Introduction
Chronic obstructive pulmonary disease (COPD) is a
chronic respiratory disease with high morbidity and mortality
rates, which can be prevented but not cured (1). The main characterization of patients
with COPD are emphysema and chronic bronchitis accompanied by
cough, dyspnea, shortness of breath and other symptoms, and the
chronic hypoxemia also occurs in moderate and severe patients
(2-4).
The main pathogenic factors of COPD are long-term smoking,
long-term exposure to air polluted environment, and bacterial
infection, among which the most important and the most wildly
studied factor is cigarette smoke (5,6).
Cigarette smoke exposure can induce oxidative stress (7,8).
Reactive substances produced by oxidative stress can activate
resident cells in the lung, such as alveolar macrophages and
epithelial cells, and then the inflammatory cells (neutrophils and
lymphocytes) are attracted to the lung by chemotaxis and release
inflammatory mediators such as tumor necrosis factor-α (TNF-α) and
various types of interleukin (1,9). The
deterioration of COPD is accompanied by other physiological
deterioration besides the weakness of respiratory system, which
causes a heavy burden for patients (10-12).
Nowadays, the treatment of COPD includes conservative treatment
with slow effect and surgical treatment with high risk. Therefore,
it is urgent to explore the pathogenesis of COPD and find effective
and safe treatment.
It was found that there are more than 500 genes
associated with COPD from several genetic studies on COPD, most of
which are associated with oxidative stress and inflammatory
response pathway, particularly TNF-α (13). TNF-α is a type of multi-effective
inflammatory factor, which plays an important role in the
physiological processes of inflammation, cytotoxicity and immune
regulation (14). The study shows
that the concentration of TNF-α in the lung of patients with COPD
is higher compared with that of healthy people, and TNF-α is
associated with the progression of COPD (15). It was also found that the
overexpression of TNF-α can not only induce emphysema and pulmonary
fibrosis, but also decrease the muscle quality and aggravate the
physiological symptoms of patients with COPD at the end of the
period, and TNF-α inhibitors are also considered as potential
therapeutic drugs for COPD (16-20).
Furthermore, the TNF-α 308 gene variation is an independent factor
to induce COPD, which has a higher association with Asians than
other populations (13,21).
Given previous studies, it is known that TNF-α
factor plays an important role in COPD. In order to study the
molecular mechanism of the effect of TNF-α on COPD, a rat model of
COPD was established by knocking down the gene of TNF-α, followed
by a series of experiments.
Materials and methods
Animal models
Twenty-four male Wister II rats (weights, 250-290 g)
were obtained from Experimental animal center of Beijing University
of traditional Chinese Medicine (Beijing, China). All animal
experiments were approved by the Experimental Animal Welfare Ethics
Review Committee of The Affiliated Hospital of Jianghan University
(grant no. AW2019091203) and adhered to the Guide for the Care and
Use of Laboratory Animals (National Institutes of Health).
After the rats were cultured for a week to adapt to
the laboratory environment, they were fasted for 12 h and divided
into four groups (six rats in each group): i) Control group, the
rats were injected saline and treated with smoke-free condition;
ii) COPD group, the rats were injected saline and treated with
lipopolysaccharide (LPS) and cigarette smoke (CS) exposure; iii)
sh-TNF-α group, these rats were given the antibody against TNF-α
and were treated with LPS and CS exposure; iv) shRNA negative
control (sh-NC) group, the rats were injected random antibody and
were treated with LPS and CS exposure. Treatment time and method:
LPS was injected into the rats' trachea on the 1st and 14th day of
the experiment, and the rats were smoked with 5% CS of CHIENMEN
cigarettes in a 72-l closed box from the 2nd to the 28th day of the
experiment. In the sh-TNF-α group, antibodies (5 µg/g) against
TNF-α-IgG (cat. no. ab6671; Abcam) were injected via caudal vein,
and LPS (Sigma-Aldrich; Merck KGaA) was injected after 0.5-1 h, and
the sh-NC group was injected with the same amount of anti-actin
antibody (cat. no. ab11003; Abcam) and LPS with the same method.
The other groups were treated with equal volume injection of saline
(Procell Life Science & Technology Co., Ltd.). A series of
tests were carried out on all experimental animals after four weeks
of feeding.
In vivo gene transfection
shRNA targeting TNF-α, random gene, SOCS3 and TRAF1
(0.1 µg, 50 pmol) and 1.5 µl Entranster-R4000 were diluted to 25 µl
with PBS. The sequences of shRNAs was as follows: TNF-α,
5'-GTAGCCCATGTTGTAGCAA-3'; random gene (control sequence),
5'-TTCTCCGAACGTGTCACGT-3'; SOCS3, 5'-CCAAGAACCTGCGCATCC A-3'; and
TRAF-1, 5'-GCCTTCTACACTGCCAAGTAT-3'. The shRNA and Entranster-R4000
were mixed fully by an oscillator to synthesize the transfection
complex and were allowed to stand for 15 min. Then,
shRNA-TNF-α-Entraster-R4000,
shRNA-SOCS3-Entraster-R4000/shRNA-TRAF1-Entraster-R4000 and
shRNA-NC-Entraster-R4000 were respectively injected into the rats
through the caudal vein. In addition, the transfection efficiencies
were detected by western blotting (Fig. S1). The MAPK activator (Asiatic
acid; cat. no. ab143120; Abcam; 50 mg/kg) was also injected into
the caudal vein of rats. Finally, the rats were executed and a
series of studies were carried out after feeding for 3 days. Each
treatment had three independent rats as receptor.
Measurement of respiratory function in
rats
AniRes2005 lung function system (Bestlab, version
2.0, China) includes a plethysmograph chamber, a
computer-controlled small animal ventilator and pressure and vacuum
reservoirs, which were used to measure the parameters of rats' lung
function.
Firstly, the peak expiratory flow-rate (PEF) and
tidal volume of the rats was recorded by the whole-body
plethysmograph when the rats were awake. Next, the rats were
anesthetized by intraperitoneal injection of 1% pentobarbital
sodium (50 mg/kg) and a cannula was inserted into the trachea, and
then the rats were connected to a small animal ventilator. Then,
adjusting the respiratory frequency of the system ventilator to
match the rat's respiratory frequency, and recording the data with
the computer connected to the ventilator for the next analysis. The
main outcome measures were forced expiratory volume (forced
expiratory volume in 1 sec, FEV1), forced vital capacity (FVC) and
forced expiratory flow at 25% (FEF 25%). Respiratory function tests
were repeated three times at each state to obtain an average.
