Introduction
X-linked retinoschisis (XLRS) [Online Mendelian Inheritance in Man (OMIM): 312700, XLRS1] is a complex ocular fundus degeneration disease manifesting as splitting of the outer plexiform and inner nuclear layers of the neurosensory retina (1). Patients exhibit varying degrees of progressive retinal splits that cause varying levels of visual handicap, at times accompanied by additional eye abnormalities, such as a macular hole, macular hemorrhage, retinal detachment and myopia (2-5). Globally, XLRS has a prevalence of ~1/25,000-1/5,000 among males and is a prominent cause of vision loss in males (6). Multiple analyses have indicated that XLRS is a developmental disorder caused by mutations in genes associated with retinoschisis (7-13). XLRS is a progressively worsening fundus disease characterized by complete or incomplete splitting of the outer plexiform and inner nuclear layers of the neurosensory retina. There is much evidence that retinoschisin 1 (RS1) mutation triggers the pathological process of retinoschisis, particularly under familial circumstances. RS1 was first described in 1898(14), the gene is located on the human Xp22.1 chromosome, consists of six exons and five introns and encodes retinoschisin containing a disk-like domain (OMIM: 300839; Xp22.1). Retinoschisin is thought to be an adhesive protein involved in cell bonding and the maintenance structural integrity (15). Numerous genes intricately orchestrate eye formation and RS1 has a key role in the developmental integrity of the retina. Males with hemizygous pathogenic RS1 mutations usually have retinoschisis, females with heterozygous RS1 mutations frequently have a normal fundus. However, one study noted binocular retinoschisis and fundus hemorrhage in a female with homozygosity who acquired the RS1 mutation due to both parents being carriers for the mutated gene (16). Almost all male patients with pathogenic RS1 variants feature splitting of the fovea; furthermore, <50% of patients have peripheral retinoschisis and the majority of peripheral splitting occurs in the subtemporal retina (17).
Whole-exome sequencing (WES) has been widely employed in the study of a variety of genetic diseases, particularly for identifying candidate genes and pathogenic variants in patients with heterogeneous genetic diseases (18-21), such as XLRS. Certain RS1 mutations have been reported in China (2,9,12,22). In the present study, a pathogenic variant, RS1:c.559C>T (p.Q187X), was identified in a three-generation Chinese family with XLRS. The present study is the first to report on RS1:c.559C>T (p.Q187X) associated with XLRS in China, although it was previously reported in Germany by Renner et al (23).
Materials and methods
Subjects
A three-generation Chinese family with XLRS were recruited from Yin Hai Eye Hospital affiliated with Chengdu University of Traditional Chinese Medicine (Chengdu, China). A total of 100 healthy controls (age, 20-55 years; 50 males and 50 females) unrelated to the family were recruited from the Yin Hai Eye Hospital (Chengdu, China). Written informed consent was obtained from all participants according to the Declaration of Helsinki. The study protocol was approved by the Yin Hai Eye Hospital affiliated with Chengdu University of Traditional Chinese Medicine Ethics Committee (Chengdu, China; no. CMEC2010-22). The pedigree's clinical history and clinical data were collected. Detailed anterior segmental and fundus examinations were performed on the participants.
DNA sampling and exome sequencing
Peripheral blood was obtained from the participants using disposable EDTA vacuum blood collection tubes (BD Biosciences), and DNA was extracted using the QIAamp DNA Blood Midi Kit (Qiagen GmbH). Qualified genomic DNA was randomly fragmented by Covaris technology (200 and 300 bp) and adapters were ligated to both ends of the resulting fragments. The DNA was amplified by ligation-mediated PCR, purified and hybridized to an exome array for enrichment. Non-hybridized fragments were then removed by washing. The exomes were captured using a MedE006 kit (MyGenostics) and each qualified captured library was loaded onto an Illumina HiSeq X-10 system (Illumina Inc.). High-throughput sequencing of the captured library was performed to ensure that the samples met the desired average sequencing coverage. Sequencing-derived raw image files were processed using the Illumina HiSeq X-10 system with default parameters and the sequencing data were generated as paired-end reads, which were defined as ‘raw data’ and stored in FASTQ format. The detailed process is depicted in Fig. 1.
