1. Introduction
Pathophysiologically, intraepidermal blistering
diseases represent a group of disorders in which the body wrongly
attacks healthy tissue with autoantibodies that attach to
structural proteins in the mucous membranes and skin, which are
components of desmosomes (desmocollins, desmogleins, plakins),
causing intraepithelial blisters (1-3).
This category of intraepidermal blistering diseases includes
pemphigus, which can be classified into the following four
entities: pemphigus vulgaris (PV), pemphigus foliaceus (PF),
paraneoplastic pemphigus (PNP), IgA pemphigus (subcorneal pustular
dermatosis and intraepidermal neutrophilic IgA dermatosis).
The specific symptoms and severity of these diseases
vary from one person to another, even among individuals with the
same disorder. The diagnosis of bullous skin diseases is based on
the typical skin manifestations, which may be objectified by
Nikolsky sign and characteristic direct immunofluorescence (DIF)
patterns in skin biopsies (1-3).
The presence of specific circulating autoantibodies guides the
diagnosis and allows a correlation between the levels of specific
autoantibodies and the severity of the disease (4).
Since the outbreak of the COVID-19 pandemic, oral
ulcerative lesions have been described, associated with SARS-COV-2
infection. Bullous dermatoses can arise with similar lesions in the
oral cavity, which is why the clinical picture must be very well
known (2-4).
Although there is no cure for autoimmune blistering
diseases, they can often be controlled with treatment. The choice
of drugs and their dosage should be based on clinical severity and
patient comorbidities. Most patients require months or even years
of immunosuppressive maintenance therapy. In other cases, untreated
autoimmune blistering diseases can cause life-threatening
complications (1-5).
In recent years, new insight into the causes and development of
these disorders has led to research into new therapies, such as the
development of drugs that target the specific autoantibodies that
cause the symptoms of these diseases (6).
2. Research methods
A literature search was conducted, using electronic
databases Key Elsevier, Medscape, PubMed, Google Scholar, for the
term ‘pemphigus’ in combination with ‘vulgaris’, ‘vegetans’,
‘herpetiformis’, ‘foliaceus’, ‘paraneoplastic’, ‘IgA’,
‘epidemiology’, ‘pathophysiology’, ‘skin manifestations’, ‘mucosal
manifestations’, ‘clinical variants’, ‘management’ and ‘evolution’
to collect reports of skin and mucosal manifestations described in
patients with different clinical variants of pemphigus. Case
reports, case series, and literature review-type articles were
included in our research. A brief report was conducted based on 98
articles found in the literature.
3. Pemphigus group
Pemphigus includes a group of potentially
life-threatening bullous autoimmune disorders of largely unknown
etiology. Clinically, they are characterized by flaccid blisters
and erosions of the skin and/or mucous membranes (1-4,7-9).
The loss of intraepidermal adhesion between keratinocytes is
attributable to the binding of autoantibodies directed against
desmosomal structural proteins, primarily desmogleins (Dsg1 and
Dsg3) and, in rare cases, also desmocollin 1-3 or plakins (1-4,10).
Pemphigus has distinct forms: pemphigus vulgaris (PV), pemphigus
foliaceus (PF), paraneoplastic pemphigus (PNP), and IgA pemphigus.
PV and PF are caused by a humoral autoimmune response, whereas PNP
is caused by both humoral and cellular autoimmune responses
(11). PV is characterized by
persistent mucosal erosions with or without skin involvement. PF
presents fragile, superficial blisters, as well as subsequent
erosions and leafy scales that exclusively affect keratinizing skin
(11). In PNP, the clinical
hallmark is painful oral mucosal lesions accompanied by
morphologically heterogeneous skin lesions (erythematous macules,
flaccid blisters, scaly plaques, or erosions) (4-8,11).
4. Pemphigus vulgaris (PV)
Epidemiology
According to several retrospective studies,
pemphigus vulgaris (PV) is the most frequent representative of the
group of pemphigus diseases, with an incidence of 0.1-0.5/100,000
population (7-9,11).
A female predominance is reported in most epidemiological studies,
with a peak age between 50-60 years, although childhood onset forms
have been described (7,12). PV is also more common in certain
ethnic groups, such as the Ashkenazi Jewish population and
Mediterranean descendants (7,12).
Pathophysiology and genetic
factors
In patients with PV, most types of antibodies are
oriented against desmosomal cadherins, Dsg1 and Dsg3, but other
autoantibodies have been identified targeting other metabolic and
structural proteins, such as Dsc1 and Dsc3 desmocolins,
mitochondrial antigens, hSPCA1, thyroid peroxidase, muscarinic and
nicotinic acetylcholine receptors, plakoglobin, E-cadherin and
plakophilin 3 (9,12-14).
The pathogenic role of these non-Dsg autoantibodies is mentioned by
some studies, which suggest that they synergistically complement
the classic effects of anti-Dsg autoantibodies in the complex
process of pemphigus pathogenesis (14). The two antigens targeted by
autoantibodies in PV are the 130-kDa glycoprotein Dsg3 and 160-kDa
glycoprotein Dsg1. Dsg1 is mainly expressed on the surface of the
epidermis, while Dsg3 accumulates predominantly in the mucous
membranes and deeper epidermal layers (9-13).
Patients with mucosal-dominant-type PV have only anti-Dsg3
antibodies, and those with mucocutaneous-type PV have both
anti-Dsg3 and anti-Dsg1 antibodies (13).
There is a genetic predisposition for developing PV;
certain major histocompatibility complex (MHC) class II molecules,
such as DR4 (DRB1*0402) and DRw6 (DQB1*0503) occurring more
frequently among those affected (7,15).
Although these alleles are rare in the European population, they
are more common in certain ethnic groups (e.g. Jewish population)
and in countries of the eastern Mediterranean and the Middle East
(Turkey, Iran, Iraq) (4-7,15).
Clinical features
PV is clinically characterized by flaccid
blisters/erosions of the mucous membranes and the skin.
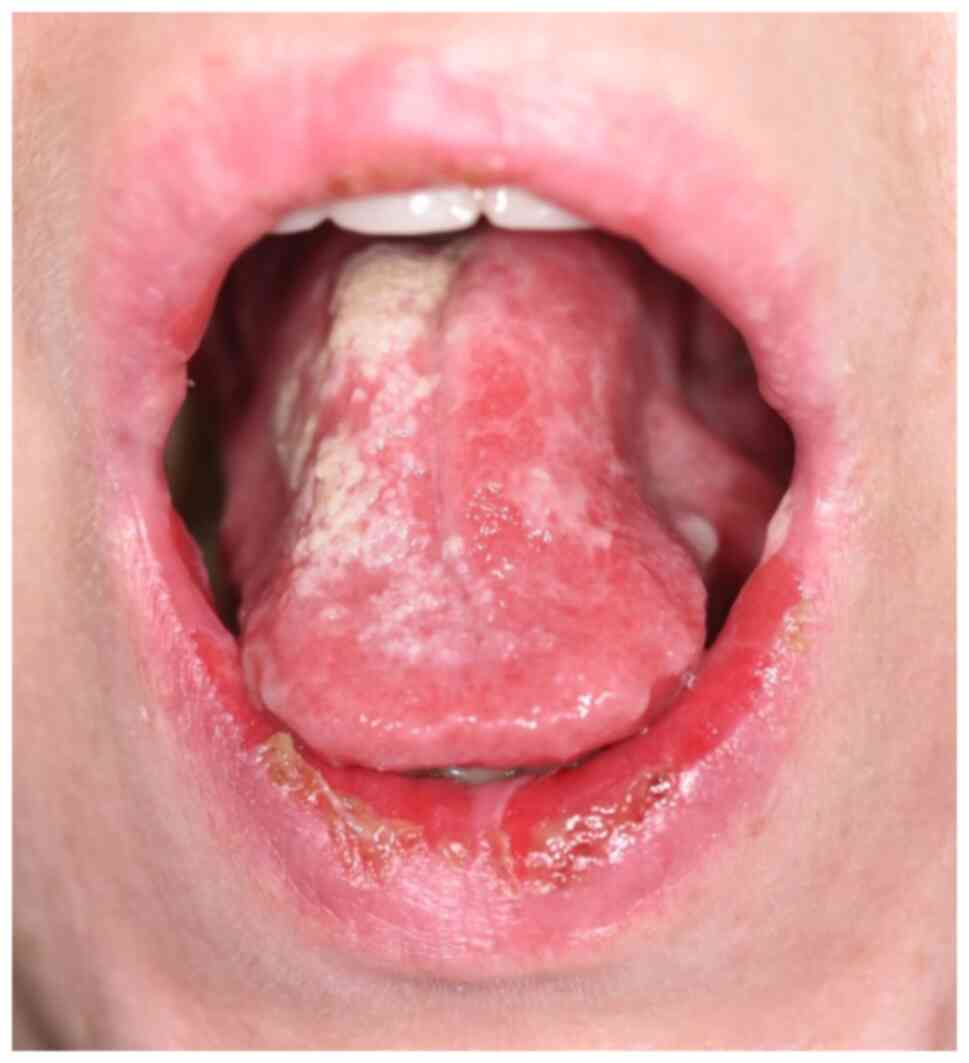
In most of the cases, the onset of the lesions
involves the oral cavity. It is often not recognized in the early
stages, thus, other oral ulcerative disorders are suspected, such
as herpetic gingivostomatitis, recurrent aphthae, erosive oral
lichen planus, or even candida stomatitis. Intact bullae are rare
in the mouth. More commonly the lesions are ill-defined, irregular,
painful erosions located on the gingiva, buccal or palatal mucosa
(8,16-18).
Other sites of the mucous membrane may be affected, including the
conjunctiva, esophagus, pharynx, larynx, urethra, penis, labia,
vagina, cervix, and anus (8)
(Fig. 1).