Cell count and enzyme activity test of
bronchoalveolar lavage fluid (BALF)
The rats were killed by using cervical dislocation
after intraperitoneally injecting with 150 mg/kg 1% phenobarbital
sodium, and the left lung was clamped to preserve architecture for
subsequent histopathological evaluation. The trachea of rats was
placed with cannula, and the right lung of rats was washed with 1.0
ml PBS for 5 times. The recovered BALF was centrifuged at 4˚C at
1500 x g for 5 min. The medium RPMI-1640 (Thermo Fisher Scientific,
Inc.) containing 10% FBS (Thermo Fisher Scientific, Inc.) was used
to resuspend the precipitate, and the cell suspension was coated on
the slide and fixed with 10% neutral formaldehyde at room
temperature for 30 min. The cells were classified and counted under
light microscope (magnification, x40) after staining with
Wright-Giemsa (solution A for 1 min and solution B for 3 min) at
room temperature. Subsequently, the level of myeloperoxidase (MPO)
was detected by ELISA kit (cat. no. SEA601Ra; Cloud-Clone Corp.)
according to the manufacturer's instructions. Three independent
experiments were conducted in each state of rats.
Western blotting
The right upper lobe of the lungs of the rats in the
experimental groups and the control groups was lysed with RIPA
buffer (10 mM Tris-HCl, pH 7.2, 0.1% SDS, 1% sodium deoxycholate,
0.15 M NaCl, 1% NP-40), and the lysates were centrifuged at 1,500 x
g for in 4˚C for 20 min. The concentration of total protein was
determined by the BCA method. The protein concentration was
measured by enzyme standard instrument at 562 nm after 30 min.
Then, 10% SDS-PAGE gel electrophoresis was performed to isolate and
transfer the protein to the PVDF membrane, and the membrane was
sealed at room temperature for 1 h with 5% skimmed milk powder.
Subsequently, the primary antibodies were incubated with the
membrane overnight at 4˚C, and the secondary antibodies were
incubated with protein for 1 h at 4˚C. Finally, the exposure and
data analysis of protein bands were carried out. Primary
antibodies, including anti-p-ERK (cat. no. ab79483; dilution,
1:1,000), p-p38 (cat. no. ab4822, dilution, 1:1,000), p38 (cat. no.
ab31828; dilution, 1:1,000), ERK (cat. no. ab17942, dilution,
1:1,000), p-JNK (cat. no. #81E11; dilution, 1:1,000), JNK (cat. no.
ab124956, dilution, 1:10,000), SOCS3 (cat. no. ab16030;
concentration, 1) and TRAF1 (cat. no. ab129279; concentration, 1
µg/ml), as well as the secondary antibodies, goat anti-rabbit IgG
H&L (HRP) (ab6721; dilution, 1:20,000); goat anti-mouse IgG
H&L (HRP) (ab6789; dilution, 1:10,000), donkey anti-goat IgG
H&L (HRP) (cat. no. ab6885, concentration, 1 µg/ml) were
purchased from Abcam. Actin was used as the internal control. Three
independent experiments were conducted in each state of rats.
Histopathological examination of
lung
The left lung of rats was fixed with 4%
paraformaldehyde at room temperature for 24 h and embedded in
paraffin. The lung tissue sections (4 µm) were stained with
hematoxylin (for 2-3 min) and eosin (for 1 min) at room
temperature. The photos were captures at the magnifications of x100
and x400 by an optical microscope, and then, the inflammatory
infiltration around alveoli or the structural damage of alveoli and
airway were observed in the photos.
Statistical analysis
The data are presented as mean ± SD. The statistical
significance of the differences among multiple groups of data was
analyzed by one-way ANOVA followed by S multiple comparisons test
in GraphPad Prism 7.0 (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of TNF-α on lung function,
inflammatory response and lung structure in rats with COPD
In order to test the effect of TNF-α on inflammatory
response and lung structure in rats with COPD, the lung function
indexes, number of inflammatory cells and structural changes in
alveoli and airway were detected in rats with COPD. After the rats
were treated with CS and LPS to establish COPD models, it was found
that the lung function indexes of the rats treated with CS and LPS,
such as FEV1, FVC, FEV1/FVC, FEF25, PEF, VE (minute ventilation at
rest) and FEV0.3 were significantly lower compared with those of
the normal rats (Fig. 1A-G).
However, the decrease in lung function caused by LPS + CS was
reversed by knocking down TNF-α (Fig.
1A-G). For inflammatory cells, the rats with COPD had a higher
level of leukocyte and neutrophils, but the number of leukocytes
and neutrophils were decreased after TNF-α knockdown (Fig. 1H-I). In addition, the number of
alveolar macrophages, lymphocytes and monocytes of the COPD rats
were decreased compared with those in normal rats, and increased
following knockdown of TNF-α (Fig.
1J-L). The high level of MPO in COPD rats was decreased by
TNF-α knockdown (Fig. 1M). Compared
with healthy rats, COPD rats showed obvious inflammatory cell
infiltration around the airway, the cystic expansion in the
disordered alveolar structure increase of pulmonary bullae
(Fig. 1N), indicating that the COPD
models were established successfully. However, the structural and
functional damage of lungs in COPD rats was decreased by the
knockdown of TNF-α (Fig. 1M). All
the experimental results showed that the damage of function and
structure of lungs in COPD rats caused by CS + LPS could be
alleviated by TNF-α knockdown.