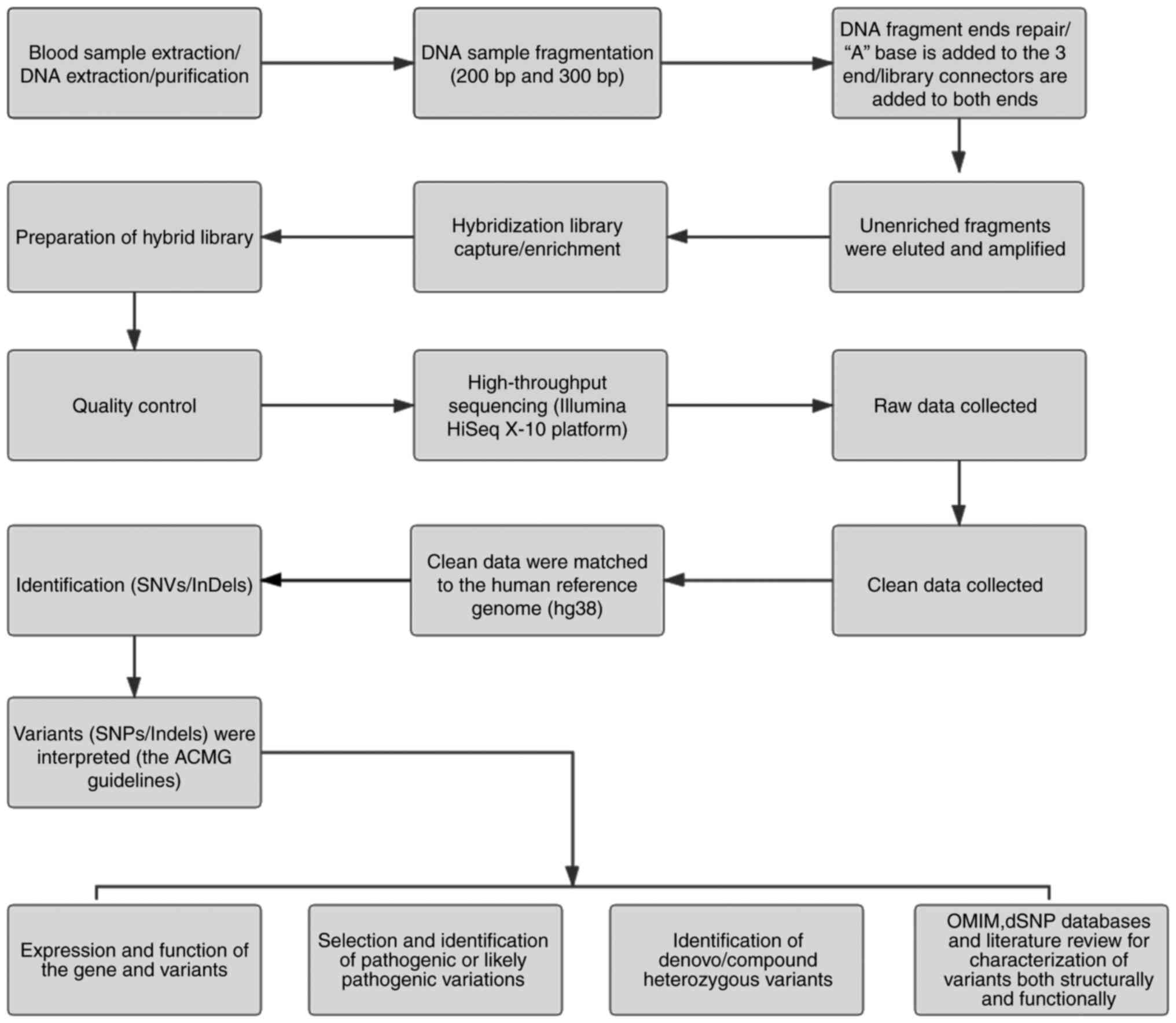 |
Figure 1
Schematic presentation of the detailed data interpretation pipeline. SNV, single-nucleotide variant; SNP, single-nucleotide polymorphism; InDel, insertion or deletion; OMIM, Online Mendelian Inheritance in Man; ACMG, American College for Medical Genetics and Genomics.
|
Sequence alignment and variant calling
WES was performed for the proband. Raw data were saved in FASTQ format. Illumina sequencing adapters and low-quality reads (<80 bp) were filtered by Cutadapt (24). After quality control, the clean reads were mapped to the UCSChg38 human reference genome using Burrows-Wheeler Aligner (BWA) (25). Duplicated reads were removed using Picard tools (http://broadinstitute.github.io/picard/) and only uniquely mapped reads were employed for variation detection. Single-nucleotide polymorphisms (SNPs) and insertion or deletions (InDels) were detected with GATK (26), further annotated by ANNOVAR (27) and evaluated with multiple databases, such as 1000 Genomes Project (ncbi.nlm.nih.gov/variation/tools/1000genomes/), ESP6500.exon.program (evs.gs.washington.edu/EVS/), dbSNP (ftp-trace.ncbi.nih.gov/snp/organisms/), ExAC (exac.broadinstitute.org/), Inhouse (MyGenostics), HGMD (hgmd.cf.ac.uk/ac/index.php), OMIM (omim.org/), ClinVar (ncbi.nlm.nih.gov/clinvar/) and gnomAD (gnomad.broadinstitute.org/about). Sorting Intolerant FromTolerant (SIFT) (28), PolyPhen-2(29), MutationTaster (30) and GERP++ (31) were used for prediction. The interpretation and evidence grading of variations were performed according to the latest edition of the standards and guidelines for the interpretation of gene variation issued by the American College for Medical Genetics and Genomics (ACMG) (32-35).
Variant filtering and annotation
After obtaining reliable SNPs and InDels, the SnpEff tool (http://snpeff.sourceforge.net/SnpEff_manual.html) was used to i) determine whether the SNPs or InDels lead to changes in the encoded protein and ii) identify whether the mutation is reported in dbSNP v.141, to recognize the subset of variants using MAF <1% in the 1000 Genomes Project, to ascertain the subset of programming non-synonymous SNPs using SIFT score <0.05 and identify intergenic variations with GERP++ >2 or a number of different annotations on particular mutations.
Variant validation with Sanger sequencing
Data analysis revealed a nonsense change in the RS1 gene. Sanger sequencing was used to assess the variant in the family members and healthy subjects. PCR primers were designed by Invitrogen (Thermo Fisher Scientific, Inc.) as follows: RS1 forward, 5'-CTGACTTTCTCTGGCCCTGA-3' and reverse, 5'-CTAGTGGCCACCCTATTGGA-3'. Sanger sequencing was performed using an ABI 3730 Genetic Analyzer (Applied Biosystems) following the manufacturer's protocol and the data were examined employing the Individual Genomic Database (ncbi.nlm.nih.gov/genbank/).
Protein modelling
3D models of wild-type (WT) RS1 (NM_000330.3) and the p.Q187X variant were generated using PyMOL and structures of RS1 were obtained from the Protein Data Bank (ID no. 3JD6). The effect of single point mutations on protein stability was simulated using PyMOL (36).
