered a non-inflammatory disease
(5,6). TNF-α, a potent proinflammatory
cytokine, is synthesized and released by infiltrating macrophages
and intrinsic kidney cells (7).
Previous studies have reported that increased urinary TNF-α, as a
pathogenic factor, may precede the appearance of pathological
albuminuria, and thus is considered a marker of kidney injury in
early stages of DN (8,9). Several alterations in renal function
occur during the initial stages of DN, including a decrease in
urinary concentrating ability; however, its molecular mechanism
remains unclear.
The thick ascending limb (TAL) of the Henle's loop
is responsible for the reabsorption of 20-25% of filtered NaCl,
which is the most important step required to establish the
hyperosmotic gradient of the medulla for the concentration of urine
(10,11). The basolateral K+
channels in the TAL play a critical role in sustaining the
transepithelial membrane transport by generating the cell membrane
potential to drive Cl- diffusion (12,13).
Previous studies have demonstrated that the basolateral
Kir4.1/Kir5.1 heterotetramers, with a conductance of 40-50 pS, are
the predominant subtype in the TAL, and indirectly influence the
tubular NaCl transportation by regulating the activity of
Na+-K+-2Cl- cotransporters
(NKCC2), which further affects urinary concentrating (14,15).
The main effects of impaired urinary concentrating ability in the
kidney include renal polyuria, or even renal diabetes insipidus,
and infection with the loss of immune substances in the urine.
The present study aimed to investigate the effect of
TNF-α on the basolateral Kir4.1/Kir5.1 channels in the TAL during
diabetes. Furthermore, the study sought to determine its underlying
regulatory mechanism to provide a theoretical basis for the
detection of impaired kidney concentrating capacity during
diabetes.
Materials and methods
Reagents
Antibodies against TNF-α (ab6671), phospholipase
A2 (PLA2, AF6329), COX2 (AF7003),
Kir4.1 (DF9260) and Kir5.1 (K009361P) were purchased from Abcam,
Affinity and Solarbio. Melittin, prostaglandin E2
(PGE2), polylysine and collagenase were purchased from
Sigma-Aldrich; Merck KGaA. TNF receptor fusion protein (TNFR:Fc)
was purchased from CPGJ Pharmaceutical Co., Ltd. (http://27919267.b2b.11467.com/). The TNF-α
(900TM73) and PGE2 ELISA kits (EK7124) were purchased
from PeproTech, Inc., and Boster Biological Technology,
respectively. The rat albumin ELISA kit (ab23564) was purchased
from Abcam.
Animals and experimental design
Male pathogen-free Sprague-Dawley rats (weight,
200±20 g, 6-7 weeks old) were obtained from the Animal Facility of
Jiamusi University, and housed at 20-25˚C, 50-65% relative humidity
and with a 12-h light/dark cycle, with free access to normal food
and water. A total of 40 rats were randomly divided into four
groups: Control rats, diabetic rats, control rats treated with
TNFR:Fc (control + TNFR:Fc) and diabetic rats treated with TNFR:Fc
(diabetic + TNFR:Fc). Diabetic rats were induced via
intraperitoneal injection of 60 mg/kg streptozotocin (STZ)
dissolved in citric acid buffer. The levels of fasting blood
glucose were monitored by drawing blood from the tail vein 72 h
after STZ injection. The rats with fasting blood glucose >16.7
mmol/l and increased drinking water, eating and urine volume were
considered to be successful diabetic models. Subcutaneous injection
of TNFR:Fc (2 mg/kg) was performed twice a week in the control and
diabetic rats before STZ injection for 3 weeks (16). All animal experiments were approved
by the Medical Ethics Committee of Jiamusi University (Jiamusi,
China; approval no. JMSU-229).
Measurement of urine output and
urinary albumin
Rats were placed in metabolic cages to collect urine
from 9 am to 9 am the next day, and the supernatant of urine
following centrifugation was the 24 h urine output. Urinary albumin
(UAlb) was measured using the ELISA kit for rat albumin, according
to the manufacturer's instructions.
Measurement of TNF-α in urine and
PGE2 in tissues
The levels of TNF-α in urine and PGE2 in
tissues were measured using the TNF-α rat ELISA and PGE2
rat ELISA kits, respectively, according to the manufacturer's
instructions. Briefly, the standard solution and samples were added
to the wells and the plates were incubated at 37˚C for 90 min.
After washing three times with washing buffer, the plates were
incubated with corresponding antibody working liquid for 60 min at
room temperature. Subsequently, the plates were re-washed and
incubated with ABC working solution at 37˚C for 30 min. Following
addition of the TMB substrate, the plates were incubated for 10 min
at 37˚C in the dark. Absorbance was measured at a wavelength of 450
nm, using a spectrophotometer (BioTek Instruments, Inc.), after
adding the stop solution. The levels of TNF-α and PGE2
were quantified according to the standard curve.
Preparation of the TAL tissues
Rats were anesthetized using pentobarbital (50
mg/kg) and sacrificed via cervical dislocation. The kidneys were
immediately removed and cut into 1-mm-thick sections after removing
the capsule and poles. The renal cortex and inner stripe of outer
medulla were carefully excised and minced with a blade under a
dissecting microscope. Samples were incubated and shaken in HEPES
buffer solution containing 10 mM HEPES, 140 mM NaCl, 5 mM KCl, 1.5
mM MgCl2 and 1.8 mM CaCl2 (pH 7.4), with
collagenase type 1A (1 mg/ml) at 37˚C for 5 min. Undigested tissues
were subjected to three treatments with collagenase (5 min each)
and the supernatants were combined. The combined supernatants were
subsequently filtered through 180 and 50 µm nylon mesh membranes,
and the TALs retained on the 50 µm mesh were collected for western
blotting.
Western blotting
Protein samples (30 µg) were extracted from the TAL
tissues using RIPA lysis buffer, separated via 10 or 12% SDS-PAGE
and transferred onto PVDF membranes. The membranes were blocked
with blocking solution containing 5% non-fat milk in TBS-0.05%
Tween (TBS-T) for 1 h at room temperature and subsequently
incubated with the corresponding primary antibody at 4˚C for 12 h.
Membranes were washed four times with TBS-T (15 min each) and
subsequently incubated with the secondary antibody (ZB-2301;
OriGene Technologies, Inc.) solution containing 5% non-fat dry milk
in TBS-T for 1 h at room temperature, prior to re-washing with
TBS-T (4x15 min). Protein bands were visualized using ECL plus
chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.) and
analyzed using ImageJ software (version 1.45s; National Institutes
of Health).