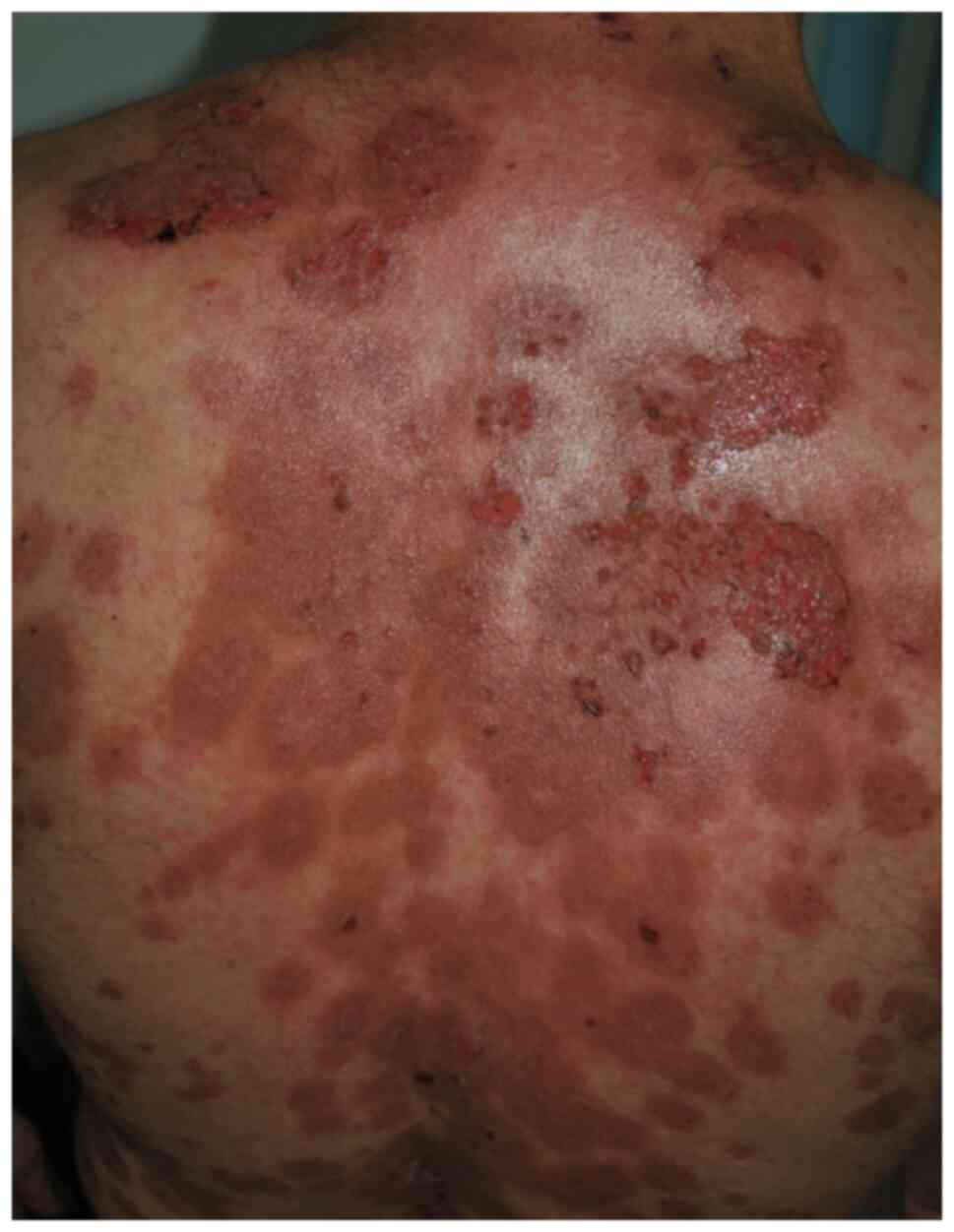
Skin lesions appear several weeks or months after
the onset of mucosal erosions and may develop anywhere on the skin,
but there are some areas of predilection that include the scalp,
face, chest, axillae, groin, and umbilicus. Blisters are flaccid,
fragile and break easily, leading to painful erosions, which bleed
easily and often become crusted, and can lead to residual pigmented
lesions after healing under immunosuppressive treatment (5,6,8-10)
(Fig. 2). The blister on the skin
may remain localized for 6 to 12 months, and then afterwards
becomes widespread. The lesions can be painful, pruritic, and
associated with a burning sensation, weakness, history of
epistaxis, malaise, weight loss, dysphagia, and hoarseness
(4,7,8). It is
uncommon that the lesions emerge as a generalized acute eruption
(9). During the active phase of PV,
Nikolsky signs can be obtained but they are not specific to PV and
can be found in other active blistering diseases (8). The direct Nikolsky appears because of
an absence of cohesion within the epidermis and its upper layers
move easily laterally with slight pressure or rubbing. Another sign
that may be present is Asboe-Hansen sign also referred to as the
‘indirect Nikolsky’ or ‘Nikolsky II’ which occurs when a gentle
pressure on intact bulla forces the fluid to spread under the skin
away from the site of pressure -‘bulla-spread phenomenon’ (1,3,4-8).
In the case of pregnant women with active pemphigus, there is a
chance that the newborn could develop neonatal pemphigus as a
result of the transmission of maternal IgG (consisting of
autoantibodies against Dsg3) through the placenta (8,19). The
clinical picture in neonatal pemphigus is not as severe compared to
the disease that caused it since it is not a systemic disease. The
symptoms and signs are reduced to skin lesions, and exanthematous;
crusted erosions erupt as a temporary phenomenon over several weeks
until the degradation of maternal autoantibodies (19-21).
Neonatal pemphigus has a good prognosis (19). In addition to classical PV, other
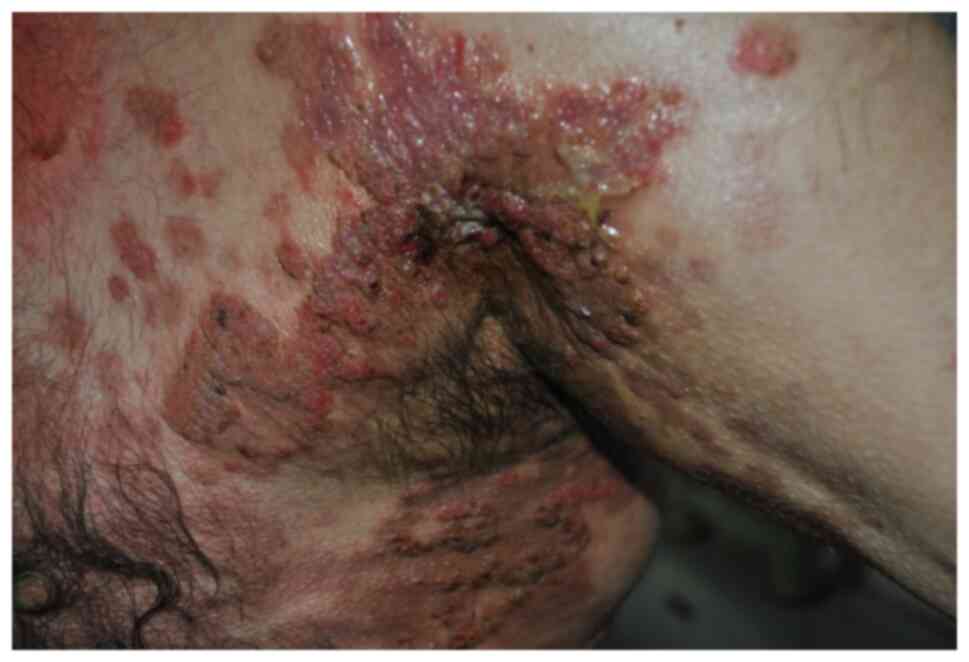
special forms exist (8). Pemphigus
vegetans is characterized by the tendency to develop papillomatous
and verrucous intertriginous vegetations (8,22,23).
Most patients initially present to their health care provider with
stomatitis or hyperkeratotic plaques in a cerebriform pattern on
the tongue (23). The cutaneous
lesions rupture and ulcerate with verrucous crusting and vegetative
plaques forming over the erosions. These hyperkeratotic lesions
characteristically present in the intertriginous areas including
the groin/inguinal folds, armpits, thighs, nasolabial and flexural
surfaces (Fig. 3) (8,23).
Depending on the clinical course, two subtypes of pemphigus
vegetans are differentiated. The Neumann subtype is characterized
by large vesiculobullous and erosive lesions, with an aggressive
course by the formation of whitish, macerated plaques. In the
Hallopeau subtype, pustules initially appear which later turn into
warty lesions and has a more indolent course (8,23-25).
Untreated, pemphigus vegetans can be fatal within 5 years due to
severe blistering, secondary infection and malnutrition. Mortality
is approximately 5 to 15% per year (8,22-24).
Pemphigus herpetiformis is a rare variant of PV,
characterized by erythematous, vesicular, bullous, pustular or
papular lesions, often in a ‘herpetiform’ pattern and with severe
pruritus, frequently located on the trunk and proximal extremities
(8,26). Skin lesions tend to present with
annular-shaped distribution, or in some cases, the main lesions can
resemble urticaria (26,27). Oral mucosa involvement is rare
(8,27). Therefore, pemphigus herpetiformis
possesses clinical similarity to dermatitis herpetiformis and must
be included in the differential diagnostic considerations (27).
Diagnosis
PV diagnosis is based on a combination of clinical
presentation (presence of recurrent blister formation, erosions and
crust, Nikolsky sign); histological detection of intraepidermal
blistering; detection of acantholytic keratinocytes by the Tzanck
test; detection of pemphigus antibodies, DIF, IDIF, ELISA (9-12).
Histopathological examination will reveal
acantholysis and a sparse inflammatory infiltrate. The acantholysis
occurs in the suprabasal layer, leaving a single layer of basal
keratinocytes attached to the dermal-epidermal basement membrane
looking like a ‘row of tombstones’ (4,7,9-12).
In the early pemphigus vulgaris. pre-bullous stage, histology also
shows eosinophilic spongiosis (12).
Direct immunofluorescence (DIF) of patients'
perilesional skin reveals a reticular fluorescence pattern, a
‘honeycomb-like pattern’, caused by the deposition of IgG
autoantibodies and C3 on the surface of epidermal keratinocytes
(4,28-30).
The detection of circulating IgG autoantibodies can be conducted
using methods such as indirect immunofluorescence (IIF), using
monkey esophagus or human skin as the substrate (29-31).
ELISA and chemiluminescent enzyme immunoassay using recombinant
Dsgs enable detection of circulating autoantibodies in pemphigus
(12,32-34).
Currently, there is no consensus on which assay should be used as a
diagnostic test for PV, but ELISA is one of the most accurate
diagnostic tests, separately measuring anti-Dsg1 and anti-Dsg3 IgG.
In a meta-analysis of 13 studies with a sample size of 1,058
patients, anti-Dsg3 ELISA demonstrated a sensitivity of 97% and
specificity of 98% in PV (34).
ELISA or chemiluminescent enzyme immunoassay are useful for both
diagnosis and monitoring of disease activity, as autoantibody
titers often fluctuate in parallel with disease activity and
decrease with clinical improvement (7,32-34).
In ~20 to 40% of patients, even after clinical remission, anti-Dsg1
and anti-Dsg3 autoantibodies remain detectable and they are
occasionally detectable in clinically healthy individuals (7,35).
There are three subtypes of PV measured by the
pattern of autoantibodies: mucosal-dominant PV, when serum is
positive for anti-Dsg3 but negative for anti-Dsg1; mucocutaneous
PV, when serum is positive for anti-Dsg3 and anti-Dsg1 and show the
implication of the epidermis in addition to the mucous membranes;
cutaneous PV is scarcely and correlated with blistering in deep
epidermal layers due to anti-Dsg1 and pathogenically weak anti-Dsg3
(12,28-31).