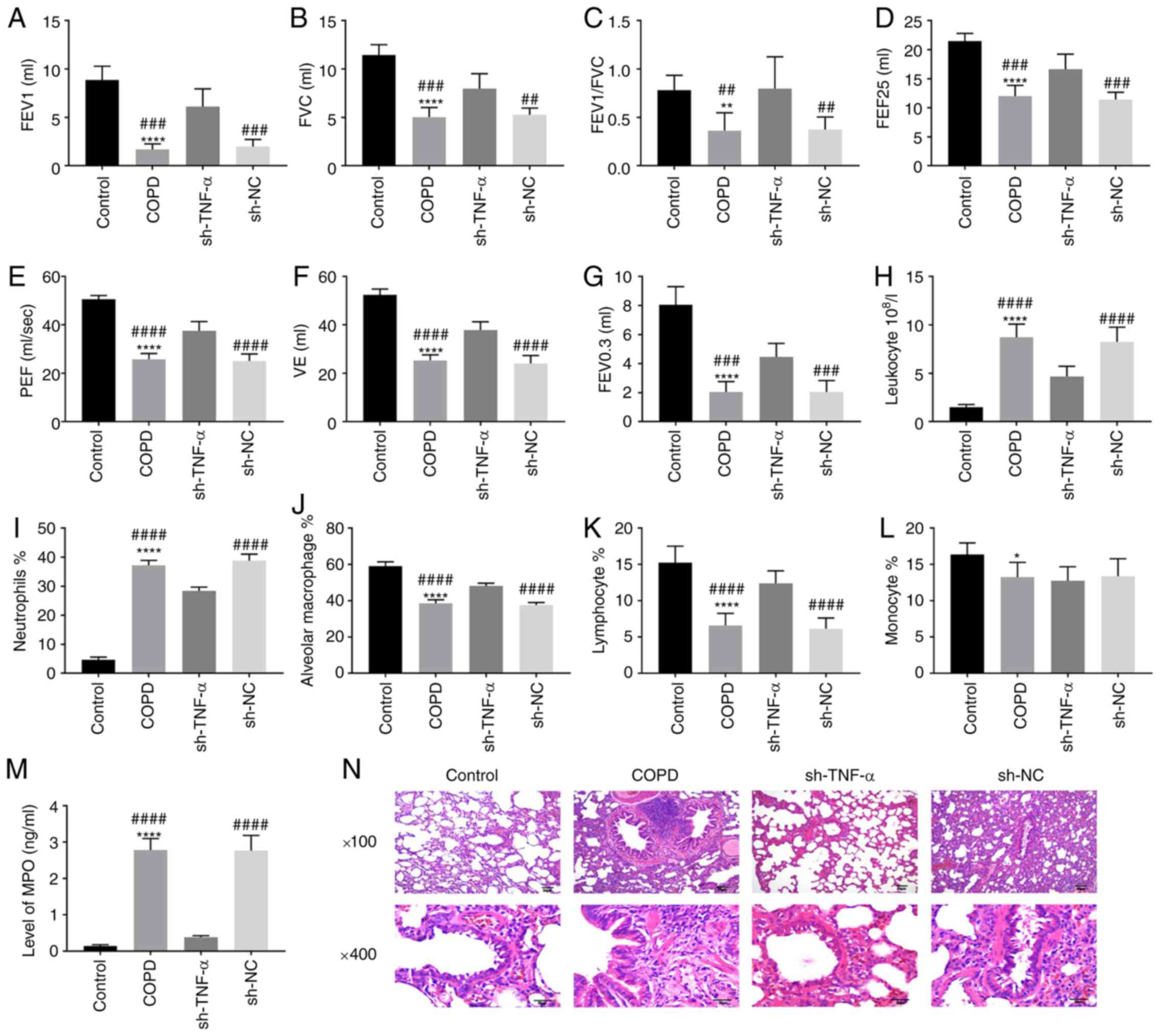 | Figure 1Effect of TNF-α on inflammatory
response and lung structure in rats with COPD. The rats were
treated with LPS + CS and transfected with sh-TNF-α. (A-G) The
pulmonary functions of rats, including (A) FEV1, (B) FVC, (C)
FEV1/FVC, (D) FEF 25, (E) PEF, (F) VE and (G) FEV0.3, were measured
by the plethysmograph. (H-L) BALF was obtained by fiberoptic
bronchoscopy and normal saline, and the cells and enzyme in BALF
such as (H) leukocyte, (I) neutrophils, (J) alveolar macrophage,
(K) lymphocyte and (L) monocyte were detected. (M) The level of MPO
in BALF was detected by enzyme-linked immunosorbent assay. (N) The
lung tissues of rats were sectioned and stained with hematoxylin
and eosin to observe the characteristics of alveoli and airway.
*P<0.05; **P<0.01;
****P<0.0001, vs. control group.
##P<0.01; ###P<0.001;
####P<0.0001, vs. sh-TNF-α group/sh-RNA group. TNF,
tumor necrosis factor; LPS, lipopolysaccharide; COPD, chronic
obstructive pulmonary disease; CS, cigarette smoke; FEV, forced
expiratory volume; FVC, forced vital capacity; FEF, forced
expiratory flow; PEF, peak expiratory flow; VE, minute ventilation
at rest; BALF, bronchoalveolar lavage fluid; MPO, myeloperoxidase;
shRNA, short interfering RNA; NC, negative control. |
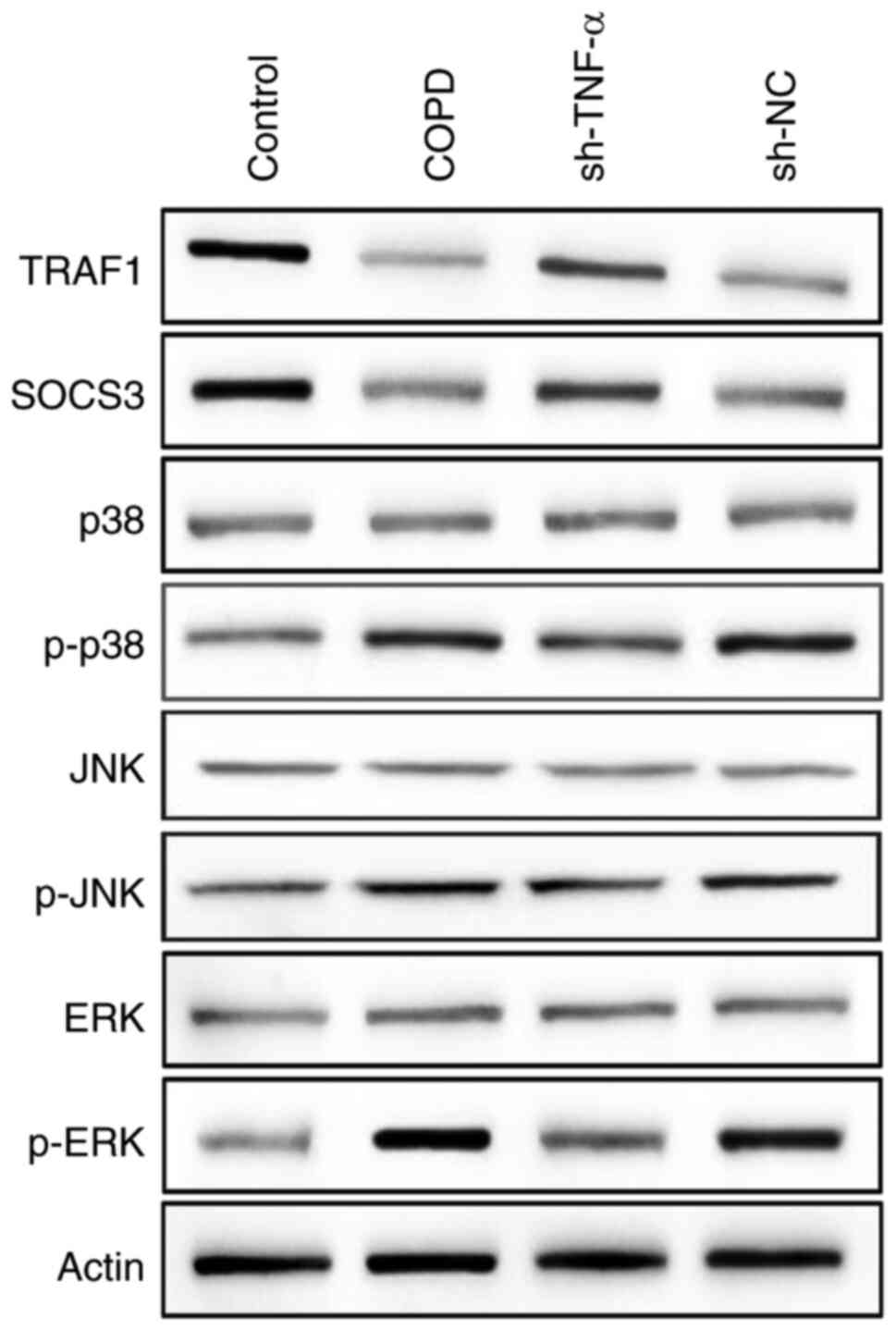
Underlying molecular mechanism of
TNF-α in COPD
TNF-α can initiate a series of molecular cascade
reaction, including the activation of the MAPK pathway and some
downstream signaling molecules, named as TNF signaling pathway.