Patch clamp technique
The 1-mm-thick sections were incubated in HEPES
buffer with collagenase type 1A at 37˚C for 40-60 min. The digested
TALs were isolated under a dissecting microscope and placed on a
cover glass (5x5 mm) coated with polylysine. The cover glass with
TALs was transferred to a chamber filled with HEPES-buffered NaCl
solution (in mM: 140 NaCl, 5 KCl, 1.5 MgCl2, 1.8
CaCl2 and 10 HEPES, pH 7.4) and mounted on an inverted
microscope (Nikon Corporation). Using a P-97 electrode-puller, the
patch clamp electrodes were filled with a pipette solution (in mM:
10 HEPES, 140 KCl and 1.8 MgCl2, pH 7.4) and fixed to
the probe to patch the treated TALs. The channel currents were
low-pass filtered at 0.5 kHz and recorded using an Axon 700B patch
clamp amplifier. The data were digitized with an Axon interface
(Digidata 1400A) and analyzed using pClamp 10.0 software (Axon
Instruments; Molecular Devices, LLC). The channel activity,
expressed as a product of channel number and open probability
(NPo), was calculated from data samples of 90 sec durations
at a steady state, as follows: NPo=Σ
(1t1+2t2+...iti),
in which ti is the fractional open time spent at
each of the observed current levels.
Melittin treatment
After patching and recording the current of the
K+ channel for 2-3 min, melittin (5 µM) was added and
the current of the K+ channel was recorded for 3-5
min.
Statistical analysis
Statistical analysis was performed using SPSS
software version 19.0 (IBM Corp.). Data were presented as the mean
± standard error of the mean and analyzed using a one-way ANOVA
followed by a Tukey's post hoc test and Student-Newman-Keuls post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
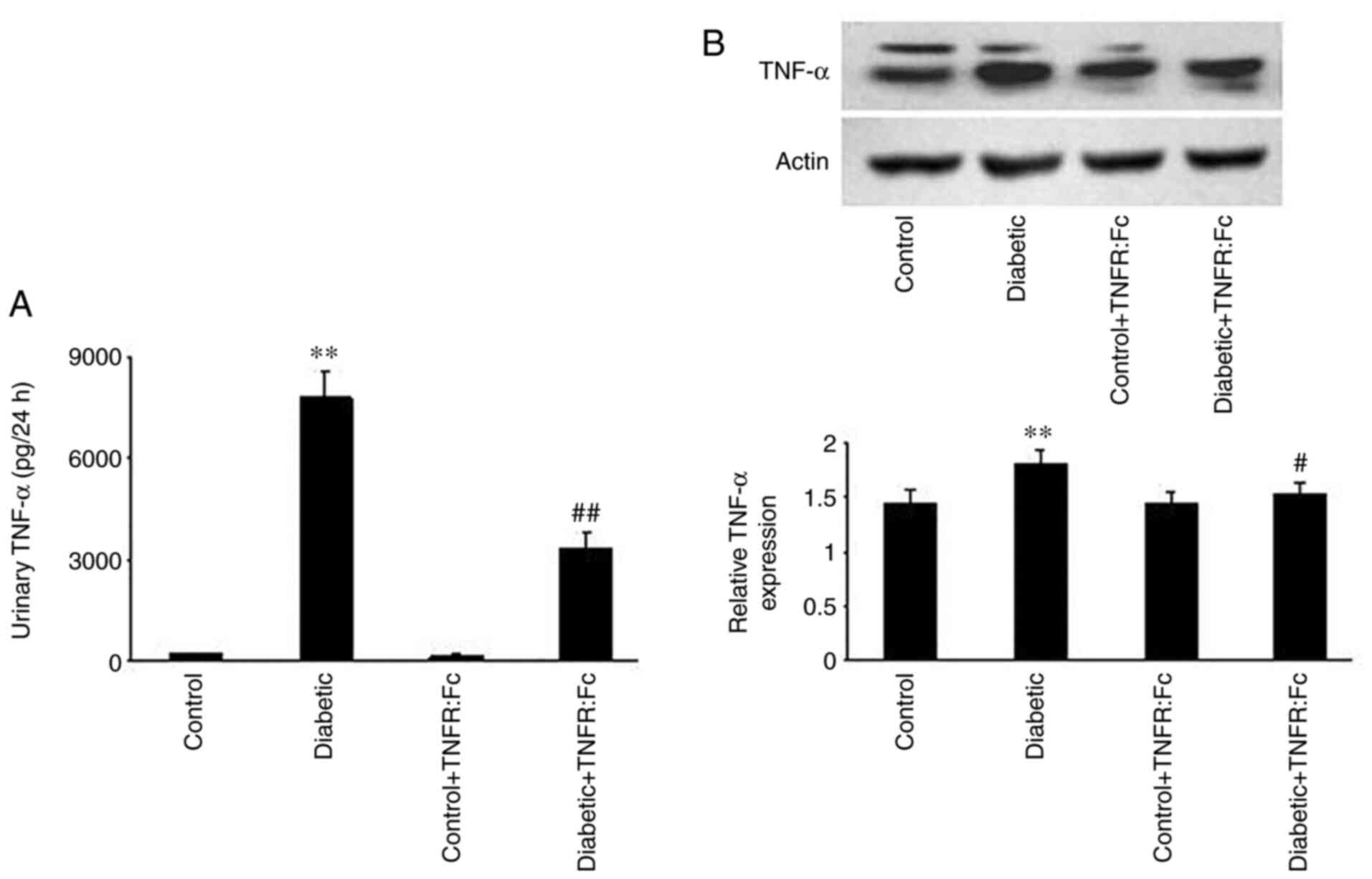
Changes in urinary TNF-α excretion and
TNF-α protein expression in the TAL during diabetes
To observe the changes in TNF-α levels during
diabetes, urinary TNF-α excretion and TNF-α protein expression in
the TAL were measured. The results demonstrated that urinary TNF-α
excretion in diabetic rats was markedly increased compared with the
control rats, and decreased following treatment with TNFR:Fc in the
diabetic + TNFR:Fc group (n=7; P<0.01; Fig. 1A). Furthermore, relative TNF-α
expression (Fig. 1B) was
significantly higher in the diabetic group compared with the
control group (n=5; P<0.01), while treatment with TNFR:Fc
decreased its level in diabetic + TNFR:Fc rats (n=5; P<0.05).
These results confirm that TNF-α expression is upregulated during
diabetes.
