Introduction
Keratosis pilaris (KP) is a common skin disorder
that can also be associated with less common variants and rare
subtypes, including keratosis pilaris rubra, erythromelanosis
follicularis faciei et colli and the spectrum of keratosis pilaris
atrophicans (1). The atrophicans
subtypes, which are even more uncommon, include keratosis pilaris
atrophicans faciei (KPAF), atrophoderma vermiculatum, and keratosis
follicularis spinulosa decalvans (1). KPAF, also known as ulerythema
ophryogenes, is a hereditary disorder characterized by altered
follicular keratinization and inflammation, which leads to
subsequent atrophy (2). Clinically,
it presents with follicular, horny papules surrounded by an
erythematous halo of the cheeks, forehead, chin and eyebrows, and
it is followed by a gradual loss of hair. These cutaneous
manifestations appear sequentially: Erythematous follicular papules
of the face are usually the first sign, followed by a gradual hair
loss on the lateral part of the eyebrows and finally follicular
scar-like follicular atrophy (3).
KPAF occurs sporadically; however, autosomal dominant and autosomal
recessive inheritance have been described (3). In some cases, an autosomal recessive
mutation in the desmoglein 4 (DSG4) gene, as well as the LDL
receptor related protein 1 (LRP1) gene, have been involved
(4,5). The onset is as early as a few months
after birth, with erythema and keratotic follicular papules
affecting the lateral third of the eyebrows (6); however, KPAF is mainly diagnosed in
children and adolescents, and it can persist through adulthood.
Most of the subjects have concomitant KP of the extremities and
trunk. Given the clinical variability of the particular cutaneous
signs, insufficient diagnosis and misdiagnosis are frequent. In the
majority of cases, the first clinical signs appeared on the face
and are attributed to other diseases, thus making the real
incidence of the disease unknown (7).
KPAF is a rare hereditary disorder with well-defined
clinical features, but with variable evolution. At present, the
natural progression of the disease is poorly understood, which
makes a correct diagnosis highly unlikely. Thus, the aim of this
observational, descriptive, retrospective study was to describe the
clinical characteristics of KPAF in patients encountered in daily
practice, in order to find common characteristics that may aid in
the earlier recognition of the disease.
Patients and methods
Cases
To identify common characteristics that may aid in
the earlier recognition of KPAF, an observational, descriptive,
retrospective clinical case series study on 14 patients with KPAF
was performed at the Dermatology Clinic of Târgu-Mures, Romania,
between January 2000 and December 2020. In this 20-year period, 14
out of 76,440 patients were diagnosed with KPAF. Mean age at
diagnosis was 17.04 years, with the youngest patient being
diagnosed at the age of 3 years and the eldest at the age of 22
years. The sex ratio was M/F=4/1. The diagnosis was established
through clinical examination. Patients were residents of the urban
area and were members of wealthy families.
One investigator evaluated the patients and
collected data and another investigator, who was blinded to
clinical cases and treatment choices, performed outcome assessment.
Data acquisition was performed by clinical examination and
telephone interview. The patients were followed-up for four years
after KPAF diagnosis. Limitations of the current study lie in the
relatively low number of patients and in confirmation bias in
reporting, since clinical assessment, diagnosis and treatment were
performed by a single investigator. Mild form of facial erythema
was defined as few papules and mild erythema, whereas severe form
was defined as predominant papules and severe erythema.
Results
In this 20-year period, 14 out of 76,440 patients
were diagnosed with KPAF. Mean age at diagnosis was 17.04 years,
with the youngest patient being diagnosed at the age of 3 years and
the eldest at the age of 22 years. The sex ratio was M/F=4/1. The
onset of clinical symptoms appeared at a mean age of 4.85 years
(2-12 years) and the time between the appearance of the first
clinical symptoms to KPAF diagnosis was 9.21 years (1-16
years).