However, the mechanism of TNF signaling pathway in COPD is not yet
well understood. To explore this issue, the Kyoto Encyclopedia of
Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/kegg2.html) was used to
identify TNF signaling pathway (Fig.
S2), and the MAPK signaling pathway was chosen as well as
downstream signaling molecules (TRAF1 and SOCS3) to investigate the
underlying molecular mechanism. Western blotting was used to detect
the changes in the expression level of TRAF1, SOCS3, as well as
major proteins in the MAPK pathway in COPD rats transfected with or
without TNF-α shRNA (sh-TNF-α). The results of western blotting
indicated that the expression of p-ERK, p-p38 and p-JNK was
increased in rats with COPD, and it was decreased after COPD model
rats were transfected with sh-TNF-α (Fig. 2). However, on the contrary, the
expression of SOCS3 and TRAF1 was decreased in the COPD rats and
increased in TNF-α knockdown rats (Fig.
2). These results indicated that TNF-α knockdown could inhibit
the activity of the MAPK pathway and upregulate the expression of
SOCS3 and TRAF1.
Effect of MAPK pathway on inflammatory
response and lung structure in rats with COPD
To detect whether the inhibition of the MAPK pathway
activity caused by TNF-α knockdown could affect the inflammatory
response and lung structure in COPD rats, western blotting,
pulmonary function test, cell count of BALF and histopathological
examination of the lungs were conducted. The results of western
blotting showed that the key proteins in MAPK pathway such as
p-ERK, p-JNK and p-p38 were decreased by the transfection of
sh-TNF-α but increased after the introduction of the activator of
MAPK pathway, while ERK and p38 had no change (Fig. 3A). The results of pulmonary function
test demonstrated that the increase in all pulmonary function
indexes induced by TNF-α knockdown was weakened after the
introduction of the activator of MAPK pathway (Fig. 3B-H). The results of cell
classification and counting showed that the decrease in the number
of leukocytes and neutrophils induced by sh-TNF-α transfection was
reversed by adding the activator of MAPK pathway (Fig. 3I-J), and the tendency of alveolar
macrophages and lymphocytes was contrary (Fig. 3K-L). And the reduction of the number
of monocytes caused by TNF-α knockdown had no significant change
after the introduction of the MAPK pathway activator (Fig. 3M). In addition, the decline in MPO
caused by knocking down the TNF-α was reversed by activating MAPK
pathway (Fig. 3N). The results of
histopathology showed that the alleviation of inflammatory
infiltration around alveoli or damage of alveoli and airway
structures caused by sh-TNF-α transfection was deteriorated by
adding the activator of MAPK pathway (Fig. 3O). Therefore, the disease
progression of COPD could be accelerated by activating the MAPK
pathway, and TNF-α could affect the inflammatory response and lung
structure in COPD through regulating MAPK pathway.
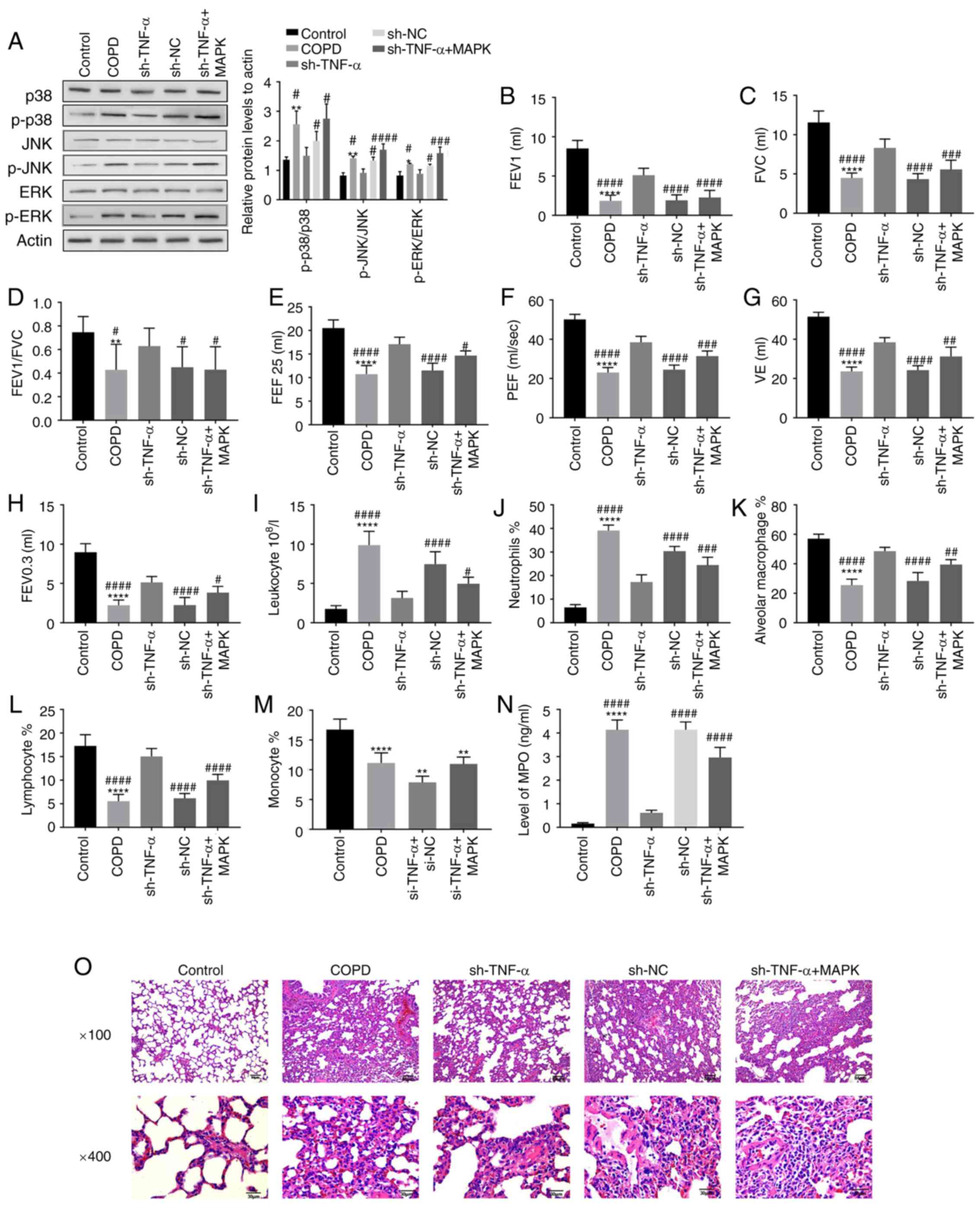 | Figure 3Effect of MAPK pathway on
inflammatory response and lung structure in rats with COPD. The
rats were treated with the activator of MAPK pathway. (A) The
expression of proteins associated with the MAPK pathway in rats
with various treatments was detected by western blotting. (B-H) The
changes in pulmonary function such as (B) FEV1, (C) FVC, (D)
FEV1/FVC, (E) FEF 25, (F) PEF, (G) VE and (H) FEV 0.3 of rats were
detected by using a plethysmograph after adding the activator of
MAPK pathway. (I-M) The number of pulmonary inflammatory cells,
such as (I) leukocyte, (J) neutrophils, (K) alveolar macrophage,
(L) lymphocyte and (M) monocyte in the lungs of rats was measured
by classifying and counting cells of BALF. (N) In addition, the
level of MPO in BALF was detected by enzyme-linked immunosorbent
assay. (O) The lung tissue of rats was sectioned and stained with
hematoxylin and eosin to observe the changes in alveoli and airway
of rats after the activator of MAPK pathway was added.