In patients suffering from PV, many autoantibodies have been found
to aim at other structural and metabolic proteins, including
desmocollins (Dsc) 1 and 3, muscarinic and nicotinic acetylcholine
receptors, mitochondrial antigens, thyroid peroxidase, hSPCA1,
plakophilin 3, plakoglobin, and E-cadherin (14,31,35-37).
Research on some of these non-Dsg autoantibodies implies that they
complement the typical effects of anti-Dsg autoantibodies in
pemphigus pathogenesis (14,37). A
cross-sectional study and meta-analysis reported a high incidence
of other coexisting autoimmune disease of patients with PV, such as
thyroid diseases (e.g hypothyroidism), rheumatoid arthritis, type 1
diabetes mellitus, inflammatory bowel disease, alopecia areata,
vitiligo, systemic lupus erythematosus, scleroderma, and rare
entities such as myasthenia gravis (38,39).
As part of PV investigations and surveillance, investigation for
these conditions should be considered.
Treatment
There is high morbidity and mortality within the
population suffering from PV. Systemic corticosteroids and
immunosuppressants remain the main therapy and have managed to
decrease the mortality rates from 75 to 10%. The purpose of initial
therapy is to control the disease by minimizing the blister
formation, stimulating the healing of present blisters, and prolong
remission with a minimum dose (e.g. oral prednisolone ≤0.2
mg/kg/day or ≤10 mg/day) (7,12,40).
The latest guidelines recommend corticosteroids
(0.5-1.5 mg/kg/day) as the first treatment in the initial phase.
Steroid-sparing immunosuppressants can be added in case there is a
high risk of an adverse reaction to CS (9-12,40).
Systemic corticosteroids are reduced in concordance with the
therapeutic response. Clinicians should be mindful of
complications, eventually relapses, of long-term CS therapy during
the maintenance period, in the likes of susceptibility to
infections and infestations, osteoporosis, secondary adrenal
insufficiency, hypertension, posterior subcapsular cataract, and
transient hyperglycemia (41).
During the steroid tapering phase around half of the patients
relapse, and the other half reach complete remission after
approximately 3 years of treatment (40,41).
Multiple immunosuppressive adjuvants have been used
to lessen the complications of high doses of CS in the long-term.
These include: azathioprine (AZA) with a recommended dose of 2.0
mg/kg/day with normal thiopurine methyltransferase activity and 1
mg/kg/day with TPMT enzyme mutations; cyclophosphamide (CYP) 2
mg/kg/day, i.v. pulse therapy or continuous oral administration;
mycophenolate mofetil (MMF) 2 g/day, divided into two doses;
methotrexate (MTX) 10-20 mg/week; dapsone, but before the
administration a measuring of serum glucose-6-phosphate
dehydrogenase (G6PD) activity is mandatory (7,42,43).
Patients who are unable to reach clinical remission
with systemic CS and/or immunosuppressant agents, or who present
moderate to severe pemphigus or refractory PV, might undergo
high-dose intravenous immunoglobulins (IVIg) treatment,
plasmapheresis, or extracorporeal immunoadsorption (IA) (7,12,44-46).
IVIg treatment is well tolerated, mostly safe, and works by quickly
decreasing the autoantibodies which are responsible for pemphigus,
targeting pathogenic antibodies (10-12,44-46).
Plasmapheresis is effective especially when it is combined with
immunosuppressant agents (e.g. pulsed IV cyclophosphamide), and
numerous clinical trials have indicated the increased efficacy and
blistering diminishing with this treatment (7,9-12,45).
IA uses affinity adsorption of pathogenic autoantibodies. These
autoantibodies attach to the adsorber through an immobilized
ligand. A quick and substantial decline in desmoglein
(Dsg)-reactive autoantibodies, along with clinical remission of
mucocutaneous erosions and blisters has been observed when applying
IA in severe PV. Systemic immunosuppressive medication can be
combined with IA and is safe and well-tolerated in general
(10,11,46).
Recently, targeted biologic therapies have been
adopted in pemphigus, such as rituximab (RTX) and tumor necrosis
factor (TNF)-α inhibitors (47).
Rituximab is a chimeric type I anti-CD20 monoclonal antibody, which
can bind to CD20 antigen and remove B-lymphocytes expelling CD20
from blood (7,12,48).
Rituximab leads to a clear reduction of circulating anti-Dsg
autoantibodies at the expense of B cells, and because of the
pathogenic role of these autoantibodies, there is a noteworthy
amelioration of the lesions (12,47-49).
Initial treatment with RTX, in combination with high potency
topical CS or IVIg, has shown to be efficient in patients with
pemphigus that have a contraindication to systemic steroids
(47-49).
A recent clinical study involving 11 patients that
had PV refractory to conventional therapy showed that three weeks
of treatment with RTX (375 mg/m2/week) followed by IVIg
(2 g/kg) for four weeks, and then four consecutive months with
monthly infusions of IVIg and RTX, resulted in remission in 9 of
the patients which lasted from 22 to 37 months (47-50).
Another study with 136 patients suffering from refractory pemphigus
coming from 4 European countries reported a 95% average response
rate, with 2/3 of patients achieving complete remission (51). The most common side effects of RTX
include infections and adverse events related to infusion.
Opportunistic infections may also arise, including cytomegalovirus
and pneumocystis jirovecii infections and theory describes the risk
of hepatitis B and C virus reactivation, as well as tuberculosis
(52-54).
Late reactions include vasculitis, hypersensitivity (serum
sickness), Steven-Johnson syndrome and some cases have shown
paradoxical pemphigus flares consequent to RTX treatment (55). RTX has revolutionized PV treatment,
but some patients remain refractory to this agent and for such
refractory cases, new drugs are being tested in clinical trials.
Ofatumumab is a fully humanized anti-CD20 mAb, which is less
immunogenic than RTX (56).
Veltuzumab is a humanized anti-CD20 mAb that can be administered
subcutaneously and shows clinical efficacy in patients with
refractory PV. Veltuzumab is a more economical alternative to
intravenous RTX, because a lower dose is required (57).
For the treatment of oral lesions, intralesional RTX
was reportedly effective in 3 patients with PV with oral lesions
refractory to systemic therapy, including intravenous RTX (58).
Evolution and complications
Most deaths associated with untreated PV occur
within the first few years of the disease onset. Considering that
the drugs used in the treatment of PV have serious side effects,
patients must be monitored carefully for infections, liver and
renal function abnormalities, electrolyte disturbances,
osteoporosis, hypertension, diabetes, anemia, and gastrointestinal
bleeding (7,10-12).
5. Paraneoplastic pemphigus (PNP)
Epidemiology
PNP represents a rare disorder with an incidence and
prevalence that remains unclear. There are ~500 cases reported in
the literature, and PNP accounts for 3-5% of all pemphigus cases
(7,12). Patients between 45 and 70 years of
age are usually affected, and a female predominance is reported in
most epidemiological studies (7,9-12,59).
However, PNP can affect also children and adolescents, particularly
in association with Castleman's disease. Clear racial, ethnic, or
geographic differences in the risk for PNP have not been
established (7,9-12,59).
Pathophysiology and genetic
factors
The etiopathogenesis of PNP is not completely known,
but it is plausible that both autoantibodies and cell-mediated
immunity play a key role.
The most common autoantibodies detected in PNP are
directed against the plakin family, such as envoplakin (210-kDa),
periplakin (190-kDa), bullous pemphigoid antigen I (230-kDa),
desmoplakin I (250-kDa), desmoplakin II (210-kDa), plectin
(500-kDa), and α2-macroglobulin-like-1 (170-kDa) (7,12,59).
Tsuchisaka reported that epiplakin is a PNP autoantigen, being
detected in 35 (72.9%) of 48 PNP sera of Japanese patients by
immunoprecipitation-immunoblotting (60). PNP antibodies are typically IgG,
although IgA has been reported in a few cases (59-61).
In a review, Czernik et al summarized that
cell-mediated immunity may also play a role in PNP, highlighting
lesional mononuclear cells and elevated IL-6 levels in the sera of
patients with PNP (61). In
addition, Wade and Black detected MHC-restricted CD8+
cytotoxic T cells, non-MHC-restricted CD56+, and
CD68+ natural killer cells within the dermoepidermal
junction of PNP lesions (62).
Regarding the genetic predisposition, an association
with HLA class II DRB1*0344 and HLA Cw *1445 confer strong
susceptibility to PNP in Caucasian and Han Chinese patients. These
conclusions were drawn by Martel et al (63) from a series of 13 Caucasian French
patients.
Clinical features
Clinical features are extremely polymorphous in PNP,
and lesions can be detected not only on the skin, but also on
different mucosae. The cross-reactivity with tumor antigens and the
presence of different autoantibodies could justify the different
manifestations in PNP patients (59,62-64).
PNP can be the first clinical manifestation that leads to the
detection of an occult tumor in ~30% of cases (7,12,59).
PNP is associated with underlying neoplasms and the most frequent
include non-Hodgkin's lymphoma (38.6%), chronic lymphocytic
leukemia (18.4%), Castleman's disease (18.4%, benign tumors,
commonly in children), adenocarcinomas (prostate, pancreas, breast,
gastric), squamous cell carcinomas (8.6%), sarcomas (6.2%), thymoma
(5,5%), Waldenström's macroglobulinemia (1.2%), Hodgkin lymphoma
(0.6%), and monoclonal gammopathy (0.6%) (12,59,62-64).
Initially, PNP typically manifests as hemorrhagic
stomatitis with extensive mucous membrane erosions accompanied by
intense pain and resistance to therapy (64,65).
The lesions are polymorphic, and symptoms such as blisters,
erosions, spots, papules, and plaques can occur, involving the
lips, vermilion and the tongue (62-65).
Painful erosions and crusting on the lips could resemble oral
lesions commonly found in erythema multiforme (EM) or
Stevens-Johnson syndrome (59). In
children, the stomatitis caused by PNP may be often mistaken for
herpetic stomatitis or toxic epidermal necrolysis (TEN), leading to
a delay in the diagnosis (66).
In addition to stomatitis, mucositis involving the
pharynx, larynx, esophagus, and anogenital region can occur.
Symptoms of oropharyngeal involvement may include a sore throat and
dysphagia. Ocular involvement occurs in ~70% of cases and the most
common symptoms and signs include painful ocular irritation,
worsening of vision, mucus discharge, conjunctival erosions, eyelid
margin thickening, corneal erosions, and pseudomembranous
conjunctivitis. In several cases, mucosal involvement is the only
sign of PNP (38,39,65-67).
Skin lesions of PNP are polymorphic and usually
appear after the onset of mucosal lesions, involving any site, but
especially the torso, head, neck, and proximal extremities
(59,62-64).