The majority of the 14 patients (71.43%) presented
with keratosis pilaris (KP) involving only the upper limb as the
first clinical sign; the remaining 28.57% had both upper and lower
limb involvement. The second clinical manifestation, erythema of
the face, appeared at a mean age of 7.21 years (2-16 years), after
a mean period of 3.66 years (1-7 years) since the onset of KP of
the extremities, in 9 out of 14 patients; for the remaining 5
patients, both KP and facial erythema appeared concomitantly. Of
the 14 patients, 7 patients had mild forms and 7 had severe forms.
The third clinical manifestation, keratotic papules on the face,
appeared at a mean age of 8.35 years (3-14 years), after a mean
period of 1.75 years (1-5 years) after facial erythema and 3.69
years (1-8 years) after KP of the extremities, in 13 out of 14
cases. In one case, though, keratotic papules on the face appeared
concomitantly with KP and preceded the appearance of facial
erythema. In addition, 8 patients presented skin atrophy. Loss of
hair in the lateral margins of the eyebrows appeared at a mean age
of 14 years (3-22 years), after a mean period of 9.14 years (1-16
years) since the onset of the first symptom, KP of the extremities,
6.78 years (1-16 years) after facial erythema and 6.07 years (1-15
years) after keratotic papules on the face, for 13 out of 14
patients. For one of the patients, loss of hair in the lateral
margins of the eyebrows was concomitant with the appearance of
keratotic papules on the face and these symptoms appeared one year
after the onset of KP and facial erythema; this case was diagnosed
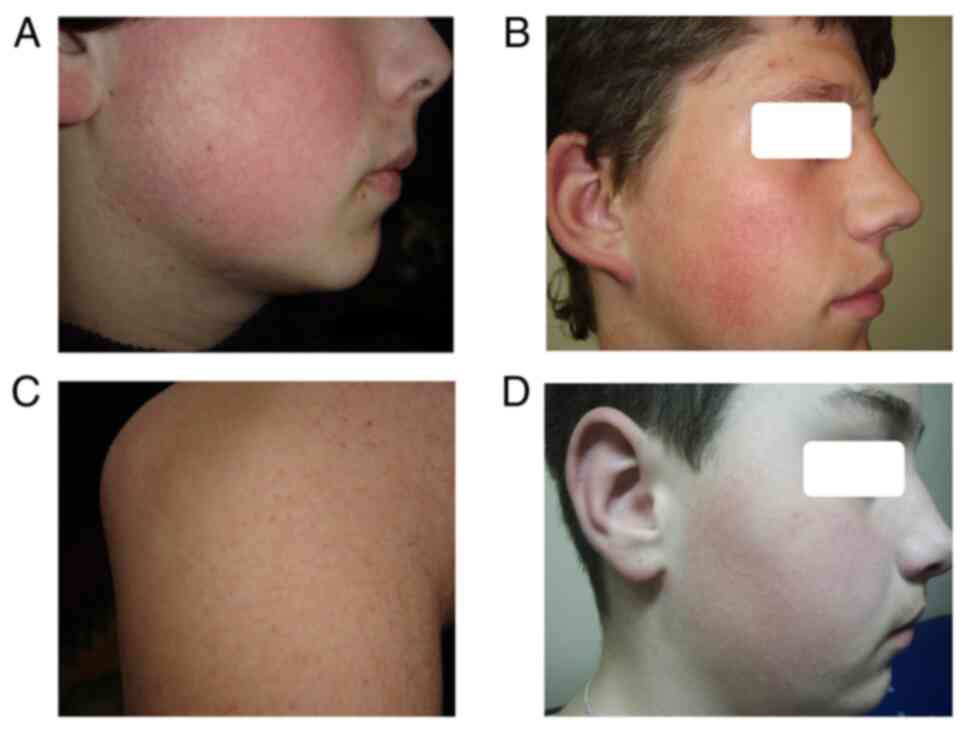
as KPAF at the age of 3 years (Fig.
1). In all of these cases, KPAF was not the first diagnosis.
Habitual erythema, keratosis pilaris, contact dermatitis, comedonal
acne, infantile acne and atopic dermatitis were the misdiagnoses.