*P<0.05, **P<0.01,
****P<0.0001, vs. Control group;
#P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001, vs. sh-TNF-α
group/sh-RNA group. COPD, chronic obstructive pulmonary disease;
FEV, forced expiratory volume; FVC, forced vital capacity; FEF,
forced expiratory flow; PEF, peak expiratory flow; VE, minute
ventilation at rest; BALF, bronchoalveolar lavage fluid;
p-phosphorylated; sh-, short interfering RNA; NC, negative
control. |
Effect of SOCS3 and TRAF1 on
inflammatory response and lung structure in rats with COPD
To detect whether the increased expression of SOCS3
and TRAF1 induced by TNF-α knockdown could affect the inflammatory
response and lung structure in COPD rats, these two molecules were
knocked down and the pulmonary function, the number of inflammatory
cells and the pathological changes in the lung were detected.
Western blotting showed that the expression of SOCS3 and TRAF1
decreased after the rats were treated with LPS + CS, but this trend
was reversed after TNF-α was knocked down, and their expression was
weakened after the rats co-transfected with sh-TNF-α and sh-SOCS3
or sh-TRAF1 (Fig. 4A). The
increased values of pulmonary function of COPD model rats caused by
sh-TNF-α was weakened after the introduction of sh-SOCS3 or
sh-TRAF1 (Fig. 4B-H). The cell
classification and counts of BALF from COPD rats indicated that the
decrease in the number of leukocytes and neutrophils by sh-TNF-α
was reversed by sh-SOCS3 and sh-TRAF1 (Fig. 4I-J). However, the increase in
alveolar macrophages and lymphocytes induced by sh-TNF-α was
decreased by sh-SOCS3 and sh-TRAF1 (Fig. 4K-L), and the number of monocytes had
no significant change (Fig. 4M). In
addition, the decline of MPO in COPD rats caused by knocking down
TNF-α was reversed by transfecting with sh-SOCS3/TRAF (Fig. 4N). It was observed through the
histopathology that knockdown of SOCS3 and TRAF1 could aggravate
the damage of alveoli and airway structure and the inflammatory
infiltration around alveoli (Fig.
4O). All results showed that the condition of COPD would be
aggravated by knocking down SOCS3 and TRAF1, and TNF-α knockdown
could delay the progression of COPD through up-regulating the
expression of SOCS3 and TRAF1.
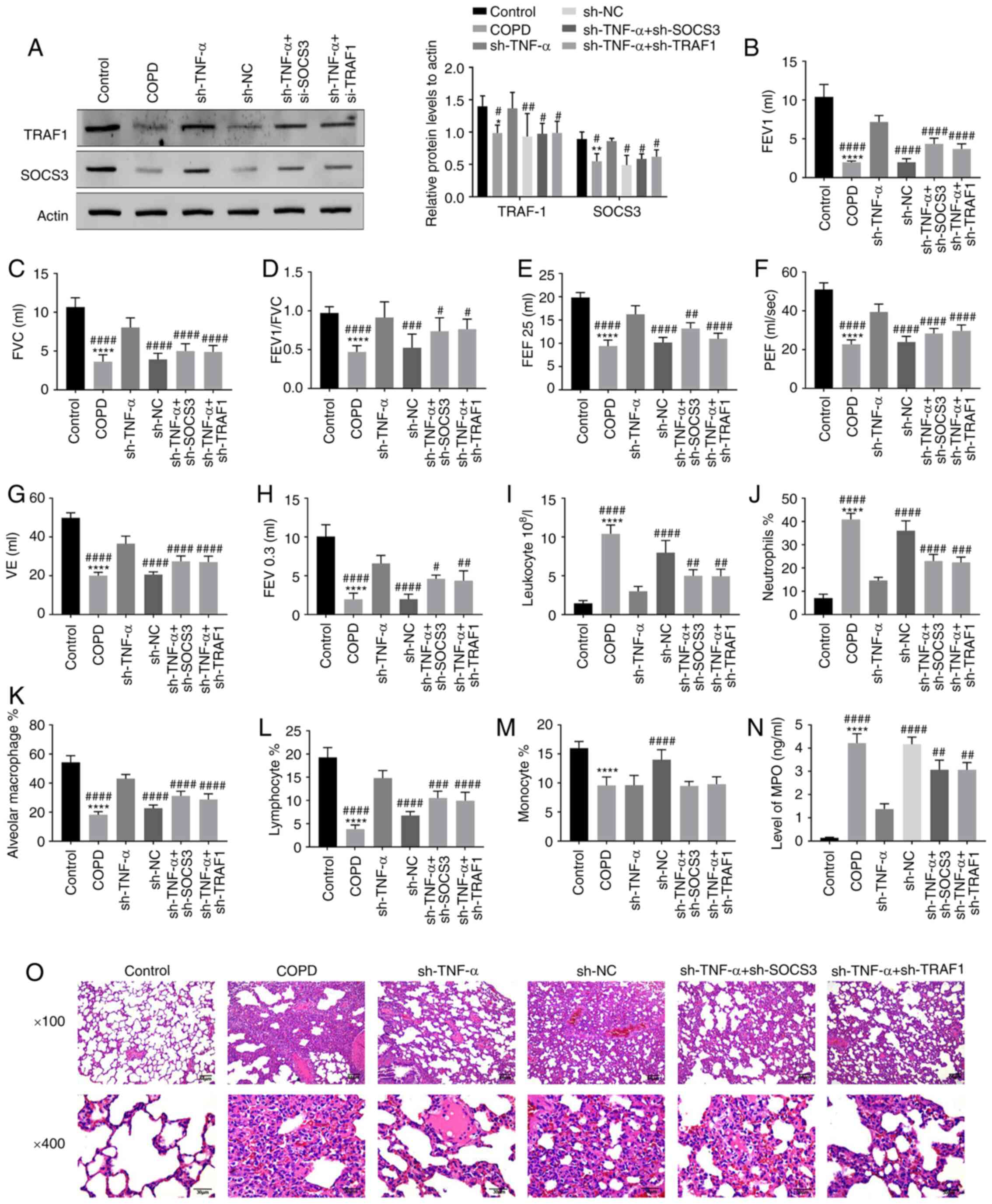 | Figure 4Effect of SOCS3 and TRAF1 on
inflammatory response and lung structure in rats with COPD. The
rats were transfected with sh-SOCS3 and sh-TRAF1. (A) The
transfection efficiency of shRNA and the expression of SOCS3/TRAF1
in rats were detected by western blotting. (B-G) The pulmonary
function of the rats, containing (B) FEV1, (C) FVC, (D) FEV1/FVC,
(E) FEF 25%, (F) PEF, (G) VE and (H) FEV0.3, were detected with a
plethysmograph. (I-M) The number of inflammatory cells such as (I)
leukocyte, (J) neutrophils, (K) alveolar macrophage, (L) lymphocyte
and (M) monocyte in the lung of rats was measured by collecting the
BALF and counting the cells. (N) The level of MPO in BALF was
detected by enzyme-linked immunosorbent assay. (O) The lung tissue
of rats was sectioned and stained with hematoxylin and eosin to
observe for changes in alveoli and airway after SOCS3 and TRAF1
were knocked down. *P<0.05, **P<0.01,
****P<0.0001, vs. Control group;
#P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001, vs. sh-TNF-α
group/sh-RNA group. COPD, chronic obstructive pulmonary disease;
FEV, forced expiratory volume; FVC, forced vital capacity; FEF,
forced expiratory flow; PEF, peak expiratory flow; VE, minute
ventilation at rest; BALF, bronchoalveolar lavage fluid; sh-, short
interfering RNA; NC, negative control. |
Discussion
The present study found that CS + LPS exposure
caused COPD in rats, and the symptoms include reduced lung
function, increased inflammatory cells and the structure of alveoli
and airway was disordered; but these symptoms could be relieved by
TNF-α knockdown. Furthermore, TNF-α knockdown could delay the
progression of COPD through regulating the activity of the MAPK