Changes in blood glucose, UAlb and
urine output during diabetes
Compared with the control rats, the levels of blood
glucose, UAlb and urine output in diabetic rats were significantly
increased (n=7; P<0.01; Table
I). Compared with the diabetic rats, UAlb levels in the
diabetic + TNFR:Fc group were markedly decreased (n=7; P<0.01;
Table I), while no significant
changes were observed in blood glucose and urine output levels
following treatment with TNFR:Fc in the diabetic + TNFR:Fc
group.
 | Table IChanges in blood glucose, UAlb and
urine output during diabetes. |
Table I
Changes in blood glucose, UAlb and
urine output during diabetes.
| Group | Blood glucose,
mmol/1 | UAlb, mg/24 h | Urine output, ml/24
h |
|---|
| Control | 5.31±0.45 | 0.29±0.04 | 9.12±1.33 |
| Diabetic |
25.47±3.09a |
1.17±0.25a |
166.75±13.40a |
| Control +
TNFR:Fc | 5.39±0.82 | 0.31±0.06 | 8.96±1.28 |
| Diabetic +
TNFR:Fc | 25.39±2.98 |
0.58±0.07b | 143.25±21.82 |
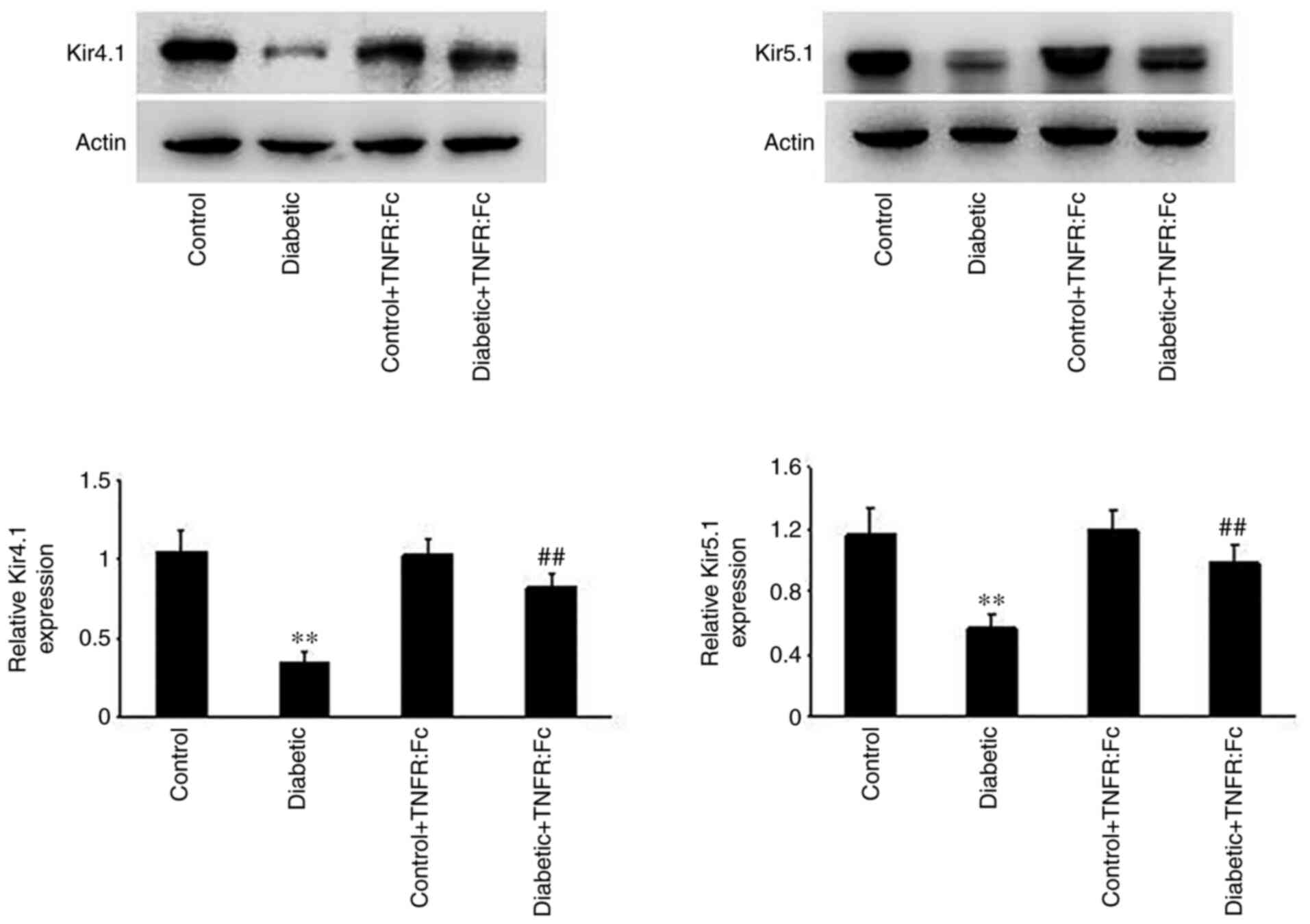
Effect of TNF-α on the basolateral
Kir4.1/Kir5.1 channels in the TAL during diabetes
Increasing evidence has suggested that basolateral
Kir4.1/Kir5.1 channels in the TAL play an important role in
determining NKCC2 activity and influencing tubular NaCl
transportation and urine concentration (10). Thus, Kir4.1/Kir5.1 protein
expression was detected in each group via western blotting to
investigate the effect of TNF-α on the basolateral K+
channel in the TAL during diabetes. As presented in Fig. 2, relative Kir4.1/Kir5.1 protein
expression was significantly decreased in diabetic rats compared
with normal rats (n=5; P<0.01), and increased in the diabetic +
TNFR:Fc group following treatment with TNFR:Fc (n=5; P<0.01).
Taken together, these results suggest that TNF-α inhibits the
activity of basolateral Kir4.1/Kir5.1 in the TAL during
diabetes.