Blisters and erosions are commonly observed and mimic those of PV,
PF or bullous pemphigoid, affecting any area of the body, but
especially the upper trunk. The erythematous maculopapular lesions
with dusky centers or central vesicles may arise on the
extremities, mimicking the erythema multiforme-like targetoid
lesions (59,64-67).
Another type of characteristic cutaneous lesions is represented by
lichenoid eruptions, which manifest as erythematous papules or
plaques, similar to that in lichen planus and graft-versus-host
disease and are frequently identified in children, predominantly on
the torso and limbs (59,62,66).
In some cases of PNP, cutaneous lesions may present as a nail or
periungual lesions (onychodystrophy, erosions, scaling) and
alopecia (59).
As for extracutaneous lesions, the involvement of
the respiratory epithelium is frequently associated with pulmonary
disease in the form of bronchiolitis obliterans, a frequently
lethal obstructive respiratory disorder (59-62,66).
The initial symptom of bronchiolitis obliterans is dyspnea, and
pulmonary function tests show obstructive lung disease.
Bronchiolitis obliterans is found in ~30% of PNP patients and
frequently develops in patients with Castleman disease (65-67).
Due to the involvement of diverse organ systems, PNP has recently
often been viewed as a mucocutaneous variant of the ‘paraneoplastic
autoimmune multiorgan syndrome’ (PAMS) (61).
Diagnosis
The diagnostic criteria for PNP include different
criteria, based on the clinical picture, histopathology, direct and
indirect immunofluorescence and immunoprecipitation.
The clinical presentation includes painful erosions
involving mucosae with or without a multiform skin eruption
producing blisters and erosions, occurring in association with an
occult or evident neoplasm.
PNP has two major clinical phenotypes, blisters and
lichenoid eruptions, and depending on the type of lesion biopsied,
the histological findings are variable, and often the diagnosis
requires multiple biopsies (62-64).
In blisters, suprabasal acantholysis and individual keratinocyte
necrosis with sparse inflammatory infiltrate are observed, while in
lichenoid eruptions, an interface and lichenoid dermatitis with a
dense band-like lymphocytic infiltrate in the upper dermis are
usually detected (59,68). Sometimes, blisters and interface
dermatitis may coappear in the same lesion (59). Histological findings show the
combination of blisters in the epidermis caused by IgG
autoantibodies (humoral autoimmune response) and interface
dermatitis caused by self-reacting T cells (cellular autoimmune
reaction) (59,64,68).
DIF reveals IgG and complement C3 deposition both in
the intercellular space and in the dermoepidermal junction, along
the basement membrane zone (28-31,59-62).
In the classic forms of pemphigus, IIF is positive only on
stratified squamous epithelial substrates, but in PNP there is
staining of other tissues, such as the bladder, heart, and liver
(28-31,59).
IIF shows the presence of circulating IgG autoantibodies that
target the intercellular proteins found in transitional or
stratified squamous epithelia (28-31,59).
Although immunoprecipitation is still the gold
standard for the demonstration of specific autoantibodies,
immunoblotting is a valuable aid for diagnosis (69). Immunoblot analysis using epidermal
extracts has been used to detect 210-kDa envoplakin and 190-kDa
periplakin, which are highly sensitive and specific for PNP
(30,59,69).
Immunoprecipitation can detect antibodies against multiple
epidermal antigens, including desmoplakin I (250 kDa), bullous
pemphigoid antigen (230 kDa), envoplakin (210 kDa), desmoplakin II
(210 kDa), periplakin (190 kDa) and 2-macroglobulin-like-1 (170
kDa) (28-31,59-61).
Enzyme-linked immunosorbent assays (ELISAs) are a
useful technique for detecting circulating autoantibodies in PNP,
especially those against Dsgs and Dscs. Approximately 80% of
patients with PNP have circulating anti-Dsg3 IgG; in 19-42% of
patients, autoantibodies have been detected against other
desmosomal cadherins (Dsg1, Dsc1, Dsc2, Dsc3), and in 40% of
patients, ELISA reveals autoantibodies against BP180 (34,59,64).
When PNP is suspected in a known patient with a
history of malignancy, thorough investigations such as blood cell
count, lactate dehydrogenase, flow cytometry, computed tomography
of the chest, abdomen, and pelvis should be performed. Up to a
third of the patients with PNP, have an underlying malignancy
discovered after the onset of PNP symptoms (7,12,59-61).
The differential diagnosis of PNP includes pemphigus
vulgaris, mucous membrane pemphigoid, erythema multiforme,
Stevens-Johnson syndrome, toxic epidermal necrolysis, major
aphthous stomatitis, oral lichen planus, graft-versus-host disease,
and herpes simplex virus infection (59,62-64).
In pediatric cases, oral manifestations may be mistaken for a
herpetic stomatitis (66).
Treatment
The rarity of PNP makes it more difficult to treat.
Even though some therapies have been proposed in the literature,
PNP has been observed to be more resilient to treatment compared to
different forms of pemphigus (59,62-64,65-67).
If there is suspicion of PNP, the six steps reported by Frew and
Murrell (70) should be followed
for better management of the patient. The six steps are as follows:
vital parameters stabilization, evaluation of any underlying
malignancy, correct diagnosis of PNP, the extirpation and medical
therapy of the trigger tumor, and PNP treatment using
immunosuppression, immunomodulation, or plasmapheresis (59,71).
Cases that are associated with benign tumors, such
as benign thymoma and localized Castleman's disease, generally
ameliorate or reach complete remission after complete tumor
resection (59,64). In patients with PNP and malignant
neoplasms, extirpating the tumor does not lead to controlling the
disease, and an agreement on the best treatment has yet to be
recognized (64).
The first line of PNP treatment is a high dose of
systemic corticosteroids (prednisolone), but many patients do not
appear to have a good response with only corticosteroids (59,64-67,71).
Corticosteroids only improve skin lesions, while mucosal lesions
are resistant to most types of therapy. Steroid-sparing agents,
namely cyclosporin, azathioprine, cyclophosphamide, and MMF, can be
used with glucocorticoid therapy to lessen the total steroid burden
(62,64,71).
IVIg and plasmapheresis manifest high efficiency, safety, and
promising effects in the treatment of PNP (71). A 2 g/kg dose per cycle is used for
IVIg; these cycles are repeated monthly. IVIg act by reducing
pathogenic autoantibodies rapidly. In addition, IVIg can be added
to the patient's existing treatment regimen without the added
concern of additional immunosuppression (44,62-64,70).
Alternative therapies are being applied notably in
patients whose malignancy is in remission. RTX and ibrutinib are
B-cell-targeting agents and they generate different outcomes among
patients suffering from PNP associated with B-cell malignant
lymphomas (70,71). Overall the efficiency of RTX in PNP
is much less consistent than PV and PF. RTX is generally well
tolerated; however, adverse effects include infusion and allergic
reactions (59,64,47-49).
According to reports, alemtuzumab, a humanized
monoclonal antibody against CD5, which is shown in most B and T
lymphocytes, has induced long-term remission in a patient with
B-cell chronic lymphocytic leukemia. Alemtuzumab administered 30 mg
i.v., 3 times a week for 12 weeks, showed recovery of cutaneous and
mucosal lesions (72). In two cases
of PNP, tocilizumab, a monoclonal antibody anti-IL-6 receptor, was
shown to quickly improve mucositis, although bronchiolitis
obliterans did not shown signs of improvement (73).
Due to the risk of sepsis followed by iatrogenic
immunosuppression and loss of skin integrity, early antimicrobial
therapy is suggested. In the case of pain caused by extensive
erosions, antalgic therapy could be beneficial (64,69-71).
Evolution and complications
The prognosis of PNP is generally poor, and the
mortality rate ranges from 75 to 90%, with a 5-year overall
survival rate of only 38%. Death is usually due to systemic
complications, including sepsis, gastrointestinal bleeding,
bronchiolitis obliterans and severe infection due to
immunosuppressive therapy. Similar to mucositis, bronchiolitis
obliterans is resistant to therapy, and lung transplantation is the
last therapeutic option for respiratory failure (59,62-64,67).
The evolution of PNP is not correlated with that of
the associated malignancy. PNP lesions may progress after removal
of the triggering malignancy or when the malignancy is under
control. However, in patients with PNP and Castleman's disease or
benign thymomas, the outcome is favorable after tumor removal
(61). Paraneoplastic pemphigus may
precede the clinical appearance of a neoplasm, which makes
screening of these patients mandatory (62,64-67).
The prognosis of PNP depends on a prompt diagnosis
and early initiation of treatment. Effective control of the oral
and skin lesions, proper treatment of the underlying neoplasm, and
prevention of bronchiolitis obliterans are of paramount importance
(70).
6. Pemphigus foliaceus (PF)
Epidemiology
PF is rare and sporadic worldwide and the incidence
varies depending on the population studied.
In Western Europe, the incidence of PF is ~0.5-1
case per million per year. In South America (Brazil, Colombia, and
Peru) and North Africa (Tunisia and neighboring countries), the
incidence of PF is higher than in other countries and this is
because of an endemic form of the disease (fogo selvagem), which
affects mostly young adults (4-7,9-12,74).
In Brazil, the incidence of endemic PF in the Terena reservation is
around 3.4% (7,12,74).
The prevalence of PF in men and women is approximately equal, but
in some regions such as the Sousse region of Tunisia, women are
more affected (6.6 cases per million per year) (9-12,75).
In El Salvador, a similar female and age predisposition may also be
evident.
The mean patient age at onset of PF is ~50-60
years, but it may occur at any age. Fogo selvagem often occurs in
children, young adults, and genetically related family members, and
the mean patient age at onset is ~20-30 years (4-7,9-12,74).
No ethnic predisposition has been reported, and
most of the patients are young rural workers living in forested
areas adjacent to rivers and streams. In these areas, some insects,
including black fly (Simulium species), trigger the disease
through insect saliva, leading to an immune reaction against Dsg1
through molecular mimicry (4-7,9-12,74).
This hypothesis is supported by high positivity rates of anti-Dsg1
IgG autoantibodies in the sera of healthy individuals living in
endemic regions of fogo selvagem and the low prevalence of endemic
PF in urbanized areas (4-7,9-12,74-76).
Pathophysiology and genetic
factors
PF is mediated by autoantibodies against desmosomal
proteins on the keratinocyte cell surface. The lesions in PF are
induced by IgG (mainly IgG4 subclass) autoantibodies directed
against Dsg1, a 160-kDa desmosomal cadherin transmembrane
glycoprotein that mediates cell adhesion, expressed mainly in the
granular layer of the epidermis (9-12,76).
Dsg1 is closely associated with plakoglobin, an 85-kDa polypeptide
found in the desmosomal plaques of keratinocytes, that links
desmoglein to the intermediate keratin filament network inside the
keratinocyte (76). Dsg1 is
expressed more strongly in skin from the upper torso than that from
the lower torso, buccal mucosa, or scalp, which may explain the
distribution of lesions (7,10-12,74-76).