pathway and the expression of SOCS3 and TRAF1.
COPD is an incurable but preventable lung disease
characterized by persistent respiratory symptoms and airflow
restriction caused by bronchitis, emphysema and apoptosis of
alveolar epithelial cells (22-24).
The diagnosis of COPD requires vital capacity measurement
(FEV1/FVC), which is at risk of over-diagnosis in older people or
under-diagnosis in younger people (25). The treatment of COPD can be divided
into conservative treatment and surgical treatment: The
conservative treatment mainly depends on bronchodilator,
anti-inflammatory agents and long-term oxygen therapy with a long
cycle and slow effect; the surgical treatment includes lung volume
reduction and lung transplant with high risk and high cost
(25). Therefore, we urgently need
a new diagnosis and treatment methods of COPD.
Overexpression of TNF-α in the lung can cause
oxidative stress and neutrophil inflammatory infiltration (4). Yao et al (26) indicated that the expression of TNF-α
is closely associated with the occurrence and development of COPD,
suggesting that TNF-α may be a potential biomarker of lung function
and inflammation in patients with COPD. TNF-α-238 G/A is associated
with airway remodeling, and its combination with TGF-β1 can
aggravate the severity of airflow limitation in patients with COPD
(27). However, the study on the
molecular mechanism of TNF-α in COPD was is insufficient, and so
the present study focused on the molecular mechanism. A COPD model
was successfully constructed, and found that TNF-α knockdown could
improve the decrease in lung function and inflammatory damage of
lung structure.
To explore the specific molecular mechanism of TNF-α
in COPD, the KEGG database was used to identify TNF signaling
pathway, and the MAPK signaling pathway, as well as downstream
signaling molecules (TRAF1 and SOCS3), were chosen to investigate
the underlying molecular mechanism. MAPKs pathways include
p38-MAPK, ERK1/2, JNK1/2, and MAPKs are the most common
inflammatory signaling pathway of TNF-α to regulate diseases
(28). For example, Chen et
al (29) found that the
inhibitor of p38-MAPK can significantly alleviate the apoptosis
induced by TNF-α. Studies also demonstrate that MAPK inhibitors
have great potential in the treatment of chronic inflammatory
diseases (30,31). For example, inhibition of p38-MAPK
can decrease the expression level of cytokines in the lung and
blood cells of COPD, suggesting that MAPK inhibitors play a role in
local and systemic inflammation (32). In agreement with previous reports,
the present study also found that the activation of MAPK pathway
could make the pathological manifestations of COPD more serious,
whilst TNF-α knockdown retarded the process of COPD.
The existing literature shows that the expression of
SOCS3 is positively correlated with TNF-α, and that SOCS3 promotes
apoptosis (33,34). However, other studies have found
that SOCS3 were transiently induced by TNF-α but inhibited over
time, therefore, it is speculated that the activation of TNF
pathway is inhibited by the combination of SOCS3 and TRAF1, thus
reverse inhibiting the expression of SOCS3 (35,36).
Furthermore, Mori et al (37) found that TNF-α can activate the
IL-6/STAS3 signaling pathway, and Huang et al (38) observed that the IL-6/STAT3 pathway
can negatively regulate the expression of SOCS3. In the present
study, it was found that the expression of SOCS3 was increased
after TNF-α knockdown. And Springer et al (39) also found that SOCS3 has a low
expression in COPD. Combined with previous research literature, it
was speculated that IL-6/STAT3 pathway was inhibited by TNF-α
knockdown and lost the inhibition for SOCS3, thus increasing the
expression of COCS3. Another possibility is that SOCS3 reversely
inhibits its own expression by combining with TRAF1 to inhibit the
activation of MAPK. These hypotheses need to be confirmed with
experiments, and thus are the future research directions.