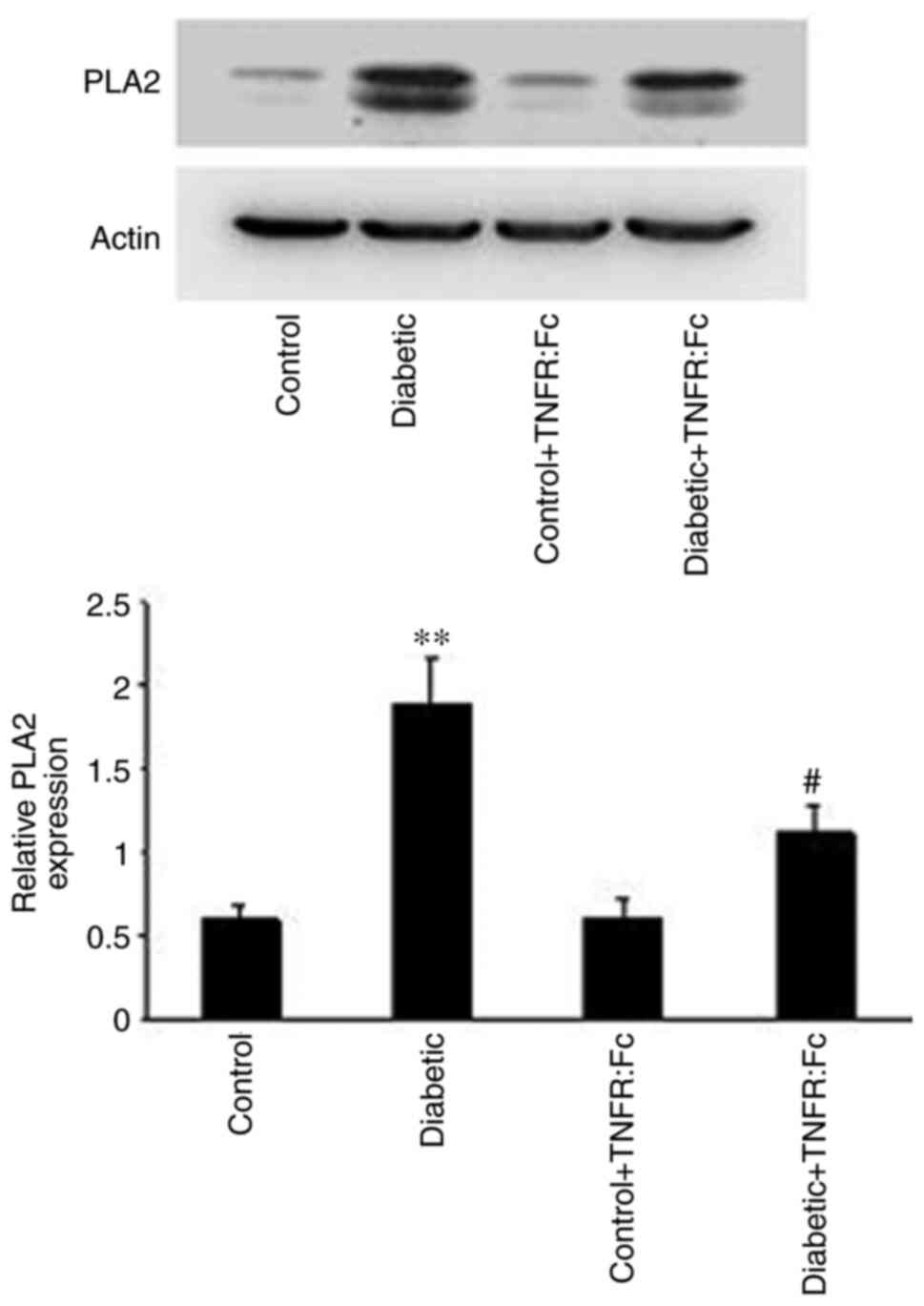
Role of the PLA2-dependent
pathway in the inhibitory effect of TNF-α on Kir4.1/Kir5.1 during
diabetes
After revealing the inhibitory effect of TNF-α on
Kir4.1/Kir5.1 during diabetes, the present study aimed to determine
its underlying molecular mechanism. Previous studies have reported
that the activity of the basolateral K+ channel is often
mediated by the PLA2-dependent pathway (12). Thus, the role of the
PLA2-dependent pathway in the inhibitory effect of TNF-α
on Kir4.1/Kir5.1 during diabetes was investigated. As presented in
Fig. 3, relative PLA2
expression was significantly higher in diabetic rats compared with
normal rats (n=5; P<0.01), but decreased in the diabetic +
TNFR:Fc group compared with diabetic rats following treatment with
TNFR:Fc (n=5; P<0.05). Thus, the PLA2-dependent
pathway may participate in the regulatory effect of TNF-α on
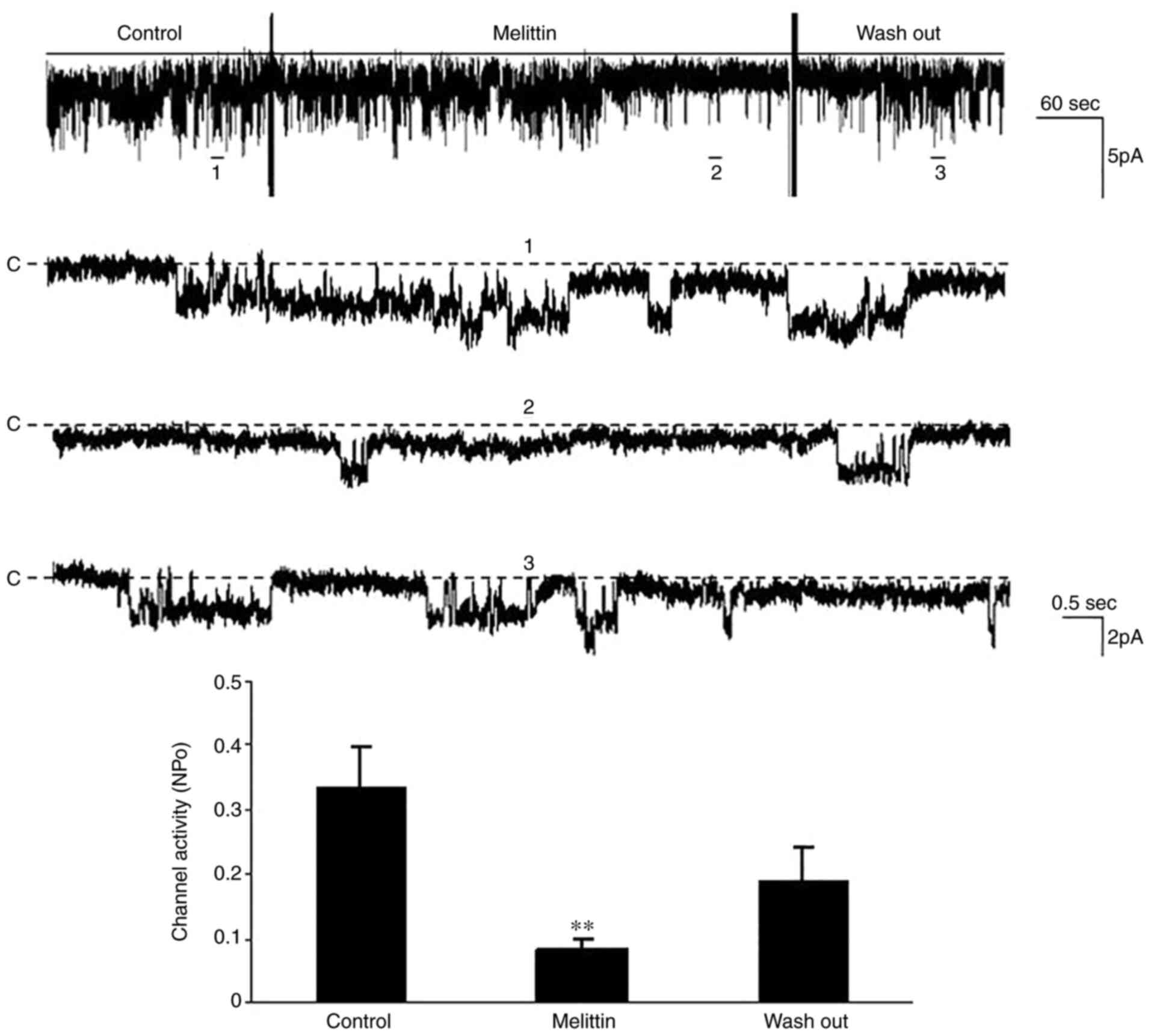
Kir4.1/Kir5.1 during diabetes. To further determine the role of the
PLA2-dependent pathway, the effect of melittin, an
agonist of PLA2 (17),
on Kir4.1/Kir5.1 in the TAL was investigated via the patch clamp
technique. The results demonstrated that addition of melittin (5
µM) decreased the channel activity (NPo) from
0.33±0.07 to 0.08±0.02 in a cell-attached patch (n=5; P<0.01,
Fig. 4). Collectively, these
results suggest that the inhibitory effect of TNF-α on
Kir4.1/Kir5.1 during diabetes is mediated by the
PLA2-dependent pathway.