Results
Clinical findings
Ocular clinical outcomes are presented in Table I. Male patients presented with varying degrees of fundus disease, including retinal splitting. As indicated in the pedigree (Fig. 2), patient III-6 was the proband, a 27-year-old male who gradually developed bilateral retinoschisis over the past three years (Fig. 3A and B). The proband's best-corrected visual acuity (BCVA) was 0.2 OD and 0.1 OS. At the age of 12 years, he went to a clinic due to decreased vision in both eyes, was diagnosed with binocular myopia and provided with spectacles for correction. However, the patient's visual acuity had undergone a decline in the previous decade, with abnormal blue-purple colour vision that clearly darkened while looking at objects. The patient visited our clinic for the first time in July 2018. Then in the following 3 years, the examination revealed that his entire retina had slowly separated until schisis. A thorough optical coherence tomography (OCT) evaluation on the patient's eyes was performed. The results demonstrated not only schisis in the fovea of the retina but also layer-like cleavage in the periphery of the retina (Fig. 4A and B). B-scan ultrasonography revealed a local retinal bulge in both eyes of the proband (Fig. 5A). Multifocal electroretinography (mERG) suggested decreased reaction of the central and outer circles on either side of the retina, with central cone malfunction affecting the 30˚ area in the centre of each eye's visual field (Fig. 5B). At the age of 27 years, the proband was diagnosed with XLRS by fundus examination and WES. Patient III-7, the proband's brother, a 20-year-old male, was also a hemizygous carrier (Fig. 6A, III-7 and B, III-7). This patient's BCVA was 0.2 OD and 0.3 OS. However, retinoschisis was not detected in his eyes and it is likely that he was still in the early stages of the disease, with only slight blue-violet colour vision abnormalities. Patient III-4, a 35-year-old male, reported that his vision acuity decreased significantly in the previous seven years; he was finally diagnosed with a macular hole in the eyes and underwent vitrectomy (Fig. 7A, III-4 and B, III-4). This patient's BCVA was 0.02 OD and 0.06 OS. The patient recollected that he also experienced abnormal blue-purple colour vision in the early stage of vision loss, seeing surrounding objects darken, and was finally diagnosed with an idiopathic macular hole, but without WES. Patient I-1, a 70-year-old male, had a BCVA of 0.01 OD and 0.01 OS. Ophthalmic examination revealed that the patient had binocular cataracts, retinoschisis, macular edema and optic nerve atrophy (Fig. 7A, I-1 and B, I-1). The remaining female heterozygous carriers did not present with any retinoschisis (dot in the circle in pedigree chart; Fig. 2). The gene mutation was also detected in them but they were carriers due to having one normal X chromosome. The OCT results of the mother of the proband (II-7) are presented in Fig. 6A, II-7 and B, II-7.
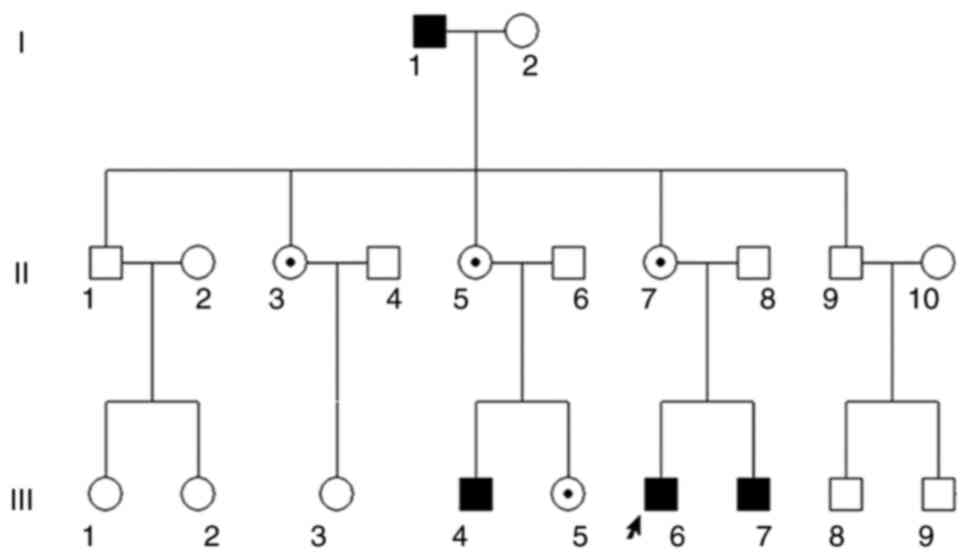 |
Figure 2
Pedigree chart of the three-generation Chinese family with X-linked retinoschisis. Males and females are indicated by squares and circles, respectively. Solid squares represent male patients. A dot in the circle represents a female carrier. Healthy individuals are displayed as empty symbols. The proband is indicated with an arrow.