Additionally, the present study also indicated that the expression
of TRAF1 was increased after TNF-α knockdown. Furthermore, the
literature shows that TRAF1 is a negative regulator of TNF
signaling pathway (40). A study
found that TRAF1 can inhibit NF-κB and JNK 1/2 pathway, and the
apoptotic induced by TNF-α can be enhanced by knocking down
TRAF1(41). Therefore, it was
postulated that TRAF1, as a reverse regulator of TNF signaling
pathway, can reverse regulate its own expression by inhibiting the
activation of MAPK pathway; this will be explored in a follow-up
study.
In conclusion, the present results indicate that
TNF-α knockdown could delay the procession of COPD. The molecular
mechanism was that TNF-α knockdown inhibited the activating of MAPK
pathway to increase the expression of downstream molecules
SOCS3/TRAF1, thus alleviating the inflammation and structural
damage of alveoli and airways. Furthermore, degenerative feedback
regulation mechanism of SOCS3 and TRAF1 in TNF signaling pathway of
COPD is the next research topic.
Supplementary Material
Transfection efficiencies of (A)
sh-TNF-α, (B) sh-TRAF-1 and (C) sh-SOCS3 by western blotting.
****P<0.0001, vs. Control group;
####P<0.0001, vs. sh-TNF-α group/sh-RNA group. TNF,
tumor necrosis factor; sh-, short interfering RNA; NC, negative
control; COPD, chronic obstructive pulmonary disease.
Signalling pathway selection. The
Kyoto Encyclopedia of Genes and Genomes database (https://www.genome.jp/kegg/kegg2.html)
was used to identify TNF signalling pathway, and the MAPK
signalling pathway as well as downstream signalling molecules
(TRAF1 and SOCS3) were chosen. TNF, tumor necrosis factor.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QF, YZY and QHM designed the experiments; YZY and
QHM carried out experiments; QF and QHM analyzed experimental
results. QF and YZY wrote the manuscript; QHM approved the
manuscript. All authors read and approved the final manuscript and
confirmed the authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experiments were approved by the
Experimental Animal Welfare Ethics Review Committee of the
Affiliated Hospital of Jianghan University (grant no. AW2019091203)
and adhered to the Guide for the Care and Use of Laboratory Animals
(National Institutes of Health).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sun X, Dong Z, Li N, Feng X, Liu Y, Li A,
Zhu X, Li C and Zhao Z: Nucleosides isolated from Ophiocordyceps
sinensis inhibit cigarette smoke extract-induced inflammation via
the SIRT1-nuclear factor-κB/p65 pathway in RAW264.7 macrophages and
in COPD mice. Int J Chron Obstruct Pulmon Dis. 13:2821–2832.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hoiland RL, Mladinov S, Barak OF, Willie
CK, Mijacika T, Stembridge M, Dujic Z and Ainslie PN: Oxygen
therapy improves cerebral oxygen delivery and neurovascular
function in hypoxaemic chronic obstructive pulmonary disease
patients. Exp Physiol. 103:1170–1177. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Kojima K, Asai K, Kubo H, Sugitani A,
Kyomoto Y, Okamoto A, Yamada K, Ijiri N, Watanabe T, Hirata K and
Kawaguchi T: Isoflavone aglycones attenuate cigarette smoke-induced
emphysema via suppression of neutrophilic inflammation in a COPD
murine model. Nutrients. 11(2023)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sun X, Feng X, Zheng D, Li A, Li C, Li S
and Zhao Z: Ergosterol attenuates cigarette smoke extract-induced
COPD by modulating inflammation, oxidative stress and apoptosis in
vitro and in vivo. Clin Sci (Lond). 133:1523–1536. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yipp BG, Petri B, Salina D, Jenne CN,
Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert
HC, et al: Infection-induced NETosis is a dynamic process involving
neutrophil multitasking in vivo. Nat Med. 18:1386–1393.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Chen L, Sun BB, Wang T, Wang X, Li JQ,
Wang HX, Zhang SF, Liu DS, Liu L, Xu D, et al: Cigarette smoke
enhances {beta}-defensin 2 expression in rat airways via nuclear
factor-{kappa}B activation. Eur Respir J. 36:638–645.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Son ES, Kim SH, Ryter SW, Yeo EJ, Kyung
SY, Kim YJ, Jeong SH, Lee CS and Park JW: Quercetogetin protects
against cigarette smoke extract-induced apoptosis in epithelial
cells by inhibiting mitophagy. Toxicol In Vitro. 48:170–178.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bhalla DK, Hirata F, Rishi AK and Gairola
CG: Cigarette smoke, inflammation, and lung injury: A mechanistic
perspective. J Toxicol Environ Health B, Criti Rev. 12:45–64.
2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Barnes PJ: New anti-inflammatory targets
for chronic obstructive pulmonary disease. Nat Rev Drug discov.
12:543–559. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Beaudin AE, Hartmann SE, Pun M and Poulin
MJ: Human cerebral blood flow control during hypoxia: Focus on
chronic pulmonary obstructive disease and obstructive sleep apnea.
J Appl Physiol (1985). 123:1350–1361. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Portegies ML, Lahousse L, Joos GF, Hofman
A, Koudstaal PJ, Stricker BH, Brusselle GG and Ikram MA: Chronic
obstructive pulmonary disease and the risk of stroke. Am J Respir
Crit Care Med. 193:251–258. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Criner GJ, Bourbeau J, Diekemper RL,
Ouellette DR, Goodridge D, Hernandez P, Curren K, Balter MS,
Bhutani M, Camp PG, et al: Prevention of acute exacerbations of
COPD: American college of chest physicians and canadian thoracic
society guideline. Chest. 147:894–942. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Salimi Asl M, Ahmadi A, Salimian J,
Shohani S, Azimzadeh Jamalkandi S and Ghanei M: TNF-α-308 G/A
variant and susceptibility to chronic obstructive pulmonary
disease: A systematic review and meta-analysis. Cytokine.
123(154763)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aggarwal BB and Natarajan K: Tumor
necrosis factors: Developments during the last decade. Eur Cytokine
Netw. 7:93–124. 1996.PubMed/NCBI
|
|
15
|
Yao Y, Zhou J, Diao X and Wang S:
Association between tumor necrosis factor-alpha and chronic
obstructive pulmonary disease: A systematic review and
meta-analysis. Ther Adv Respir Dis.
13(1753466619866096)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lundblad LK, Thompson-Figueroa J, Leclair
T, Sullivan MJ, Poynter ME, Irvin CG and Bates JH: Tumor necrosis
factor-alpha overexpression in lung disease: A single cause behind
a complex phenotype. Am J Respir Crit Care Med. 171:1363–1370.