Role of the
cyclooxygenase-2 (COX2)/PGE2
pathway in the inhibitory effect of TNF-α on Kir4.1/Kir5.1 during
diabetes
As a downstream pathway of the
PLA2-dependent pathway, the
COX2/PGE2 pathway is associated with the
development of diabetes (18).
However, whether it is involved in the regulation of TNF-α on
Kir4.1/Kir5.1 during diabetes remains unclear. Thus, the role of
the COX2/PGE2 pathway in the inhibitory
effect of TNF-α on Kir4.1/Kir5.1 during diabetes was also
investigated in the present study. Western blot analysis was
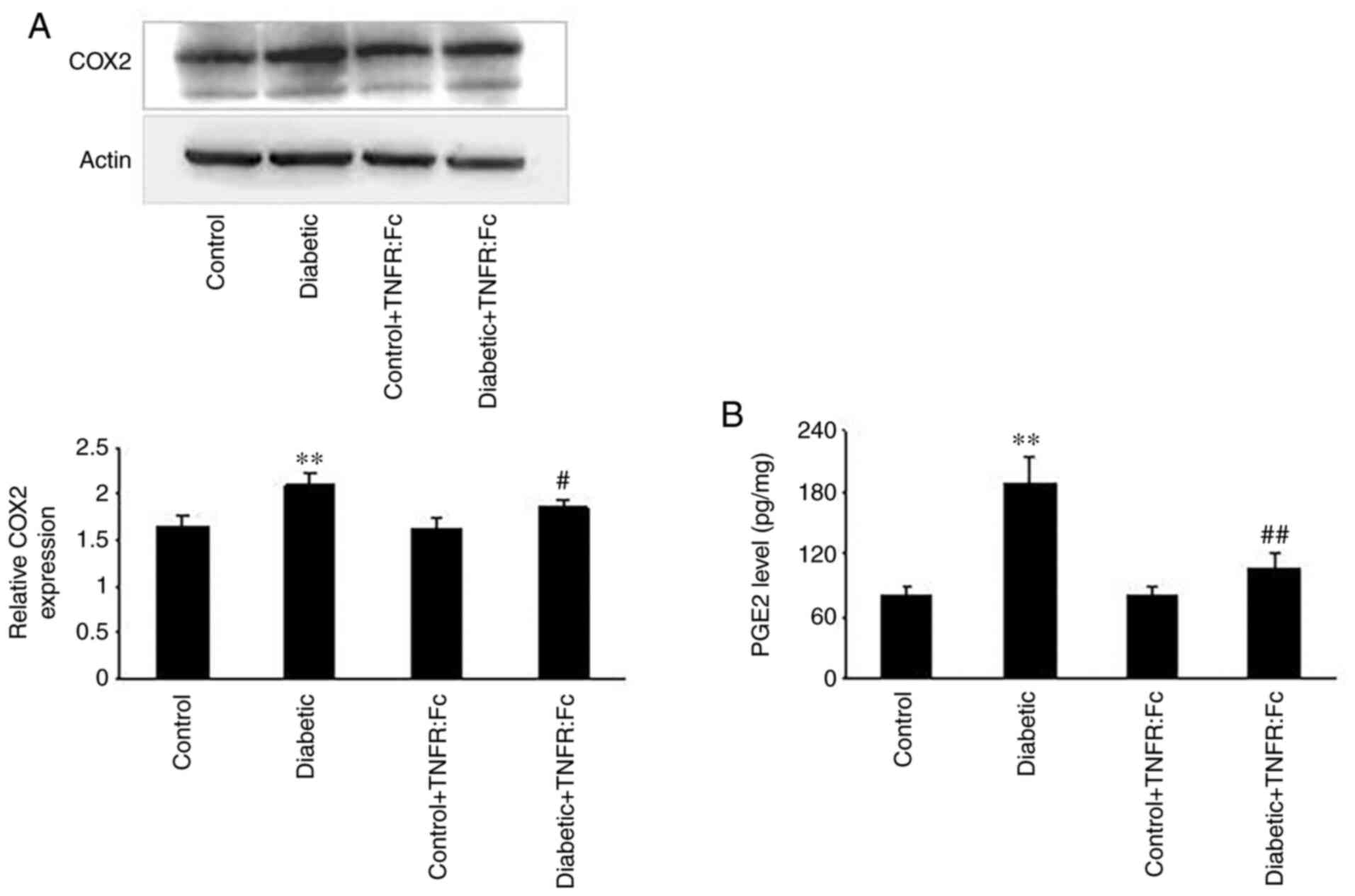
performed to detect COX2 protein expression in the TAL.
As presented in Fig. 5A, relative
COX2 protein expression was significantly higher in
diabetic rats compared with normal rats (n=5; P<0.01), but
decreased in the diabetic + TNFR:Fc group compared with diabetic
rats following treatment with TNFR:Fc (n=5; P<0.05).
Subsequently, PGE2 expression was measured in the TAL
via ELISA. As presented in Fig.