|
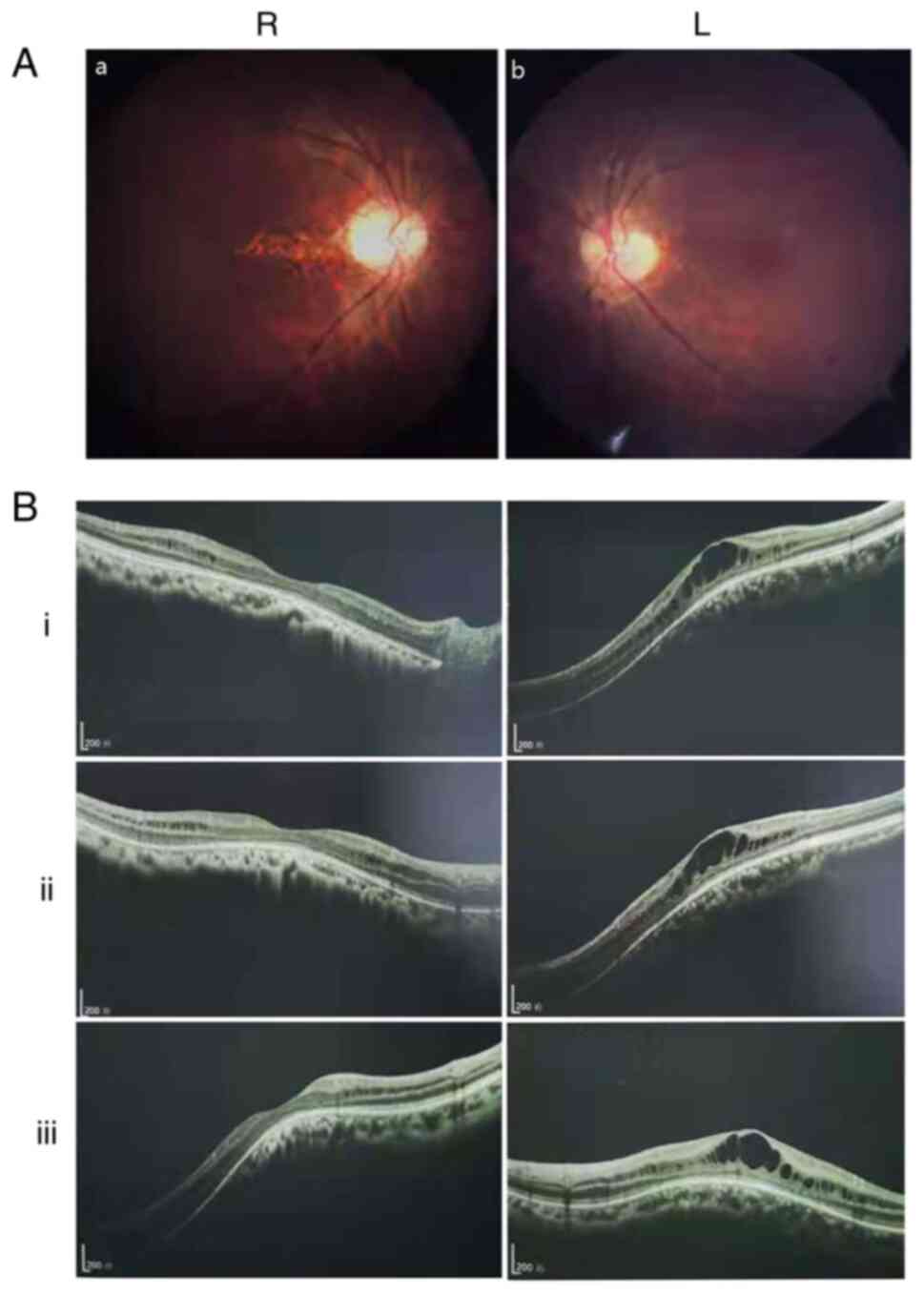 |
Figure 3
(A) Fundus photograph of the proband (III-6). (a) R eye and (b) L eye. (B) Retinal optical coherence tomography indicated that the binocular macular area of the proband gradually split within 3 years [(i) July 2018, (ii) October 2019 and (iii) September 2020]. Scale bar, 200 um; R, right; L, left.
|
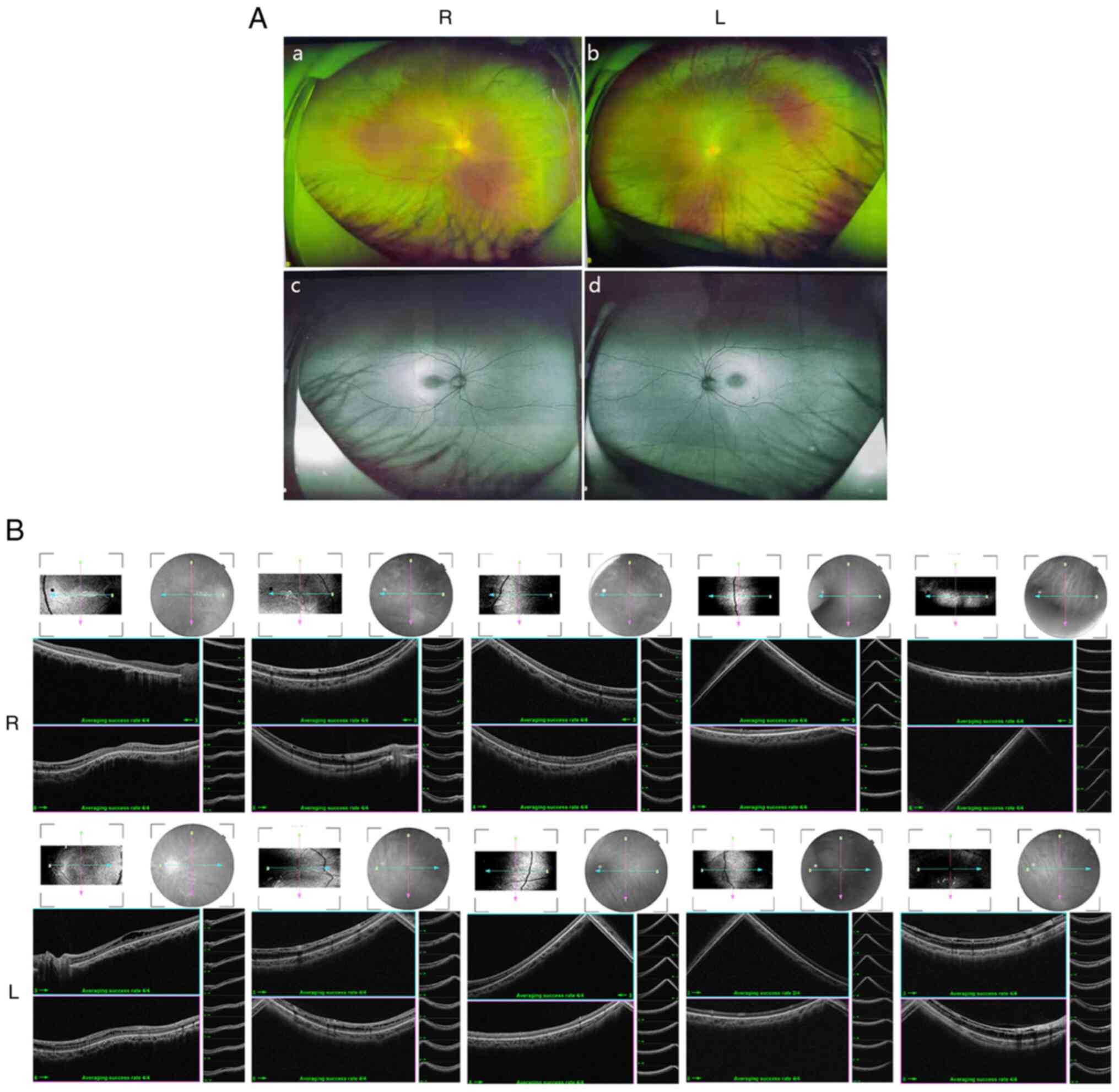 |
Figure 4
(A) Fundus examination and autofluorescence results of both eyes of proband III-6. (a and c) R eye and (b and d) L eye. (B) Detailed