Furthermore, 78.57% of the patients had associated xerosis cutis,
35.71% also had multiple mole syndrome, 14.28% had acne, 7.14% had
contact dermatitis and 7.14% had Laugier-Hunziker syndrome
(Table I). Topical treatments were
recommended: emollients, keratolytics (containing lactic acid,
salicylic acid, or urea), benzoyl peroxide, with mild, temporary
improvement.
 | Table IAnamnestic and clinical findings. |
Table I
Anamnestic and clinical findings.
| Case | Age at diagnosis
(years) | Sex | Onset of clinical
signs (age); KP/location | Onset of clinical
signs (age); erythema of the face/severity | Onset of clinical
signs (age); keratotic papules on the face | Onset of clinical
signs (age); loss of hair on eyebrow area/atrophy yes/no | Previous
diagnosis | Associated
diseases |
|---|
| 1 | 16 | Male | 6 years; Upper and
lower limbs | 10 years; Severe | 11 years | 16 years; No | Habitual erythema;
KP; Contact dermatitis; Comedonal acne | Xerosis cutis |
| 2 | 17 | Male | 8 years; Upper
limb | 13 years; Severe | 14 years | 17 years; Yes | Habitual erythema;
KP; Comedonal acne | Xerosis cutis |
| 3 | 11 | Male | 7 years; Upper
limb | 8 years; Mild | 10 years | 11 years; No | Atopic dermatitis;
Comedonal acne | Xerosis cutis
Multiple mole syndrome |
| 4 | 22 | Female | 8 years; Upper
limb | 15 years; Mild | 14 years | 22 years; Yes | Habitual erythema;
KP; Contact dermatitis; Comedonal acne | Xerosis cutis |
| 5 | 19 | Female | 12 years; Upper
limb | 16 years; Mild | 12 years | 19 years; Yes | KPAF (at 18 years);
KP | Xerosis cutis, mild
acne |
| 6 | 7 | Male | 3 years; Upper
limb | 3 years; Mild | 5 years | 7 years; No | KP; Habitual
erythema; Atopic dermatitis | Xerosis cutis
Infantile acne |
| 7 | 14 | Male | 4 years; Upper and
lower limbs | 8 years; Severe | 10 years | 13 years; No | KP; Habitual
erythema; Atopic dermatitis | Xerosis cutis
Multiple mole syndrome |
| 8 | 9 | Male | 3 years; Upper
limb | 3 years; Severe | 5 years | 9 years; No | KP; Atopic
dermatitis | Contact
dermatitis |
| 9 | 3 | Male | 2 years; Upper
limb | 2 years; Mild | 3 years; | 3 years; No | Atopic dermatitis;
Infantile acne | Xerosis cutis |
| 10 | 15 | Female | 4 years; Upper
limb | 7 years; Mild | 12 years | 15 years; No | Atopic dermatitis;
Comedonal acne | Xerosis cutis
Multiple mole syndrome |
| 11 | 16 | Male | 3 years; Upper and
lower limbs | 3 years; Severe | 5 years | 16 years; Yes | Atopic dermatitis;
Comedonal acne; Contact dermatitis | Multiple mole
syndrome |
| 12 | 19 | Male | 3 years; Upper and
lower limbs | 4 years; Severe | 5 years | 19 years; Yes | Habitual erythema;
Atopic dermatitis | Xerosis cutis |
| 13 | 10 | Male | 2 years; Upper
limb | 6 years; Mild | 7 years | 10 years; No | Atopic dermatitis;
Infantile acne | Xerosis cutis |
| 14 | 19 | Male | 3 years; Upper
limb | 3 years; Severe | 4 years | 1 year; Yes | Atopic dermatitis;
Comedonal acne | Multiple mole
syndrome; Laugier-Hunziker syndrome |
Discussion
Keratosis pilaris atrophicans faciei (KPAF) is an
early-onset disease with an elusive diagnosis, due to the long-term
progression of the symptoms. Results of the present study revealed
that from the appearance of the first symptom to a correct
diagnosis 9.21 years had passed, with a mean age at diagnosis of
17.04 years. Considering that the first clinical signs appeared as
early as the age of 1 year, misdiagnosis delayed a correct
assessment of the clinical image. The majority of the cases
(64.28%) had a sequential progression; first, they presented KP
involving the limbs, then, after a mean of 3.66 years facial
erythema appeared; the third symptom, keratotic papules on the
face, occurred after a mean of 1.75 years and 6.07 years later loss
of hair appeared on the lateral margin of the eyebrows. Patients
included in the present study also had associated xerosis cutis,
multiple mole syndrome, acne, contact dermatitis and
Laugier-Hunziker syndrome. In the literature, more severe
afflictions have been described in association with KPAF including