5B, PGE2 expression was markedly increased in
diabetic rats compared with normal rats, while treatment with
TNFR:Fc decreased PGE2 expression in the diabetic +
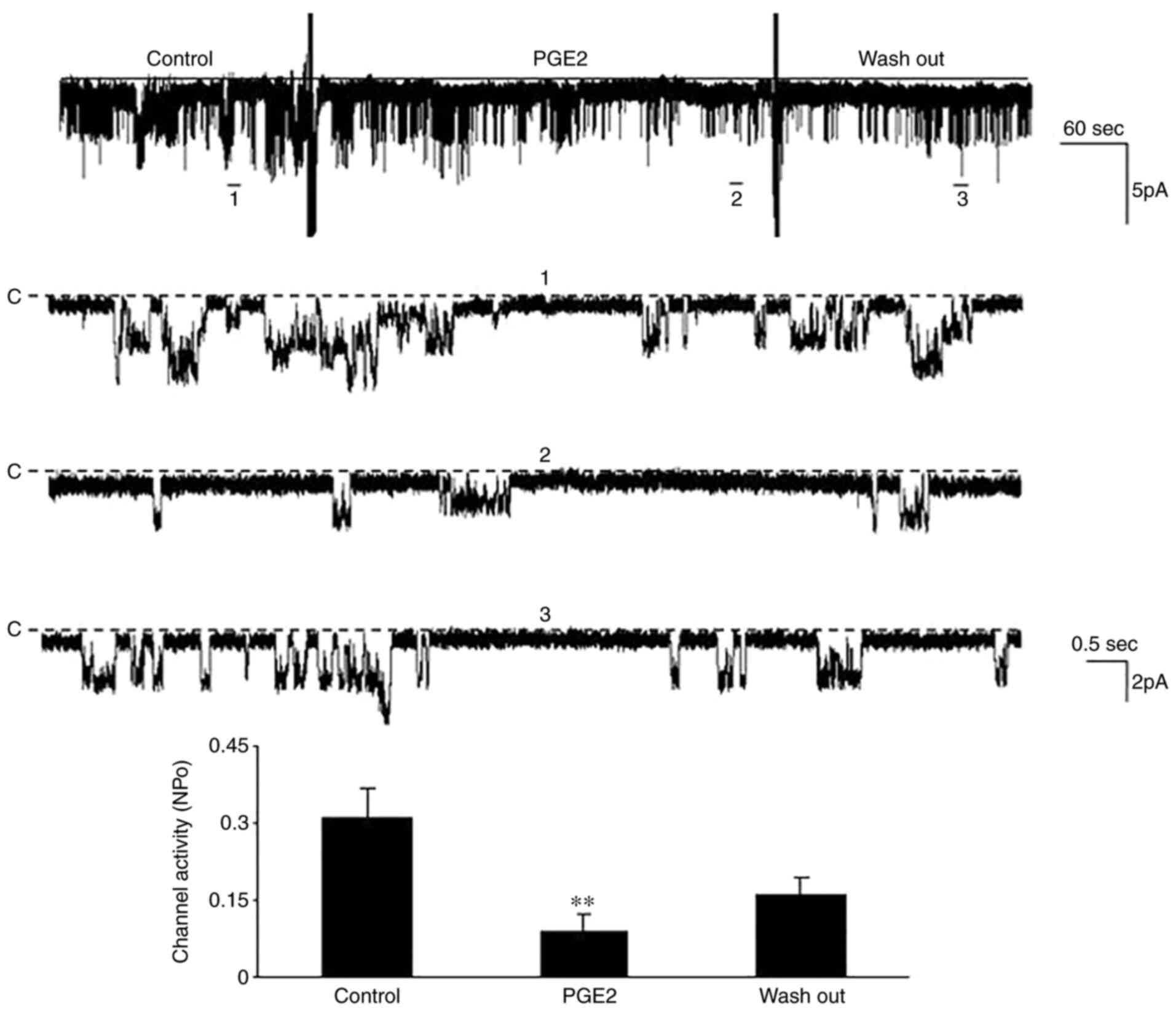
TNFR:Fc group (n=5; P<0.01). The patch clamp technique was
performed to determine the effect of PGE2 on
Kir4.1/Kir5.1 during diabetes. The results demonstrated that the
NPo decreased from 0.31±0.06 to 0.09±0.03
following treatment with 10 µM PGE2 (n=5; P<0.01;
Fig. 6). Taken together, these
results suggest that the COX2/PGE2 pathway is
involved in the inhibitory effect of TNF-α on Kir4.1/Kir5.1 during
diabetes.
Discussion
The present study aimed to investigate the effect of
TNF-α, elevated by diabetes, on the Kir4.1/Kir5.1 in the TAL. The
results demonstrated that TNF-α inhibited the activity of
Kir4.1/Kir5.1 via the PLA2/COX/PGE2 pathway
during diabetes. Currently, three lines of evidence support this
concept. First, TNF-α expression in the TAL was significantly
increased in diabetic rats, and decreased following treatment with
TNFR:Fc. Secondly, the protein expression levels of PLA2
and COX2 were higher in the TAL of diabetic rats, and
were decreased following treatment with TNFR:Fc. Thirdly, addition
of melittin or PGE2 inhibited the channel activity of
Kir4.1/Kir5.1 in a cell-attached patch. Thus, it was hypothesized
that increased TNF-α expression during diabetes activates the
PLA2/COX2/PGE2 pathway, thereby
inhibiting basolateral Kir4.1/Kir5.1 channel activity in the
TAL.
The Na+ and Cl- load filtered
from the glomerulus is reabsorbed in the TAL via two steps: First,
they enter epithelial cells via apical NKCC2 and then, they leave
the cells via basolateral Na-K+ pump and
Cl- channels, respectively (19). It is well-known that basolateral
K+ channels play an important role in the modulation of
NaCl transportation by affecting cell membrane potential under
physiological conditions (20).
Activation of basolateral K+ channels increases the
negativity of the cell membrane potential, thereby augmenting the
driving force for Cl- exit, while inhibition of
basolateral K+ channel activity depolarizes the cell
membrane potential, thereby diminishing the driving force for the
diffusion of Cl- across the basolateral membrane
(21). Consequently, inhibition of
Cl- diffusion leads to an increase in intracellular
Cl- concentration, which suppresses the interaction
between WNK lysine deficient protein kinase 3 and serine/threonine
kinase 39, and inhibits NKCC2 activity by decreasing the
phosphorylation of NKCC2(22).
Given that the active reabsorption of NaCl in the water-impermeable
TAL is essential for the urinary concentrating mechanism,
inhibition of NaCl reabsorption in the TAL under pathological
conditions decreases the urinary concentrating ability (13). It has been reported that diabetic
nephropathy impairs urinary concentrating ability (23).