binocular retinal optical coherence tomography examination results of the proband revealed binocular retinoschisis, including the binocular macular area and peripheral retinas. R, right; L, left.
|
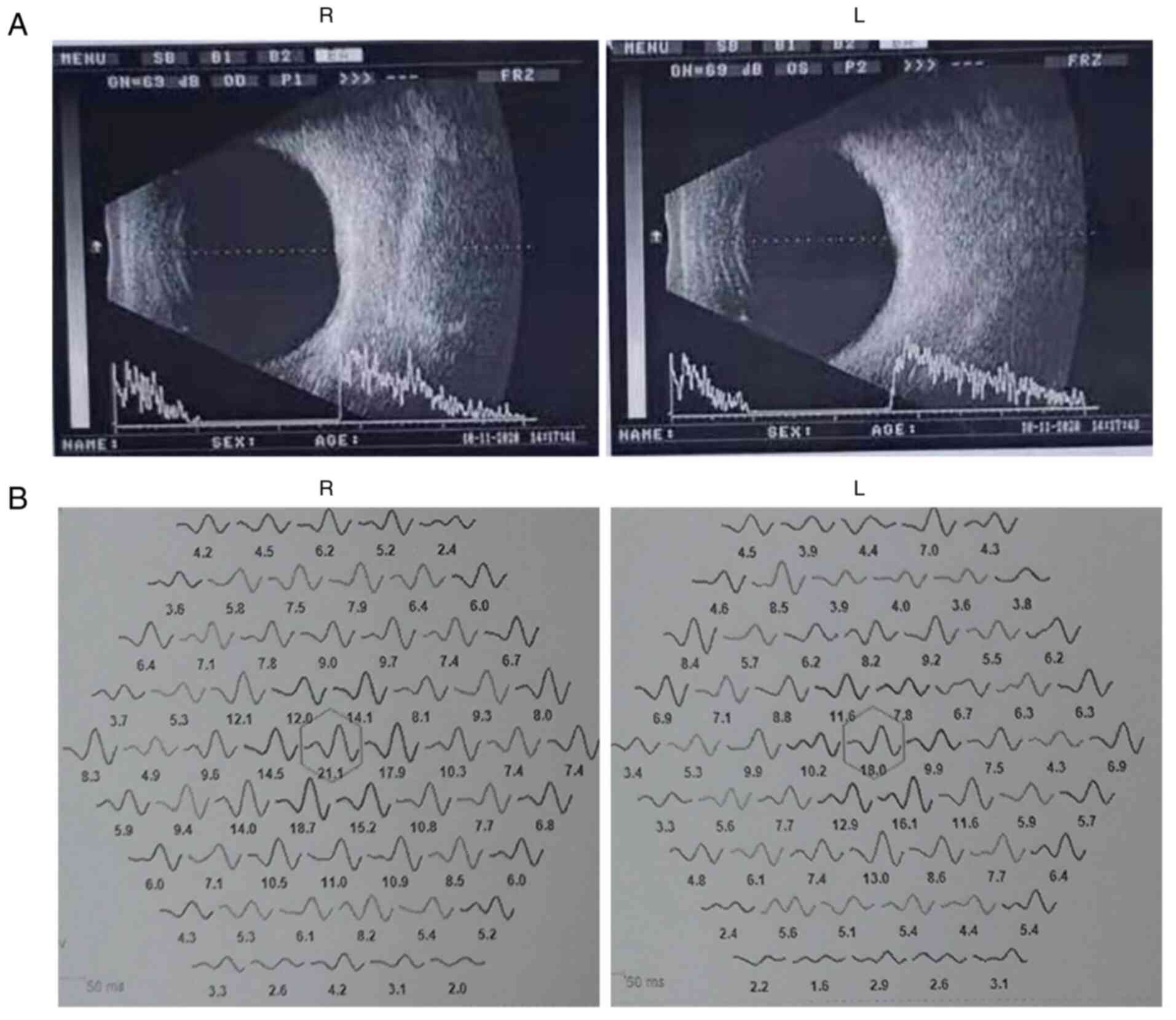 |
Figure 5
(A) Binocular B-ultrasound images of the proband revealed that the macular area of both eyes is locally raised. (B) Binocular electroretinogram analysis of the proband revealed that binocular responses were reduced in the central and outer rings (central 30 of the visual field). R, right; L, left.