Noonan syndrome (8), Zouboulis
syndrome (2), Cornelia de la Lange
syndrome (6), Rubinstein-Taybi
syndrome (9), and woolly hair
syndrome (10). Furthermore, Wang
and Orlow summarized KP associations with various
neuro-cardio-facial-cutaneous syndromes, ectodermal dysplasias, and
neuro developmental disorders, as well as KP-induced drug reactions
(11). Differential diagnosis
includes a large spectrum of diseases that affect children and
adolescents, and which progresses with facial erythema, from
habitual erythema, rosacea, atopic dermatitis, comedonal acne, to
KP subtypes, including keratosis pilaris rubra faciei,
erythromelanosis follicularis faciei et colli, atrophoderma
vermiculatum, Darier-White disease, and pityriasis folliculorum
(12-14).
An early diagnosis allows for a targeted treatment
option. The patients in the current study were recommended to
receive emollients, benzoyl peroxide and keratolytics containing
lactic acid, salicylic acid, or urea, with only mild temporary
improvement. Other therapies have been described, with encouraging
results including pulsed dye laser (PDL) (15) and intense pulsed laser (16). Additionally, Apalla et al
described a case of atrophoderma vermiculatum which responded to
systemic isotretinoin (17).
Furthermore, Wang et al summarized various topical
treatments with reasonably adequate effect in patients with KP.
Thiese included lactic acid, salicylic acid, retinoids, aquaphor,
fractional prickle coral calcium, spray-on nitrosomonas eutropha
mist, chlorine dioxide complex cleanser, as well as various lasers:
PDL, 532-nm potassium titanyl phosphate laser (KTPL), alexandrite
laser, long-pulsed diode laser, Q-switched Nd:YAG laser, and
fractional carbon dioxide laser (11,18,19).
There are no available treatments to prevent or reduce the atrophy
in KPAF.
Limitations of the current study lie in the
relatively low number of patients and in confirmation bias in
reporting, since clinical assessment, diagnosis and treatment were
performed by a single investigator.
In summary, KP is a very common skin condition,
often dismissed as a cosmetic matter, which frequently leads to
missed diagnoses of associated diseases, hereditary syndromes or
even adverse events of certain medications. Evidence of disease
progression, associations, as well as efficacious treatment
measures is lacking. Further case series investigating the
chronology of symptoms, as well as the efficacy, tolerability and
recurrence rates are necessary in deciphering KPAF.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GLF was responsible for the clinical management of
the cases, the evaluation and analysis of data, and writing of the
manuscript. LF was responsible for corrections and preparation of
the manuscript. NN, MD and BV were responsible for data search and
revision of the manuscript. IB contributed to writing the
manuscript. The final version of the article has been read and
approved by all authors. GLF and NN are responsible for confirming
the authenticity of the raw data.
Ethics approval and consent to
participate
Written informed consent from the patients was
obtained.
Patient consent for publication
Written informed consent from the patients was
obtained.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
GLF is an Associate Professor of Dermatology,
Dermatology Department, Dermatology Clinic, ‘George Emil Palade’
University of Medicine, Pharmacy, Science and Technology, Târgu
Mureş, Romania.
References
|
1
|
Egan O, Luther EM and Oakley A: Keratosis
pilaris atrophicans. Edited Gus Mitchel, 2020 https://dermnetnz.org/topics/keratosis-pilaris-atrophicans-faciei/.
|
|
2
|
Liakou AI, Esteves de Carvalho AV and
Nazarenko LP: Trias of keratosis pilaris, ulerythema ophryogenes
and 18p monosomy: Zouboulis syndrome. J Dermatol. 41:371–376.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Callaway SR and Lesher JL Jr: Keratosis
pilaris atrophicans: Case series and review. Pediatr Dermatol.