The mechanism of acantholysis induction by specific autoantibodies
may involve phosphorylation of intracellular proteins associated
with desmosomes.
Other target antigens, including the acetylcholine
receptor and desmoglein 3 (Dsg3), have been postulated to be
relevant in the pathogenesis of PF (4-7,75-77).
The regulation of keratinocyte cell-to-cell and cell-matrix
adhesion is an important biological function of cutaneous
acetylcholine and the progress in therapy of pemphigus using
cholinergic drugs supports this concept (7,9-12,74).
Patients with both sporadic and endemic forms of PF
have anti-Dsg1 antibodies, their titer correlating with the extent
and activity of the disease (76).
The prevalence of anti-Dsg1 antibodies is high in people living in
endemic areas of Brazil, and a Tunisian study found that anti-Dsg1
IgG antibodies were generally against pre-Dsg1 domains and/or
C-terminals of Dsg1 (7,9-12,74-77).
Some cases have been associated with the use of certain drugs, such
as penicillamine (78). In patients
who were treated with penicillamine, PF is more frequent than PV,
with a ratio of 4:1. Penicillamine and captopril contain sulfhydryl
groups that are speculated to interact with the sulfhydryl groups
in Dsg1 and Dsg3 (7,12,76-78).
Most patients with drug-induced pemphigus go into remission after
the offending drug is discontinued.
Genetic factors predispose to the development of
PF. The HLA-DRB1*04:01, HLA-DRB1*04:06, HLA-DRB1*14,
HLA-DRB1*01:01, have been associated with a higher risk of PF
(7,12,77-79).
In the Brazilian population HLA-DRB1 alleles *04:04, *14:02,
*14:06, and *01:02 have been reported as risk factors for fogo
selvagem (12,79). In France, people with DRB1*0102 and
0404 are at an increased risk of PF (77). It has been suggested that
polymorphisms in the 2q33 and 3q21 chromosomal regions increase
susceptibility to PF (80).
Clinical features
Unlike PV, PF only affects the skin. Mucosal
lesions do not usually occur, because Dsg1 is only expressed in
skin (11,74). There are two versions of PF: an
endemic version (fogo selvagem), and a localized version (pemphigus
erythematosus or Senear-Usher syndrome), that typically share the
same clinical findings (9-12,74-76).
Initial circumscribed lesions appear in seborrheic
areas, such as the scalp, face, and chest (presternal and
interscapular regions). Blisters appear slowly and are not obvious,
because the cleavage is superficial, and small flaccid blisters
break easily. The scales separate leaving painful erosions,
surrounded by erythema and small vesicles along the edges (4-8,10-12,74-76).
In the endemic version (fogo selvagem), the erosions are intensely
painful, like ‘wild fire’, and predominantly affect young women in
endemic regions (8,74). In the localized version (pemphigus
erythematosus/Senear-Usher syndrome), the lesions are similar to
the malar erythema present in lupus erythematosus (strongly scaled
erythematous plaques) that appear on sun-exposed areas such as the
scalp, face and upper torso (8,12,74-76,81).
Pemphigus erythematosus mainly affects elderly patients, and
medications, sun exposure and trauma are considered possible
triggers (8,80,81).
In approximately 80% of these cases, immunoreactive deposits along
the basement membrane and a mean titer of antinuclear antibodies
can be detected, usually without the presence of anti-ds-DNA
antibodies, which may suggest an association with lupus
erythematosus (8,80,81).
In PF a common clinical finding is a positive Nikolsky sign, which
is very specific in the diagnosis of pemphigus.
In the most severe form of PF, the skin lesions can
dramatically progress, leading to exfoliative erythroderma,
characterized by generalized erythema and diffuse scaling involving
90% or more of the cutaneous surface (8,74). In
cases of erythroderma of unknown origin, PF must be considered as a
possible cause. These patients require prompt hospitalization to
prevent serious and sometimes fatal complications from metabolic
instability (8,12,74-76).
Unusual presentations of PF have also been
described, such as an acute rash with multiple hyperpigmented and
hyperkeratotic lesions similar to seborrheic keratoses, lesions
resembling impetigo, and scaly erythema on the scalp that may be
confused with seborrheic dermatitis (74,80-82).
In cases of sporadic PF in children, patients have the same primary
lesions (blisters) and secondary lesions (erosions), but with a
distinct configuration that has been described as arcuate,
circinate and/or polycyclic (81-83).
Pemphigus seborrhoicus is a special form of PF, with very
superficial blisters, extensive erythematous plaques and erosions
that develop in the seborrheic areas (4,12).
Diagnosis
The diagnosis of PF is based on the following
criteria: the overall clinical picture, including the patient's
history and physical examination; the histopathological findings;
the presence of autoantibodies as detected by direct and indirect
immunofluorescence studies.
The histologic changes of pemphigus foliaceus,
pemphigus erythematosus, and fogo selvagem are identical. The
histopathological examination of early blisters demonstrates
acantholysis of the upper epidermis, often resulting in a
subcorneal cleft and leading to detachment of the epidermis in its
midlevel. Subcorneal pustules contain neutrophils, fibrin and
scattered acantholytic keratinocytes. The stratum corneum is often
lost from the surface, the deeper epidermis usually remains intact,
eosinophilic spongiosis and a mixed inflammatory infiltrate of
neutrophils and eosinophils in the superficial dermis may be
present (4-7,12,74).
These superficial blisters are histologically indistinguishable
from those seen in staphylococcal scalded skin syndrome or bullous
impetigo, because Dsg1 is targeted in both of these diseases, thus
the histological features may not be diagnostic in the early stages
(74-76).
Chronic persistent lesions are acanthotic, papillomatous, and
hyperkeratotic with focal parakeratosis. Dyskeratotic cells in the
granular layer of older lesions distinguish PF from PV (4-7,12,74-76).
The DIF biopsy must be performed on the skin with a
normal appearance, immediately adjacent to a lesion because
inflamed and blistered skin can lead to the destruction of immune
deposits (28,29,74).
DIF reveals IgG and C3 deposition in intercellular space staining
(ICS), this model being called ‘chicken wire’ (4,12,74-76).
This is a result of the antibody bound to Dsg1 on desmosomes on the
surface of keratinocyte cells. The intensity of this fluorescent
stain in PF may be greater in the upper epidermis due to the
increased density of Dsg1 (28,74).
IIF is positive in over 85% of PF cases and detects circulating IgG
antibodies against epithelial cell surfaces, using monkey or guinea
pig esophagus as substrate (28-30,74).
Staining of the IgG subclass for PF shows both IgG1 and IgG4
subclasses are produced against Dsg1, IgG4 being the predominant
autoantibody subclass. IIF titers can be used to estimate the
disease activity (28-30,74).
ELISA detects anti-Dsg1 antibodies in 71% of patients with PF,
using purified recombinant human Dsg1 to detect IgG autoantibodies
in patient serum (32-34,74,84).
It was found that the sensitivity and specificity of detecting
anti-Dsg1 antibodies by ELISA is 97.9%, respectively 98.9%
(84-86).
In endemic regions with FS, ELISA specificity is relatively lower
because more normal individuals in these areas test positive
(false-positive increase) for total anti-Dsg1 IgG autoantibodies
(85). ELISA titers have been found
to correlate with disease activity, and are considered the best
laboratory test for monitoring a patient's response to therapy
(32-34,84-86).
Trichoscopy has proven to be a useful tool in the differential
diagnosis of scalp damage in pemphigus. Extravasations and yellow
hemorrhagic crusts were the most common findings and the ‘fried egg
sign’ (yellow dots with a whitish halo) was identified as a
trichoscopy feature in pemphigus (87).
The differential diagnosis of PF includes other
forms of pemphigus, bullous impetigo, subcorneal pustular
dermatosis, subacute cutaneous lupus erythematosus, and seborrheic
dermatitis (74-76,84-86).
Treatment
The purpose of therapy in the handling of PF is to
heal the existing lesions and to stop the surfacing of new ones.
Before the advent of steroid therapy, PF was fatal in approximately
60% of patients, and almost always fatal in elderly patients with
concurrent medical problems (11,74-76,88).
With corticosteroids, immunosuppressive therapy, and other
therapeutic options, mortality has been dramatically reduced.
There are several factors to consider when deciding
on a therapy such as the severity of the disease at introduction,
associated medical illnesses in the likes of diabetes or
tuberculosis, the patient's general health and age, hypertension,
the speed of onset, efficacy, adverse effects, and the cost of the
therapy (4,10-12,74).
The initial treatment in PF is topical and oral
corticosteroids. If the condition is not responsive to topical
corticosteroid, systemic corticosteroid therapy may be initiated
with prednisone at a dose of 0.5-1.5 mg/kg daily or prednisolone
20-40 mg daily (40,88). If there are no signs of remission in
the first 2 weeks, a higher dose of prednisone is recommended.
Nearly all patients reach total remission in 4-12 weeks, afterwards
the dose of prednisone is reduced gradually. If no recurrence
happens, the dose is maintained at 5 or 7.5 mg/day, the reason
being that low doses help prevent recurrences (88).
In patients who fail treatment with
corticosteroids, have contraindications to systemic
corticosteroids, or that have serious adverse effects, an
immunosuppressant agent can be added, In cases of severe PF, an
immunosuppressant and prednisone combined treatment can be used
(10-12,40,88).
Immunosuppressants used include azathioprine, cyclophosphamide, and
mycophenolate mofetil. Azathioprine (AZA) is a synthetic, quite
potent, anti-inflammatory immunosuppressant.
Thiopurine-methyltransferase dosing is required before the
administration of AZA. The standard recommended dose is 1-3
mg/kg/day (7,10-12,88).
Mycophenolate mofetil (MMF) works by reducing the production of
antibodies and inhibiting purine synthesis in stimulated T and B
lymphocytes, blocking their proliferative response. The recommended
dose of MMF is 1g x 2 daily. It should be noted that the onset of
action with MMF is slow and evidence of response occurs between 2
and 12 months of continued use (10-12,40,88).
Cyclophosphamide (CP), an alkylating agent, is an immunosuppressive
and cytotoxic drug that binds DNA regardless of the cell cycle
phase. The dose of CP ranges from 1-3 mg/kg per day, generally
given as 50-200 mg per day in doses equally divided or as a single
dose in the morning (10-12,40,88).
Other treatment options for refractory disease, or
if there are contraindications to immunosuppressive agents include
hydroxychloroquine 200 mg x2 daily, dapsone 100 mg daily or up to
1.5 mg/kg daily, methotrexate 10-20 mg weekly, IVIg 2 g/kg monthly,
or RTX, given as 4 weekly infusions of 375 mg/m2
(10,11,47).
Plasmapheresis and IVIg are therapeutic options in patients with
recalcitrant disease (44,45,88).