|
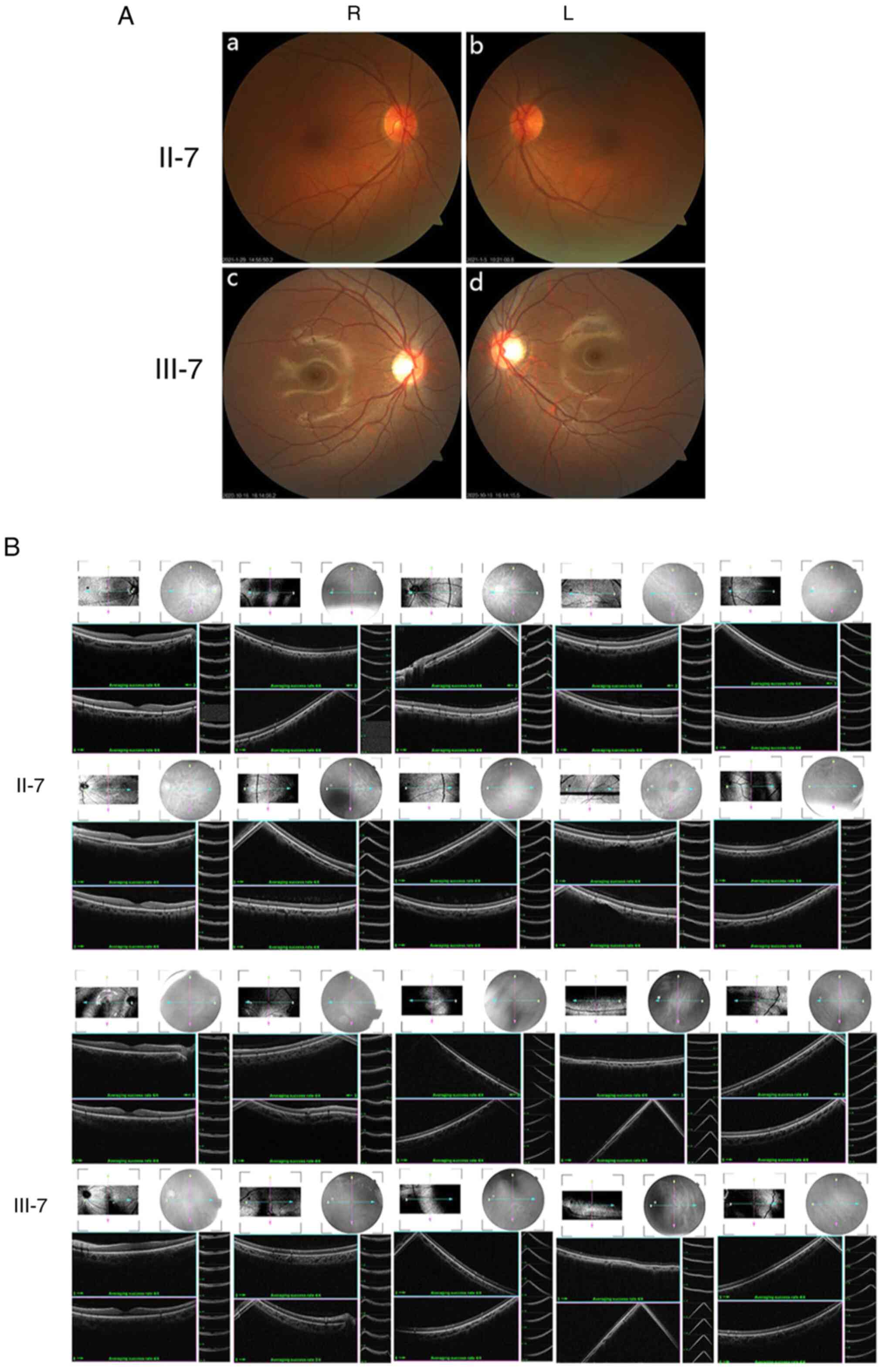 |
Figure 6
(A) Fundus photography of family members II-7 and III-7. (a) II-7, R eye; (b) II-7, L eye; (c) III-7, R eye; (d) III-7, L eye. (B) Retinal optical coherence tomography revealed that retinoschisis was not present in these two individuals (II-7, III-7). R, right; L, left.