21:14–17. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cohen-Barak E, Danial-Farran N, Hammad H,
Aleme O, Krauz J, Gavishi E, Khayat M, Zivi M and Shalev S:
Desmoglein 4 mutation underlies autosomal recessive keratosis
pilaris atrophicans. Acta Derm Venereol. 98:809–810.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Klar J, Schuster J, Khan TN, Jameel M,
Mäbert K and Forsberg L: Whole exome sequencing identifies LRP1 as
a pathogenic gene in autosomal recessive keratosis pilaris
atrophicans. J Med Genet. 52:599–606. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Flórez A, Fernández-Redondo V and Toribio
J: Ulerythema ophryogenes in Cornelia de Lange syndrome. Pediatr
Dermatol. 19:42–45. 2002.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Morton CM, Bhate C, Janniger CK and
Schwartz RA: Ulerythema ophryogenes: Updates and insights. Cutis.
93:83–87. 2014.PubMed/NCBI
|
|
8
|
Li K, Ann Thomas M and Haber RM:
Ulerythema ophryogenes, a rarely reported cutaneous manifestation
of Noonan syndrome: Case report and review of the literature. J
Cutan Med Surg. 17:212–218. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gómez Centeno P, Rosón E, Peteiro C,
Mercedes Pereiro M and Toribio J: Rubinstein-Taybi syndrome and
ulerythema ophryogenes in a 9-year-old boy. Pediatr Dermatol.
16:134–136. 1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chien AJ, Valentine MC and Sybert VP:
Hereditary woolly hair and keratosis pilaris. J Am Acad Dermatol.
54 (Suppl 2):S35–S39. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang JF and Orlow SJ: Keratosis pilaris
and its subtypes: Associations, new molecular and pharmacologic
etiologies, and therapeutic options. Am J Clin Dermatol.
19:733–757. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fekete GL, Boda D, Căruntu C and Fekete L:
Paraneoplastic pityriasis rubra pilaris in association with
prostate carcinoma: A case report and literature review. Exp Ther
Med. 18:5052–5055. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kanakpur SH and Caculo DU: Rare ocular
manifestations in keratosis follicularis (Darier-White disease).
Indian J Ophthalmol. 65:874–876. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tatu AL and Violeta VC: Pityriasis
folliculorum of the back thoracic area: Pityrosporum, keratin
plugs, or Demodex involved? J Cutan Med Surg. 21:441–447.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Alcántara González J, Boixeda P, Truchuelo
Díez MT and Fleta Asín B: Keratosis pilaris rubra and keratosis
pilaris atrophicans faciei treated with pulsed dye laser: Report of
10 cases. J Eur Acad Dermatol Venereol. 25:710–714. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rodríguez-Lojo R, Pozo JD, Barja JM,
Pineyro F and Perez-Varela L: Keratosis pilaris atrophicans:
Treatment with intense pulsed light in four patients. J Cosmet
Laser Ther. 12:188–190. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Apalla Z, Karakatsanis G, Papageorgiou M,
Kastoridou C and Chademenos G: A case of atrophoderma vermiculatum
responding to systemic isotretinoin. J Dermatol Case Rep. 3:62–63.
2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bacârea PF, Neda I, Daniliuc CG, Bacârea A
and Silaghi DL: Chiral selectivity in the basic or acid α-amino
acids homomeric Cu(II) complexes range. Rev Chim. 63:489–498.
2012.
|
|
19
|
Ianosi SL, Batani A, Ilie MA, Tampa M,
Georgescu SR, Zurac S, Boda D, Ianosi NG, Neagoe D, Calina D, et
al: Non-invasive imaging techniques for the in vivo diagnosis of
Bowen's disease: Three case reports. Oncol Lett. 17:4094–4101.
2019.PubMed/NCBI View Article : Google Scholar
|