2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Vuillemenot BR, Rodriguez JF and Hoyle GW:
Lymphoid tissue and emphysema in the lungs of transgenic mice
inducibly expressing tumor necrosis factor-alpha. Am J Respir Cell
Mol Biol. 30:438–448. 2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Churg A, Wang RD, Tai H, Wang X, Xie C and
Wright JL: Tumor necrosis factor-alpha drives 70% of cigarette
smoke-induced emphysema in the mouse. Am J Respir Crit Care Med.
170:492–498. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kochetkova EA, Nevzorova VA, Ugai LG,
Maistrovskaia YV and Massard G: The role of tumor necrosis factor
Alpha and TNF superfamily members in bone damage in patients with
end-stage chronic obstructive lung disease prior to lung
transplantation. Calcif Tissue Int. 99:578–587. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Matera MG, Calzetta L and Cazzola M:
TNF-alpha inhibitors in asthma and COPD: We must not throw the baby
out with the bath water. Pulm Pharmacol Ther. 23:121–128.
2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang S, Wang C, Xi B and Li X:
Association between the tumour necrosis factor-α-308G/A
polymorphism and chronic obstructive pulmonary disease: An update.
Respirology. 16:107–115. 2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li A, Liu Y, Zhu X, Sun X, Feng X, Li D,
Zhang J, Zhu M and Zhao Z: Protective effect of methylallyl sulfone
in the development of cigarette smoke extract-induced apoptosis in
rats and HFL-1 cells. Biochem Biophys Res Commun. 498:627–632.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vogelmeier CF, Criner GJ, Martinez FJ,
Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M,
Fabbri LM, et al: Erratum to ‘Global strategy for the diagnosis,
management, and prevention of chronic obstructive lung disease 2017
report: GOLD executive summary’ [Arch Bronconeumol.
2017;53:128-149]. Arch Bronconeumol. 53:411–412. 2017.PubMed/NCBI View Article : Google Scholar : (In English,
Spanish).
|
|
24
|
Tsai MJ, Chang WA, Jian SF, Chang KF, Sheu
CC and Kuo PL: Possible mechanisms mediating apoptosis of bronchial
epithelial cells in chronic obstructive pulmonary disease-A
next-generation sequencing approach. Pathol Res Pract.
214:1489–1496. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang IA, Brown JL, George J, Jenkins S,
McDonald CF, McDonald VM, Phillips K, Smith BJ, Zwar NA and
Dabscheck E: COPD-X australian and New Zealand guidelines for the
diagnosis and management of chronic obstructive pulmonary disease:
2017 update. Med J Aust. 207:436–442. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yao Y, Zhou J, Diao X and Wang S:
Association between tumor necrosis factor-α and chronic obstructive
pulmonary disease: A systematic review and meta-analysis. Ther Adv
Respir Dis. 13(1753466619866096)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chiang CH, Chuang CH and Liu SL:
Transforming growth factor-β1 and tumor necrosis factor-α are
associated with clinical severity and airflow limitation of COPD in
an additive manner. Lung. 192:95–102. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Plomgaard P, Bouzakri K, Krogh-Madsen R,
Mittendorfer B, Zierath JR and Pedersen BK: Tumor necrosis
factor-alpha induces skeletal muscle insulin resistance in healthy
human subjects via inhibition of Akt substrate 160 phosphorylation.
Diabetes. 54:2939–2945. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen NN, Wei F, Wang L, Cui S, Wan Y and
Liu S: Tumor necrosis factor alpha induces neural stem cell
apoptosis through activating p38 MAPK pathway. Neurochem Res.
41:3052–3062. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gaffey K, Reynolds S, Plumb J, Kaur M and
Singh D: Increased phosphorylated p38 mitogen-activated protein
kinase in COPD lungs. Eur Respir J. 42:28–41. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bosquet A, Girona J, Guaita-Esteruelas S,
Heras M, Saavedra-García P, Martínez-Micaelo N, Masana L and
Rodríguez-Calvo R: FABP4 inhibitor BMS309403 decreases
saturated-fatty-acid-induced endoplasmic reticulum
stress-associated inflammation in skeletal muscle by reducing p38
MAPK activation. Biochim Biophys Acta Mol Cell Biol Lipids.
1863:604–613. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Armstrong J, Harbron C, Lea S, Booth G,
Cadden P, Wreggett KA and Singh D: Synergistic effects of p38
mitogen-activated protein kinase inhibition with a corticosteroid
in alveolar macrophages from patients with chronic obstructive
pulmonary disease. J Pharmacol Exp Ther. 338:732–740.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhao X, Qi R, Sun C and Xie Y: Silencing
SOCS3 could inhibit TNF-α induced apoptosis in 3T3-L1 and mouse
preadipocytes. Mol Biol Rep. 39:8853–8860. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chhabra JK, Chattopadhyay B and Paul BN:
SOCS3 dictates the transition of divergent time-phased events in
granulocyte TNF-α signaling. Cell Mol Immunol. 11:105–106.
2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Collins AS, Ahmed S, Napoletano S,
Schroeder M, Johnston JA, Hegarty JE, O'Farrelly C and Stevenson
NJ: Hepatitis C virus (HCV)-induced suppressor of cytokine
signaling (SOCS) 3 regulates proinflammatory TNF-α responses. J
Leukoc Biol. 96:255–263. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhu X, Bai J, Liu P, Wang X and Jiang P:
Suppressor of cytokine signaling 3 plays an important role in
porcine circovirus type 2 subclinical infection by downregulating
proinflammatory responses. Sci Rep. 6(32538)2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mori T, Miyamoto T, Yoshida H, Asakawa M,
Kawasumi M, Kobayashi T, Morioka H, Chiba K, Toyama Y and Yoshimura
A: IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in
mediating inflammatory cytokines and RANKL expression in
inflammatory arthritis. Int Immunol. 23:701–712. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Huang L, Hu B, Ni J, Wu J, Jiang W, Chen
C, Yang L, Zeng Y, Wan R, Hu G and Wang X: Transcriptional
repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1
promotes pancreatic cancer growth and metastasis. J Exp Clin Cancer
Res. 35(27)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Springer J, Scholz FR, Peiser C, Dinh QT,
Fischer A, Quarcoo D and Groneberg DA: Transcriptional
down-regulation of suppressor of cytokine signaling (SOCS)-3 in
chronic obstructive pulmonary disease. J Occup Med Toxicol.
8(29)2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tsitsikov EN, Laouini D, Dunn IF,
Sannikova TY, Davidson L, Alt FW and Geha RS: TRAF1 is a negative
regulator of TNF signaling. enhanced TNF signaling in
TRAF1-deficient mice. Immunity. 15:647–657. 2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wen X, Wang B, Feng T, Yuan W, Zhou J and
Fang T: TNF receptor-associated factor 1 as a biomarker for
assessment of non-small cell lung cancer metastasis and overall
survival. Clin Respir J. 12:2197–2203. 2018.PubMed/NCBI View Article : Google Scholar
|