|
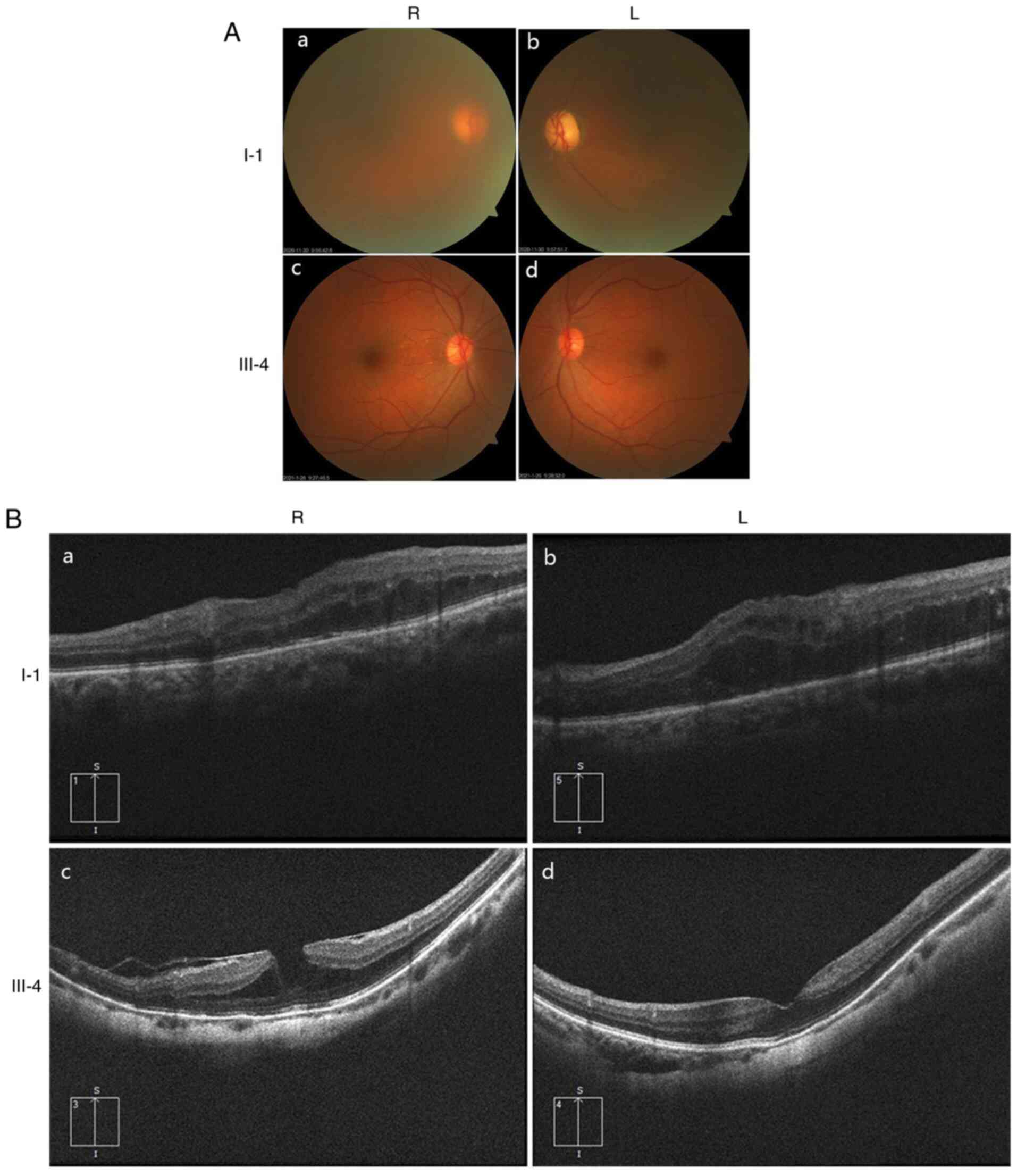 |
Figure 7
(A) Fundus photography of family members I-1 and III-4. (a) I-1, R eye; (b) I-1, L eye; (c) III-4, R eye; (d) III-4, L eye. (B) Retinal OCT revealed that patient I-1 had binocular retinoschisis, macular enema and optic nerve atrophy. Retinal OCT indicated that patient III-4 has a macular hole in both eyes with retinoschisis. (a) I-1, R eye; (b) I-1, L eye; (c) III-4, R eye; (d) III-4, L eye. R, right; L, left; OCT, optical coherence tomography.
|
 |
Table I
Clinical information of the subjects.
|
Table I
Clinical information of the subjects.
| Patient |
Sex |
Age of onset (years) |
BCVA R/L |
Color vision |
Retinoschisis |
Surgery |
Gene type |
Other abnormalities |
| III-6 |
M |
24 |
0.2/0.1 |
Blue-purple anomaly |
Bilateral macular and peripheral retinal |
- |
Hemizygous |
- |
| III-7 |
M |
20 |
0.2/0.3 |
Blue-purple anomaly |
Not found |
- |
Hemizygous |
- |
| III-4 |
M |
28 |
0.02/0.06 |
Blue-purple anomaly |
Macular hole |
Vitrectomy |
Hemizygous |
- |
| I-1 |
M |
NA |
0.01/0.01 |
Blue-purple anomaly |
Bilateral retinoschisis, macular edema |
- |
Hemizygous |
Cataracts, optic nerve atrophy |
| II-7 |
F |
- |
0.8/1.0 |
- |
- |
- |
Heterozygous |
- |
| II-3 |
F |
- |
0.6/0.8 |
- |
- |
- |
Heterozygous |
- |
| II-5 |
F |
- |
0.8/1.0 |
- |
- |
- |
Heterozygous |
- |
| III-5 |
F |
- |
0.8/1.0 |
- |
- |
- |
Heterozygous |
- |
Sequencing and analysis
WES of the proband (III-6) was performed using an Illumina HiSeq X-10 system (Illumina Inc.) with 2X 150 paired-end sequencing. An average of 10,570.65 Mb raw bases was obtained. After filtration, an average of 10,309.65 clean reads was obtained. The clean reads had a high quality value (Q30, 0.91) and the average GC content was 47.07%. The results of the analysis of the clean data suggested that the coverage of the target area was 99.94%, the average sequencing depth was 439.56 and the coverage of the sequencing depth >20X was 98.96%. Thus, the target area had been effectively covered, with high sequencing quality (Table II). After bioinformatics analysis, 117,403 SNPs were identified in the proband, 97.66% of which were annotated in dbSNP and 93.47% were annotated in the 1000 Genomes Project database. Of these, 2,229 SNPs were novel. Among all SNPs, 11,182 were synonymous, 10,306 were missense, 37 were stop-loss, 85 were stop-gain, 19 were start-loss and 90 were splice-site mutations. A total of 20,902 InDels were called. Of these, 75.77% were listed in dbSNP and 58.56% in the 1000 Genomes Project. The number of novel InDels was 3,840. Of the overall InDels, 290 were frameshift, 7 were stop-loss, 4 were start-loss and 76 were splice site mutations (Table III). After comparison with mutation sites in dbSNP, The 1000 Genomes Project, ExAC, OMIM, HGMD, ClinVar and gnomAD, variation sites with a frequency not higher than 0.01 were selected; genes related to hereditary eye diseases were analysed and the following candidate mutation gene loci were screened: AAGAB:c.871-2A>G; SIX3:c.278C>G; GLUD2:c.94C>G; HMGB3:c.573_574insGAG; MYH7:c.5561C>T; MYH7:c.2456G>A; MYO6:c.2743dupA; RS1:c.559C>T. According to ACMG guidelines (33,34), the variant grading evidence is as follows: PVS1 (pathogenic, very strong), this is a nonsense mutation that may cause loss of gene function; PS4 (pathogenic, strong), the prevalence in affected individuals is significantly increased over controls, case reports on this locus are present in the literature and the variant label is DM (disease mutation); PM2 (pathogenic, moderate), this is a low-frequency variant in the normal database. Analysis of the family and verification suggested that the proband's father did not carry a variant at this locus, whereas the mother was a heterozygous carrier. Based on the X-linked genetic model, the filtered data eventually revealed this variant to be RS1:c.559C>T (p.Q187X) in exon 6.