Considering the possible side effects of therapy,
patients should be monitored closely. After the interruption of
systemic corticosteroids in patients with total remission, adjuvant
immunosuppression can be decreased over 6 to 12 months (41). The interruption of therapy depends
on the clinical picture that shows no active cutaneous lesions over
several months. Negative or low ELISA-Dsg1 values or negative
immunofluorescence are useful to support the discontinuation of
therapy (84-86).
Evolution and complications
PF tends to persist for months or years and is
regarded as a benign disease that responds well to treatment. PF
may be associated with thymoma, myasthenia gravis, lupus
erythematosus, and other autoimmune bullous diseases (88,89).
New cutaneous lesions, changes in primary
morphology, rapid disease progression, constitutional symptoms, or
failure to respond to appropriate therapies may suggest a
concomitant viral skin infection, such as herpes simplex or
cytomegalovirus (89).
7. IgA pemphigus
Epidemiology
IgA pemphigus is one of the rarest forms of the
autoimmune blistering disease. The frequency of IgA pemphigus is
not well defined but has been reported in Asia (Japan and India),
South America (Brazil), Europe (Scandinavian countries), and in the
US (1,3,4-7,90).
The distribution by sex and race is unknown, with cases being
reported in all age groups, with a mean onset age of 53 years
(90).
Pathophysiology and genetic
factors
The exact pathomechanism of IgA pemphigus is not
well defined, but it is related to IgA autoantibodies that target
desmosomal and non-desmosomal keratinocyte cell surface components.
These components are cell-to-cell-adhering molecules, including
Dsg1, Dsg3, and Dsc1 (10-12,90,91).
Desmogleins and desmocollins are glycoproteins that
belong to a superfamily of cadherin molecules. The subcorneal
pustular dermatosis subtype exhibits IgA autoantibodies targeting
the transmembrane glycoprotein Dsc1, while the antigen of the
intraepidermal neutrophilic dermatosis has been found to interact
with both Dsg1 and Dsg3 (90,91).
In IgA pemphigus, autoantibodies bind to sites
containing the monocyte/granulocyte IgA-Fc receptor (CD89), causing
a massive inflammatory reaction and neutrophil infiltration of the
epidermis, which clinically presents as blistering and pustule
(4,7,12,90).
Although the targets of IgA antibodies have been identified, the
direct pathogenic effects of the IgA autoantibodies and the exact
mechanism of blister formation have not been established, thus a
clinical picture of IgA pemphigus is not well known and requires
additional investigations (90).
Clinical features
IgA pemphigus is a rare entity among the pemphigus
diseases. It is considered to be a distinct entity that includes 2
clinical subtypes with different histologic features and different
IgA deposition patterns in the epidermis: intraepidermal
neutrophilic IgA dermatosis (IEN) and the subcorneal pustular
dermatosis (SPD) (4-7,10-12,90).
IgA pemphigus is characterized by fragile blisters
and intraepidermal pustules or vesicles with neutrophilic
infiltration in the erythematous skin located in flexural areas
(axilla and groin), distal trunk, proximal limbs, and
intertriginous sites. The lesions have a distinct tendency to
coalesce, so that clinically often annular infiltrated plaques with
accentuated margins and collarette-like scaling are seen. Mucosal
involvement is rare (4-8,10-12,90).
Diagnosis
Investigations in patients suspected of IgA
pemphigus often include a skin biopsy to detect histological and
immunological changes. Histologic examination of IgA pemphigus
demonstrates subcorneal blisters with massive neutrophilic
infiltration and with a mild loss of cohesion between
keratinocytes. Histopathology is useful in differentiating the two
major subtypes of IgA pemphigus (4-7,12,90).
In the SPD subtype, there are subcorneal pustules and increased
intensity of IgA autoantibodies in the upper surface of the
epidermis. In contrast, the IEN type is histologically
characterized by suprabasal pustules and inflammatory infiltrates,
located in the entire or lower part of the dermis (7,12,90).
DIF is considered an early screening tool for the
diagnosis of IgA pemphigus, detecting the absence or presence of
IgA autoantibodies on epidermal cell surfaces (4,28-31,90).
DIF can be used to differentiate IgA pemphigus from PF because the
clinical differentiation between IgA pemphigus and PF is nearly
impossible (90-92).
DIF of PF identifies IgG autoantibodies against Dsg1 in contrast to
the IgA deposits against Dsc1 found in IgA pemphigus (29,90).
Moreover, in contrast to IgA pemphigus, DIF in Sneddon-Wilkinson
disease will be negative for IgA deposits against adhesion
molecules, such as Dsc1, which is key in the diagnosis of the SPD
subtype of IgA pemphigus (92).
IIF reveals circulating IgA antibodies in
intraepidermal structures in half of the cases (28,30,90).
In the SPD type, Dsc1, one of the desmosomal cadherins, has been
identified as a target autoantigen (91). In the IEN type, the autoantigen is
not fully characterized, but there are several cases in which Dsg1
and Dsg3 have been demonstrated to be targets of the autoantibodies
in this variant (12,90-93).
An association of IgA pemphigus with monoclonal IgA
gammopathies, multiple myeloma, HIV infection, Sjogren's syndrome,
rheumatoid arthritis, and inflammatory bowel diseases (Crohn's
disease, ulcerative colitis) have been reported (90,94).
While the direct relationship between these diseases and IgA
pemphigus is still unclear, a thorough survey of hematologic and
infectious disorders is advised for patients presenting with IgA
pemphigus (90-92).
Differential diagnoses of IgA pemphigus include:
classic subcorneal pustular dermatosis (Sneddon-Wilkinson disease),
dermatitis herpetiformis, PF, eosinophilic pustular folliculitis,
pemphigus herpetiformis and bacterial skin infections (90).
Treatment
The treatment for IgA pemphigus must be directed to
reduce the inflammation because IgA pemphigus represents a group of
autoimmune blistering skin diseases manifested clinically as
chronic inflammation.
Generally, oral and topical corticosteroids with a
suggested daily dose of 0.5-1 mg/kg, are the mainstay of treatment
for IgA pemphigus (40,90-92).
To minimize adverse effects, slow tapering of corticosteroids is
advised in order to identify the lowest efficacious dose, and
patients should be aware of the complications associated with
long-term use of steroids, including osteoporosis, diabetes,
cataracts, and infection (41).
Many studies have found that dapsone may also be helpful in IgA
pemphigus due to its antineutrophilic effects, but long-term
treatment can determine hemolysis and methemoglobinemia (95,96).
Other drugs that have been reported to be successful in the
treatment of IgA pemphigus include colchicine, retinoids,
mycophenolate mofetil, and adalimumab (95,96).
One case report described lesion regression after the addition of
azithromycin to a local steroid and a keratolytic agent, while
another case report described rapid response in SPD-type IgA
pemphigus with oral isotretinoin treatment (97,98).
Evolution and complications
IgA pemphigus presents as a milder and more limited
disease, and by using appropriate treatment and follow-up, IgA
pemphigus usually heals without scarring. Open wounds should be
cared for in order to avoid infections and scarring. For patients
diagnosed with IgA pemphigus, it is recommended that they undergo
screening for hematological diseases, especially the elderly
patients and those with systemic symptoms (90-93).
Complications that can occur during the disease
are: infections (secondary to open wounds or drugs), malignancies
(secondary to the chronic inflammatory process), growth retardation
is possible secondary to medications used to treat IgA pemphigus
during childhood (90-93).
8. Conclusions
The pathophysiology and autoantigen profile of
bullous autoimmune diseases, especially pemphigus and its subforms,
are more complex than previously assumed. Although the
pathophysiology of blistering autoimmune diseases has been
elucidated, there are still unanswered questions, including
determination of the mechanism of the autoantibody production, or
if there are any predictive factors of response to therapy.
Pemphigus is a heterogeneous condition, and further studies are
needed to assess the complexity of the disease.
As most patients require long-term
immunosuppressive therapy, health care providers must establish
effective and interdisciplinary management of the side effects of
therapy.
The treatment of pemphigus should target the cells,
autoantibodies, and/or factors directly involved in pathogenesis to
avoid general immune suppression. New treatments, including
B-cell-directed therapy, are the new therapeutic frontier for this
kind of disease.
In this review, we summarized the process of
establishing and revising the diagnostic criteria, and the clinical
and therapeutic aspects of the main types of intraepidermal
blistering diseases from the pemphigus group.
Acknowledgements
Professional editing, linguistic and technical
assistance performed by Irina Radu, Individual Service
Provider.
Funding
Funding: No funding was received.
Availability of data and materials
All information provided in this review is
documented by relevant references.
Authors' contributions
VVC and MPT conceived and supervised the research.
CP, MFH and EPA analyzed the literatue data. VVC, CP, MFH, EPA and
MPT contributed to data acquisition and interpretation and wrote
the manuscript. All authors contributed to acquisition, analysis
and systematization of the literature data, manuscript writing and
critical revision of it for important intellectual content. All
authors reviewed the results and read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Consent was provided by the patient to use any
clinical image that did not reveal personal identity.
Competing interests
The authors declare that they have no competing
interests and they have no financial relationships to disclose.
References
|
1
|
Lakoš Jukić I, Jerković Gulin S and
Marinović B: Blistering diseases in the mature patient. Clin
Dermatol. 36:231–238. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kasperkiewicz M: COVID-19 outbreak and
autoimmune bullous diseases: A systematic review of published
cases. J Am Acad Dermatol. 84:563–568. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chaudhari P and Marinkovich MP: What's new
in blistering disorders? Curr Allergy Asthma Rep. 7:255–263.
2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hofmann SC, Juratli HA and Eming R:
Bullous autoimmune dermatoses. J Dtsch Dermatol Ges. 16:1339–1358.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bickle K, Roark TR and Hsu S: Autoimmune
bullous dermatoses: A review. Am Fam Physician. 65:1861–1870.
2002.PubMed/NCBI
|
|
6
|
Patrício P, Ferreira C, Gomes MM and
Filipe P: Autoimmune bullous dermatoses: A review. Ann NY Acad Sci.
1173:203–210. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Egami S, Yamagami J and Amagai M:
Autoimmune bullous skin diseases, pemphigus and pemphigoid. J
Allergy Clin Immunol. 145:1031–1047. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kneisel A and Hertl M: Autoimmune bullous
skin diseases. Part 1: Clinical manifestations. J Dtsch Dermatol
Ges. 9:844–856; quiz 857. 2011.PubMed/NCBI View Article : Google Scholar : (In English,
German).
|
|
9
|
Kasperkiewicz M, Ellebrecht CT, Takahashi
H, Yamagami J, Zillikens D, Payne AS and Amagai M: Pemphigus. Nat
Rev Dis Primers. 3(17026)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pollmann R, Schmidt T, Eming R and Hertl
M: Pemphigus: A comprehensive review on pathogenesis, clinical
presentation and novel therapeutic approaches. Clin Rev Allergy
Immunol. 54:1–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Melchionda V and Harman KE: Pemphigus
vulgaris and pemphigus foliaceus: An overview of the clinical
presentation, investigations and management. Clin Exp Dermatol.