DN is considered a form of ‘microinflammation',
whereby several cytokines are involved in its underlying
immunopathological mechanisms (24). Among these, TNF-α is an important
mediator of inflammatory tissue damage and a major participant in
the pathogenesis of DN (25).
Consistent with experimental models, clinical investigations have
reported that serum and urinary concentrations of TNF-α in diabetic
patients with nephropathy are higher than non-diabetic subjects or
diabetic patients without renal involvement (16). Enhanced TNF-α is cytotoxic to renal
cells and can cause direct renal injury by promoting inflammation
and the accumulation of extracellular matrix, decreasing glomerular
blood flow, inducing apoptosis and damaging glomerular permeability
barrier (26,27). TNF-α can also indirectly disrupt
the barrier function of the glomerular capillary wall and enhance
the albumin permeability by inducing the production of reactive
oxygen species in diverse cells, including mesangial cells
(28). In addition, Battula et
al (29) demonstrated that
increased TNF-α production in response to hypercalcemia inhibits
NKCC2 activity and NaCl reabsorption via the
COX2/PGE2 pathway, which contributes to
polyuria and concentration defects.
There is a distinct association between
PLA2 and the COX-PG system (30). PLA2 enzymes are the
upstream regulators of liberating free arachidonic acid from the
sn-2 position of membrane phospholipids (31,32).
Arachidonic acid is released from phospholipids via the action of
PLA2 and converted into PGs via COXs (33). PGE2 is a prominent
prostanoid produced in the kidney, which is involved in diverse
renal functions regulating hemodynamics and tubular salt and water
transport (34). COXs, including
COX-1 and COX-2, are rate-limiting enzymes in
the PGE2 synthesis pathway (35). While no major renal pathology has
been reported for COX-1 knockout mice,
COX-2-lacking mice display abnormalities in renal
development and severe nephropathy (36,37).
Previous studies have reported that renal COX-2 activity
and PGE2 production are elevated in diabetes mellitus,
which contributes to the pathogenesis of DN (38). Inhibition of COX-2 has
been demonstrated to reverse some of the renal complications of
STZ-diabetes, such as attenuating glomerulosclerosis and glomerular
hypertrophy, thereby slowing the development of proteinuria
(36,39).
The results of the present study and previous
findings suggest that the COX2/PGE2 pathway
is involved in the pathogenesis of diabetic nephropathy and the
impairment of urinary concentrating ability; however, its
underlying molecular mechanisms remain unclear. The findings of the
current study demonstrated that the inhibitory effect of TNF-α on
the basolateral Kir4.1/Kir5.1 channels in the TAL during diabetes
occurred via regulation of the
PLA2/COX2/PGE2 pathway. Given that
the basolateral K+ channels determine the driving force
for Cl- diffusion across the basolateral membrane
(23), a TNF-α-induced decrease in
channel activity of Kir4.1/Kir5.1 during diabetes may be associated
with a decrease in NaCl reabsorption in the TAL and urine
concentration. Thus, the results presented in the present study
provide a novel mechanism by which TNF-α impairs urinary
concentrating ability during diabetes, which occurs via stimulation
of the PLA2/COX2/PGE2 pathway to
inhibit the activity of the basolateral Kir4.1/Kir5.1 channels in
the TAL.
Only some pathological changes in early stage of
diabetic nephropathy were observed in the present study, others in
middle or late stages of diabetic nephropathy will be done in the
future study in order to comprehensively explore the pathogenesis
of diabetes and find effective prevention and treatment
methods.
Acknowledgements
The authors would like to thank Dr Jiaqi Wang
(Statistics Department at the Public Health College of Jiamusi
University) for providing constructive suggestions on the
statistical analysis in the manuscript.
Funding
Funding: This work was supported by the Chinese National Nature
Science Foundation (grant no. 31400994), Heilongjiang Natural
Science Foundation (grant no. LH2019C065), Heilongjiang Provincial
Department of Health Project (grant no. 2018104), National Basic
Medical Science Team Project (grant no. JDXKTD-2019002) and North
Medicine and Functional Food Characteristic Construction
Project.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GZ and ZL performed the experiments. YZ and RC
acquired and analyzed data. XZ and WW analyzed data and drafted the
manuscript. HS designed the project and revised the manuscript. All
authors have read and approved the final manuscript. GZ and HS
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of Jiamusi University (Jiamusi, China; approval no.
JMSU-229).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jenny L, Melmer A, Laimer M, Hardy ET, Lam
WA and Schroeder V: Diabetes affects endotelial cell function and
alters fibrin clot formation in a microvascular flow model: A pilot
study. Diab Vasc Dis Res: Feb 10, 2020 (Epub ahead of print).
|
|
2
|
Iwai T, Miyazaki M, Yamada G, Nakayama M,
Yamamoto T, Satoh M, Sato H and Ito S: Diabetes mellitus as a cause
or comorbidity of chronic kidney disease and its outcomes: The
Gonryo study. Clin Exp Nephrol. 22:328–336. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kanwar YS, Sun L, Xie P, Liu FY and Chen
S: A glimpse of various pathogenetic mechanisms of diabetic
nephropathy. Annu Rev Pathol. 6:395–423. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kashihara N, Haruna Y, Kondeti VK and
Kanwar YS: Oxidative stress in diabetic nephropathy. Curr Med Chem.
17:4256–4269. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mora C and Navarro J: Inflammation and
pathogenesis of diabetic nephropathy. Metabolism. 53:265–266.
2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wada J and Makino H: Inflammation and the
pathogenesis of diabetic nephropathy. Clin Sci. 124:139–152.
2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang B, Ramesh G, Norbury CC and Reeves
WB: Cisplatin-induced nephrotoxicity is mediated by tumor necrosis
factor-alpha produced by renal parenchymal cells. Kidney Int.
72:37–44. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kalantarinia K, Awad AS and Siragy HM:
Urinary and renal interstitial concentrations of TNF-α increase
prior to rise in albuminuria in diabetic rats. Kidney Int.
64:1208–1213. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Navarro J, Milena FJ, Mora C, León C,
Claverie F, Flores C and García J: Tumor necrosis factor-alpha gene
expression in diabetic nephropathy: Relationship with urinary
albumin excretion and effect of angiotensin-converting enzyme
inhibition. Kidney Int Suppl. 68:S98–S102. 2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kong S, Zhang C, Li W, Wang L, Luan H,
Wang WH and Gu R: Stimulation of Ca2+-sensing receptor
inhibits the basolateral 50-pS K channels in the thick ascending
limb of rat kidney. Biochim Biophys Acta. 1823:273–281.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hebert SC: Roles of Na-K-2Cl and Na-Cl
cotransporters and ROMK potassium channels in urinary concentrating
mechanism. Am J Physiol. 275:F325–F327. 1998.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang M, Sui H, Li W, Wang J, Liu Y, Gu L,
Wang WH and Gu R: Stimulation of A(2a) adenosine
receptor abolishes the inhibitory effect of arachidonic acid on the
basolateral 50-pS K channel in the thick ascending limb. Am J
Physiol Renal Physiol. 300:F906–F913. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hebert SC, Desir G, Giebisch G and Wang W:
Molecular diversity and regulation of renal potassium channels.