44:740–746. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Didona D, Maglie R, Eming R and Hertl M:
Pemphigus: Current and future therapeutic strategies. Front
Immunol. 10(1418)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Oktarina DA, van der Wier G, Diercks GF,
Jonkman MF and Pas HH: IgG-induced clustering of desmogleins 1 and
3 in skin of patients with pemphigus fits with the desmoglein
nonassembly depletion hypothesis. Br J Dermatol. 165:552–562.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Amber KT, Valdebran M and Grando SA:
Non-desmoglein antibodies in patients with pemphigus vulgaris.
Front Immunol. 9(1190)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yan L, Wang JM and Zeng K: Association
between HLA-DRB1 polymorphisms and pemphigus vulgaris: A
meta-analysis. Br J Dermatol. 167:768–777. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sultan AS, Villa A, Saavedra AP, Treister
NS and Woo SB: Oral mucous membrane pemphigoid and pemphigus
vulgaris-a retrospective two-center cohort study. Oral Dis.
23:498–504. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mustafa MB, Porter SR, Smoller BR and
Sitaru C: Oral mucosal manifestations of autoimmune skin diseases.
Autoimmun Rev. 14:930–951. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rashid H, Lamberts A, Diercks GFH, Pas HH,
Meijer JM, Bolling MC and Horváth B: Oral lesions in autoimmune
bullous diseases: An Overview of clinical characteristics and
diagnostic algorithm. Am J Clin Dermatol. 20:847–861.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Carvalho AA, Santos Neto DAD, Carvalho
MADR, Eleutério SJP and Xavier AREO: Neonatal pemphigus in an
infant born to a mother with pemphigus vulgaris: A case report. Rev
Paul Pediatr. 37:130–134. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Panko J, Florell SR, Hadley J, Zone J,
Leiferman K and Vanderhooft S: Neonatal pemphigus in an infant born
to a mother with serologic evidence of both pemphigus vulgaris and
gestational pemphigoid. J Am Acad Dermatol. 60:1057–1062.
2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Amer YB and Al Ajroush W: Pemphigus
vulgaris in a neonate. Ann Saudi Med. 27:453–455. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Huang YH, Wang SH, Kuo TT and Chi CC:
Pemphigus vegetans occurring in a split-thickness skin graft.
Dermatol Surg. 31:240–243. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sillevis Smitt JH, Mulder TJ, Albeda FW
and Van Nierop JC: Pemphigus vegetans in a child. Br J Dermatol.
127:289–291. 1992.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Jain V, Jindal N and Imchen S: Localized
pemphigus vegetans without mucosal involvement. Indian J Dermatol.
59(210)2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Son YM, Kang HK, Yun JH, Roh JY and Lee
JR: The Neumann type of pemphigus vegetans treated with combination
of dapsone and steroid. Ann Dermatol. 23 (Suppl 3):S310–S313.
2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Peterman CM, Vadeboncoeur S, Schmidt BA
and Gellis SE: Pediatric pemphigus herpetiformis: Case report and
review of the literature. Pediatr Dermatol. 34:342–346.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shimizu Y, Wakabayashi K, Hayashi Y, Hara
K, Aoyama R, Niimi T, Tomino Y, Wada R, Hata M and Suzuki Y: MPGN
type 3 associated with pemphigus herpetiformis mimicking PGNMID and
dermatitis herpetiformis. Case Rep Nephrol Dial. 9:15–24.
2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mihai S and Sitaru C: Immunopathology and
molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med.
11:462–481. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aoki V, Sousa JX Jr, Fukumori LM, Périgo
AM, Freitas EL and Oliveira ZN: Direct and indirect
immunofluorescence. An Bras Dermatol. 85:490–500. 2010.PubMed/NCBI View Article : Google Scholar : (In English,
Portuguese).
|
|
30
|
Mihályi L, Kiss M, Dobozy A, Kemény L and
Husz S: Clinical relevance of autoantibodies in patients with
autoimmune bullous dermatosis. Clin Dev Immunol.
2012(369546)2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Saschenbrecker S, Karl I, Komorowski L,
Probst C, Dähnrich C, Fechner K, Stöcker W and Schlumberger W:
Serological diagnosis of autoimmune bullous skin diseases. Front
Immunol. 10(1974)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Abasq C, Mouquet H, Gilbert D, Tron F,
Grassi V, Musette P and Joly P: ELISA testing of anti-desmoglein 1
and 3 antibodies in the management of pemphigus. Arch Dermatol.
145:529–535. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mortazavi H, Shahdi M, Amirzargar AA,
Naraghi ZS, Valikhani M, Daneshpazhooh M, Vasheghani-Farahani A,
Sedaghat M and Chams-Davatchi C: Desmoglein ELISA in the diagnosis
of pemphigus and its correlation with the severity of pemphigus
vulgaris. Iran J Allergy Asthma Immunol. 8:53–56. 2009.PubMed/NCBI
|
|
34
|
Tampoia M, Giavarina D, Di Giorgio C and
Bizzaro N: Diagnostic accuracy of enzyme-linked immunosorbent
assays (ELISA) to detect anti-skin autoantibodies in autoimmune
blistering skin diseases: A systematic review and meta-analysis.
Autoimmun Rev. 12:121–126. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Naseer SY, Seiffert-Sinha K and Sinha AA:
Detailed profiling of anti-desmoglein autoantibodies identifies
anti-Dsg1 reactivity as a key driver of disease activity and
clinical expression in pemphigus vulgaris. Autoimmunity.
48:231–241. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Di Zenzo G, Di Lullo G, Corti D, Calabresi
V, Sinistro A, Vanzetta F, Didona B, Cianchini G, Hertl M, Eming R,
et al: Pemphigus autoantibodies generated through somatic mutations
target the desmoglein-3 cis-interface. J Clin Invest.
122:3781–3790. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lakshmi MJD, Jaisankar TJ, Rajappa M,
Thappa DM, Chandrashekar L, Divyapriya D, Munisamy M and Revathy G:
Correlation of antimuscarinic acetylcholine receptor antibody
titers and antidesmoglein antibody titers with the severity of
disease in patients with pemphigus. J Am Acad Dermatol. 76:895–902.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Heelan K, Mahar AL, Walsh S and Shear NH:
Pemphigus and associated comorbidities: A cross-sectional study.
Clin Exp Dermatol. 40:593–599. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Parameswaran A, Attwood K, Sato R,
Seiffert-Sinha K and Sinha AA: Identification of a new disease
cluster of pemphigus vulgaris with autoimmune thyroid disease,
rheumatoid arthritis and type I diabetes. Br J Dermatol.
172:729–738. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Almugairen N, Hospital V, Bedane C,
Duvert-Lehembre S, Picard D, Tronquoy AF, Houivet E, D'incan M and
Joly P: Assessment of the rate of long-term complete remission off
therapy in patients with pemphigus treated with different regimens
including medium- and high-dose corticosteroids. J Am Acad
Dermatol. 69:583–588. 2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Satyanarayanasetty D, Pawar K, Nadig P and
Haran A: Multiple adverse effects of systemic corticosteroids: A
case report. J Clin Diagn Res. 9:FD01–FD2. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ahmed AR, Kaveri S and Spigelman Z:
Long-term remissions in recalcitrant pemphigus vulgaris. N Engl J
Med. 373:2693–2694. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Beissert S, Werfel T, Frieling U, Böhm M,
Sticherling M, Stadler R, Zillikens D, Rzany B, Hunzelmann N,
Meurer M, et al: A comparison of oral methylprednisolone plus
azathioprine or mycophenolate mofetil for the treatment of
pemphigus. Arch Dermatol. 142:1447–1454. 2006.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Amagai M, Ikeda S, Shimizu H, Iizuka H,
Hanada K, Aiba S, Kaneko F, Izaki S, Tamaki K, Ikezawa Z, et al:
Pemphigus study group. A randomized double-blind trial of
intravenous immunoglobulin for pemphigus. J Am Acad Dermatol.
60:595–603. 2009.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Higashihara T, Kawase M, Kobayashi M, Hara
M, Matsuzaki H, Uni R, Matsumura M, Etoh T and Takano H: Evaluating
the efficacy of double-filtration plasmapheresis in treating five
patients with drug-resistant pemphigus. Ther Apher Dial.
21:243–247. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Eming R and Hertl M: Immunoadsorption in
pemphigus. Autoimmunity. 39:609–616. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Du FH, Mills EA and Mao-Draayer Y:
Next-generation anti-CD20 monoclonal antibodies in autoimmune
disease treatment. Auto Immun Highlights. 8(12)2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wang HH, Liu CW, Li YC and Huang YC:
Efficacy of rituximab for pemphigus: A systematic review and
meta-analysis of different regimens. Acta Derm Venereol.
95:928–932. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Seyfizadeh N, Seyfizadeh N, Hasenkamp J
and Huerta-Yepez S: A molecular perspective on rituximab: A
monoclonal antibody for B cell non Hodgkin lymphoma and other
affections. Crit Rev Oncol Hematol. 97:275–290. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ahmed AR, Spigelman Z, Cavacini LA and
Posner MR: Treatment of pemphigus vulgaris with rituximab and
intravenous immune globulin. N Engl J Med. 355:1772–1779.
2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Schmidt E, Goebeler M and Zillikens D:
Rituximab in severe pemphigus. Ann N Y Acad Sci. 1173:683–691.
2009.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Chiu HY, Chang CY, Hsiao CH and Wang LF:
Concurrent cytomegalovirus and herpes simplex virus infection in
pemphigus vulgaris treated with rituximab and prednisolone. Acta
Derm Venereol. 93:200–201. 2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Wei KC, Chen W, Tang PL and Huang YT:
Pneumocystis jirovecii pneumonia infection in pemphigus patients
treated with rituximab: An observational nationwide epidemiological
study in Taiwan. Eur J Dermatol. 28:713–715. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Amber KT, Kodiyan J, Bloom R and Hertl M:
The controversy of hepatitis C and rituximab: A multidisciplinary
dilemma with implications for patients with pemphigus. Indian J
Dermatol Venereol Leprol. 82:182–183. 2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Frampton JE: Rituximab: A review in
pemphigus vulgaris. Am J Clin Dermatol. 21:149–156. 2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Rapp M, Pentland A and Richardson C:
Successful treatment of pemphigus vulgaris with ofatumumab. J Drugs
Dermatol. 17:1338–1339. 2018.PubMed/NCBI
|
|
57
|
Ellebrecht CT, Choi EJ, Allman DM, Tsai
DE, Wegener WA, Goldenberg DM and Payne AS: Subcutaneous
veltuzumab, a humanized anti-CD20 antibody, in the treatment of
refractory pemphigus vulgaris. JAMA Dermatol. 150:1331–1335.
2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Vinay K, Kanwar AJ, Mittal A, Dogra S,
Minz RW and Hashimoto T: Intralesional rituximab in the treatment
of refractory oral pemphigus vulgaris. JAMA Dermatol. 151:878–882.
2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Paolino G, Didona D, Magliulo G, Iannella
G, Didona B, Raffaele S, Moliterni E, Donati M, Ciofalo A, Granata
G, et al: Paraneoplastic pemphigus: Insight into the autoimmune
pathogenesis, clinical features and therapy. Int J Mol Sci.
18(2532)2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Tsuchisaka A, Numata S, Teye K, Natsuaki
Y, Kawakami T, Takeda Y, Wang W, Ishikawa K, Goto M, Koga H, et al:
Epiplakin is a paraneoplastic pemphigus autoantigen and related to
bronchiolitis obliterans in Japanese patients. J Investig Dermatol.
136:399–408. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Czernik A, Camilleri M, Pittelkow MR and
Grando SA: Paraneoplastic autoimmune multiorgan syndrome: 20 years
after. Int J Dermatol. 50:905–914. 2011.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wade MS and Black MM: Paraneoplastic
pemphigus: A brief update. Australas J Dermatol. 46:1–8; quiz 9-10.
2005.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Martel P, Loiseau P, Joly P, Busson M,
Lepage V, Mouquet H, Courville P, Flageul B, Charron D, Musette P,
et al: Paraneoplastic pemphigus is associated with the DRB1*03
allele. J Autoimmun. 20:91–95. 2003.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Kim JH and Kim SC: Paraneoplastic
pemphigus: Paraneoplastic autoimmune disease of the skin and
mucosa. Front Immunol. 10(1259)2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Healy WJ, Peters S and Nana-Sinkam SP: A
middle-aged man presenting with unexplained mucosal erosions and
progressive dyspnoea. BMJ Case Rep.
2015(bcr2014208677)2015.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Mar WA, Glaesser R, Struble K,
Stephens-Groff S, Bangert J and Hansen RC: Paraneoplastic pemphigus
with bronchiolitis obliterans in a child. Pediatr Dermatol.
20:238–242. 2003.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wieczorek M and Czernik A: Paraneoplastic
pemphigus: A short review. Clin Cosmet Investig Dermatol.
9:291–295. 2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Cummins DL, Mimouni D, Tzu J, Owens N,
Anhalt GJ and Meyerle JH: Lichenoid paraneoplastic pemphigus in the
absence of detectable antibodies. J Am Acad Dermatol. 56:153–159.
2007.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Hashimoto T, Amagai M, Watanabe K,
Chorzelski TP, Bhogal BS, Black MM, Stevens HP, Boorsma DM, Korman
NJ, Gamou S, et al: Characterization of paraneoplastic pemphigus
autoantigens by immunoblot analysis. J Invest Dermatol.
104:829–834. 1995.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Frew JW and Murrell DF: Current management
strategies in paraneoplastic pemphigus (paraneoplastic autoimmune
multiorgan syndrome). Dermatol Clin. 29:607–612. 2011.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lee A, Sandhu S, Imlay-Gillespie L,
Mulligan S and Shumack S: Successful use of Bruton's kinase
inhibitor, ibrutinib, to control paraneoplastic pemphigus in a
patient with paraneoplastic autoimmune multiorgan syndrome and
chronic lymphocytic leukaemia. Australas J Dermatol. 58:e240–e242.
2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Hohwy T, Bang K, Steiniche T, Peterslund
NA and d'Amore F: Alemtuzumab-induced remission of both severe
paraneoplastic pemphigus and leukaemic bone marrow infiltration in
a case of treatment-resistant B-cell chronic lymphocytic leukaemia.
Eur J Haematol. 73:206–209. 2004.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Gu L and Ye S: Tocilizumab cannot prevent
the development of bronchiolitis obliterans in patients with
Castleman disease-associated paraneoplastic pemphigus. J Clin
Rheumatol. 25:e77–e78. 2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
James KA, Culton DA and Diaz LA: Diagnosis
and clinical features of pemphigus foliaceus. Dermatol Clin.
29:405–412, viii. 2011.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Bastuji-Garin S, Souissi R, Blum L, Turki
H, Nouira R, Jomaa B, Zahaf A, Ben Osman A, Mokhtar I, Fazaa B, et
al: Comparative epidemiology of pemphigus in Tunisia and France:
Unusual incidence of pemphigus foliaceus in young Tunisian women. J
Invest Dermatol. 104:302–305. 1995.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Shirakata Y, Amagai M, Hanakawa Y,
Nishikawa T and Hashimoto K: Lack of mucosal involvement in
pemphigus foliaceus may be due to low expression of desmoglein 1. J
Invest Dermatol. 110:76–78. 1998.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Abida O, Kallel-Sellami M, Joly P, Ben
Ayed M, Zitouni M, Masmoudi A, Mokni M, Fezzaa B, Ben Osman A,
Kammoun MR, et al: Franco-Tunisian group of survey and research on
pemphigus. Anti-desmoglein 1 antibodies in healthy related and
unrelated subjects and patients with pemphigus foliaceus in endemic
and non-endemic areas from Tunisia. J Eur Acad Dermatol Venereol.
23:1073–1078. 2009.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Matkaluk RM and Bailin PL:
Penicillamine-induced pemphigus foliaceus. A fatal outcome. Arch
Dermatol. 117:156–157. 1981.PubMed/NCBI
|
|
79
|
Aoki V, Millikan RC, Rivitti EA,
Hans-Filho G, Eaton DP, Warren SJ, Li N, Hilario-Vargas J, Hoffmann
RG and Diaz LA: Cooperative Group for Fogo Selvagem Research.
Environmental risk factors in endemic pemphigus foliaceus (fogo
selvagem). J Investig Dermatol Symp Proc. 9:34–40. 2004.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Dalla-Costa R, Pincerati MR, Beltrame MH,
Malheiros D and Petzl-Erler ML: Polymorphisms in the 2q33 and 3q21
chromosome regions including T-cell coreceptor and ligand genes may
influence susceptibility to pemphigus foliaceus. Hum Immunol.
71:809–817. 2010.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Bruckner N, Katz RA and Hood AF: Pemphigus
foliaceus resembling eruptive seborrheic keratoses. Arch Dermatol.
116:815–816. 1980.PubMed/NCBI
|
|
82
|
Jacyk WK and Simson IW: Pemphigus
erythematosus resembling multiple seborrheic keratoses. Arch
Dermatol. 126:543–544. 1990.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Metry DW, Hebert AA and Jordon RE:
Nonendemic pemphigus foliaceus in children. J Am Acad Dermatol.
46:419–422. 2002.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Gomi H, Kawada A, Amagai M and Matsuo I:
Pemphigus erythematosus: Detection of anti-desmoglein-1 antibodies
by ELISA. Dermatology. 199:188–189. 1999.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Qaqish BF, Prisayanh P, Qian Y, Andraca E,
Li N, Aoki V, Hans-Filho G, dos Santos V, Rivitti EA and Diaz LA:
Cooperative Group on Fogo Selvagem Research. Development of an
IgG4-based predictor of endemic pemphigus foliaceus (fogo
selvagem). J Invest Dermatol. 129:110–118. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Schmidt E, Dähnrich C, Rosemann A, Probst
C, Komorowski L, Saschenbrecker S, Schlumberger W, Stöcker W,
Hashimoto T, Bröcker EB, et al: Novel ELISA systems for antibodies
to desmoglein 1 and 3: Correlation of disease activity with serum
autoantibody levels in individual pemphigus patients. Exp Dermatol.
19:458–463. 2010.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Sar-Pomian M, Kurzeja M, Rudnicka L and
Olszewska M: The value of trichoscopy in the differential diagnosis
of scalp lesions in pemphigus vulgaris and pemphigus foliaceus. An
Bras Dermatol. 89:1007–1012. 2014.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Sami N, Qureshi A and Ahmed AR: Steroid
sparing effect of intravenous immunoglobulin therapy in patients
with pemphigus foliaceus. Eur J Dermatol. 12:174–178.
2002.PubMed/NCBI
|
|
89
|
Sawamura S, Kajihara I, Makino K, Makino
T, Fukushima S, Jinnin M, Oyama B, Hashimoto T and Ihn H: Systemic
lupus erythematosus associated with myasthenia gravis, pemphigus
foliaceus and chronic thyroiditis after thymectomy. Australas J
Dermatol. 58:e120–e122. 2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Kridin K, Patel PM, Jones VA, Cordova A
and Amber KT: IgA pemphigus: A systematic review. J Am Acad
Dermatol. 82:1386–1392. 2020.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Düker I, Schaller J, Rose C, Zillikens D,
Hashimoto T and Kunze J: Subcorneal pustular dermatosis–type IgA
pemphigus with autoantibodies to desmocollins 1, 2, and 3. Arch
Dermatol. 145:1159–1162. 2009.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Watts PJ and Khachemoune A: Subcorneal
pustular dermatosis: A review of 30 years of progress. Am J Clin
Dermatol. 17:653–671. 2016.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Kuan YZ, Chiou HT, Chang HC, Chan HL and
Kuo TT: Intraepidermal neutrophilic IgA dermatosis. J Am Acad
Dermatol. 22:917–919. 1990.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Adam Z, Krejcí M, Pour L, Feit J, Büchler
T and Hájek R: IgA pemphigus associated with monoclonal gammopathy
completely resolved after achievement of complete remission of
multiple myeloma with bortezomib, cyclophosphamide and
dexamethasone regimen. Wien Klin Wochenschr. 122:311–314.
2010.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Hirata Y, Abe R, Kikuchi K, Hamasaka A,
Shinkuma S, Ujiie H, Nomura T, Nishie W, Arita K and Shimizu H:
Intraepidermal neutrophilic IgA pemphigus successfully treated with
dapsone. Eur J Dermatol. 22:282–283. 2012.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Howell SM, Bessinger GT, Altman CE and
Belnap CM: Rapid response of IgA pemphigus of the subcorneal
pustular dermatosis subtype to treatment with adalimumab and
mycophenolate mofetil. J Am Acad Dermatol. 53:541–543.
2005.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Bliziotis I, Rafailidis P, Vergidis P and
Falagas ME: Regression of subcorneal pustular dermatosis type of
IgA pemphigus lesions with azithromycin. J Infect. 51:E31–E34.
2005.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Gruss C, Zillikens D, Hashimoto T, Amagai
M, Kroiss M, Vogt T, Landthaler M and Stolz W: Rapid response of
IgA pemphigus of subcorneal pustular dermatosis type to treatment
with isotretinoin. J Am Acad Dermatol. 43:923–926. 2000.PubMed/NCBI View Article : Google Scholar
|