Physiol Rev. 85:319–371. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang C, Wang L, Su X, Lin DH and Wang WH:
KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the
cortical thick ascending limb. Am J Physiol Renal Physiol.
308:F1288–F1296. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fan L, Wang X, Zhang D, Duan X, Zhao C, Zu
M, Meng X, Zhang C, Su XT, Wang MX, et al: Vasopressin-induced
stimulation of the Na(+)-activated K(+) channels is responsible for
maintaining the basolateral K(+) conductance of the thick ascending
limb (TAL) in EAST/SeSAME syndrome. Biochim Biophys Acta.
1852:2554–2562. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dipetrillo K, Coutermarsh B and Gesek FA:
Urinary tumor necrosis factor contributes to sodium retention and
renal hypertrophy during diabetes. Am J Physiol Renal Physiol.
284:F113–F121. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ling BN, Webster CL and Eaton DC:
Eicosanoids modulate apical Ca(2+)-dependent K+ channels
in cultured rabbit principal cells. Am J Physiol. 263:F116–F126.
1992.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mouchlis VD and Dennis EA: Phospholipase
A2 catalysis and lipid mediator lipidomics. Biochim
Biophys Acta Mol Cell Biol Lipids. 1864:766–771. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang G, Gui S, Wang W, Meng D, Meng Q,
Luan H, Zhao R, Zhang J and Sui H: Acute stimulatory effect of
tumor necrosis factor on the basolateral 50 pS K channels in the
thick ascending limb of the rat kidney. Mol Med Rep. 18:4733–4738.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Paulais M, Lourdel S and Teulon J:
Properties of an inwardly rectifying K(+) channel in the
basolateral membrane of mouse TAL. Am J Physiol Renal Physiol.
282:F866–F876. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang W, Hebert SC and Giebisch G: Renal
K+ channels: Structure and function. Annu Rev Physiol.
59:413–436. 1997.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ponce-Coria J, San Cristobal P, Kahle KT,
Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juárez P, Muñoz E,
Michel G, Bobadilla NA, et al: Regulation of NKCC2 by a
chloride-sensing mechanism involving the WNK3 and SPAK kinases.
Proc Natl Acad Sci. 105:8458–8463. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ma SZ, Du J and Ma LR: The change of
urinary osmolalities and plasma osmolalities in diabetic rats
induced by STZ. Chin Lab Diagn. 12:976–978. 2008.
|
|
24
|
Zhang Y, Liu T, Chen Y, Dong Z, Zhang J,
Sun Y, Jin B, Gao F, Guo S and Zhuang R: CD226 reduces endothelial
cell glucose uptake under hyperglycemic conditions with
inflammation in type 2 diabetes mellitus. Oncotarget.
7:12010–12023. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Darwish NM, Elnahas YM and Alqahtany FS:
Diabetes induced renal complications by leukocyte activation of
nuclear factor κ-B and its regulated genes expression. Saudi J Biol
Sci. 28:541–549. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ortiz A, González-Cuadrado S, Bustos C,
Alonso J, Gómez-Guerrero C, López-Armada MJ, González-Arana E,
Plaza JJ and Egido J: Tumor necrosis factor as a mediator of
glomerular damage. J Nephrol. 8:27–34. 1995.
|
|
27
|
Xu C, Chang A, Hack BK, Eadon MT, Alper SL
and Cunningham PN: TNF-mediated damage to glomerular endothelium is
an important determinant of acute kidney injury in sepsis. Kidney
Int. 85:72–81. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mccarthy E, Sharma R, Sharma M, Li JZ, Ge
XL, Dileepan KN and Savin VJ: TNF-alpha increases albumin
permeability of isolated rat glomeruli through the generation of
superoxide. J Am Soc Nephrol. 9:433–438. 1998.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Battula S, Hao S, Pedraza PL, Stier CT and
Ferreri NR: Tumor necrosis factor-alpha induces renal
cyclooxygenase-2 expression in response to hypercalcemia.
Prostaglandins Other Lipid Mediat. 99:45–50. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Nasrallah R, Hassouneh R and Hebert RL:
PGE2, kidney disease, and cardiovascular risk: Beyond
hypertension and diabetes. J Am Soc Nephrol. 27:666–676.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Murakami M, Nakatani Y, Atsumi GI, Inoue K
and Kudo I: Regulatory functions of Phospholipase A2. Crit Rev
Immunol. 37:127–195. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Murakami M and Kudo I: Phospholipase A2. J
Biochem. 131:285–292. 2002.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Kudo I and Murakami M: Prostaglandin E
synthase, a terminal enzyme for prostaglandin E2 biosynthesis. J
Biochem Mol Biol. 38:633–638. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Quilley J, Santos M and Pedraza P: Renal
protective effect of chronic inhibition of COX-2 with SC-58236 in
streptozotocin-diabetic rats. Am J Physiol Heart Circ Physiol.
300:H2316–H2322. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nasrallah R, Landry A, Singh S, Sklepowicz
M and Hébert RL: Increased expression of cyclooxygenase-1 and -2 in
the diabetic rat renal medulla. Am J Physiol Renal Physiol.
285:F1068–F1077. 2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Langenbach R, Morham SG, Tiano HF, Loftin
CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH,
Kluckman KD, et al: Prostaglandin synthase 1 gene disruption in
mice reduces arachidonic acid-induced inflammation and
indomethacin-induced gastric ulceration. Cell. 83:483–492.
1995.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Morham SG, Langenbach R, Loftin CD, Tiano
HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A,
Lee CA and Smithies O: Prostaglandin synthase 2 gene disruption
causes severe renal pathology in the mouse. Cell. 83:473–482.
1995.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jia Z, Sun Y, Liu S, Liu Y and Yang T:
COX-2 but not mPGES-1 contributes to renal PGE2 induction and
diabetic proteinuria in mice with type-1 diabetes. PLoS One.
9(e93182)2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Komers R, Lindsley JN, Oyama TT and
Anderson S: Cyclo-oxygenase-2 inhibition attenuates the progression
of nephropathy in uninephrectomized diabetic rats. Clin Exp
Pharmacol Physiol. 34:36–41. 2007.PubMed/NCBI View Article : Google Scholar
|