|
Table II
Quality control data statistics for the proband.
|
Table II
Quality control data statistics for the proband.
| Sample |
Value |
| Raw_data_bases, Mb |
10,570.65 |
| Clean_data_bases, Mb |
10,309.65 |
| Aligned_bases, Mb |
10,308.92 |
| Aligned, % |
97.52 |
| Initial bases on target |
11,339,163 |
| Base covered on target |
11,332,246 |
| Coverage of target region, % |
99.94 |
| Effective bases on target |
4,984,289,108 |
| Fraction of effective bases on target, % |
58.21 |
| Average sequencing depth on target |
439.56 |
| Fraction of target covered with at least 4X, % |
99.78 |
| Fraction of target covered with at least 10X, % |
99.51 |
| Fraction of target covered with at least 20X, % |
98.96 |
| Duplication rate, % |
15.11 |
| Quality value |
0.91 |
| Average GC content, % |
47.07 |
| Qualified |
Yes |
| Reported average sequencing depth on target |
529.19 |
| Reported fraction of target covered with at least 4X, % |
99.79 |
| Reported fraction of target covered with at least 10X, % |
99.55 |
| Reported fraction of target covered with at least 20X, % |
99.04 |
|
Table III
Summary statistics for sequencing data for the proband (III-6).
|
Table III
Summary statistics for sequencing data for the proband (III-6).
| Samples |
Value |
| Total SNPs |
117,403 |
| Fraction of SNPs in dbSNP, % |
97.66 |
| Fraction of SNPs in 1000 genomes, % |
93.47 |
| Novel |
2,229 |
| Homozygous |
53,126 |
| Heterozygous |
64,277 |
| Intron |
76,291 |
| 5'UTRs |
2,077 |
| 3'UTRs |
3,524 |
| Upstream |
3,346 |
| Downstream |
1,995 |
| Intergenic |
4,651 |
| X-chromosome |
7 |
| Synonymous |
11,182 |
| Missense |
10,306 |
| Stopgain |
85 |
| Stoploss |
37 |
| Startloss |
19 |
| Splicing |
90 |
| Total InDels |
20,902 |
| Fraction of InDels in dbSNP, % |
75.77 |
| Fraction of InDels in 1000genomes, % |
58.56 |
| Novel |
3.840 |
| Homozygous |
7.550 |
| Heterozygous |
13,191 |
| Intron |
16,351 |
| 5'UTRs |
382 |
| 3'UTRs |
797 |
| Upstream |
422 |
| Intergenic |
760 |
| X-chromosome |
1 |
| Frameshift |
290 |
| Non-frameshift insertion |
183 |
| Non-frameshift Deletion |
181 |
| Stoploss |
7 |
| Startloss |
4 |
| Splicing |
76 |
Variant detection and evaluation
Sanger sequencing was used to examine the RS1 variant in family members of the pedigree and healthy control participants (Fig. 8A). The variant was not detected in 100 standard controls and the site is highly conserved in several species (Fig. 8B). The mutation was assessed in the RS1 mutation database (http://www.dmd.nl/rs), with >230 variants reported to date. Although RS1:c.559C>T (p.Q187X) has been previously reported (23), it was discovered for the first time in Chinese individuals in the present study. In summary, the results comprising clinical information and the gene mutation dates, as stated by the ACMG guidelines, indicate that the nonsense change c.559C>T (p.Q187X) in exon 6 of the RS1 gene is a causal variant responsible for XLRS in this Chinese family. The structure of the RS1 variant protein and the protein surface's electrostatic capacity were predicted in PyMOL and analyzed. The amino acid alterations were indicated to affect highly conserved residues, which may influence the integrity of structural domains. The mutation causes a truncation of the RS1 protein and the secondary structure is obviously changed (Fig. 9A). The structure of the RS1 protein polymer is illustrated in Fig. 9B. The electrostatic potential of the protein surface was also indicated to be changed, which may affect normal folding and interaction with other proteins (Fig. 9C).
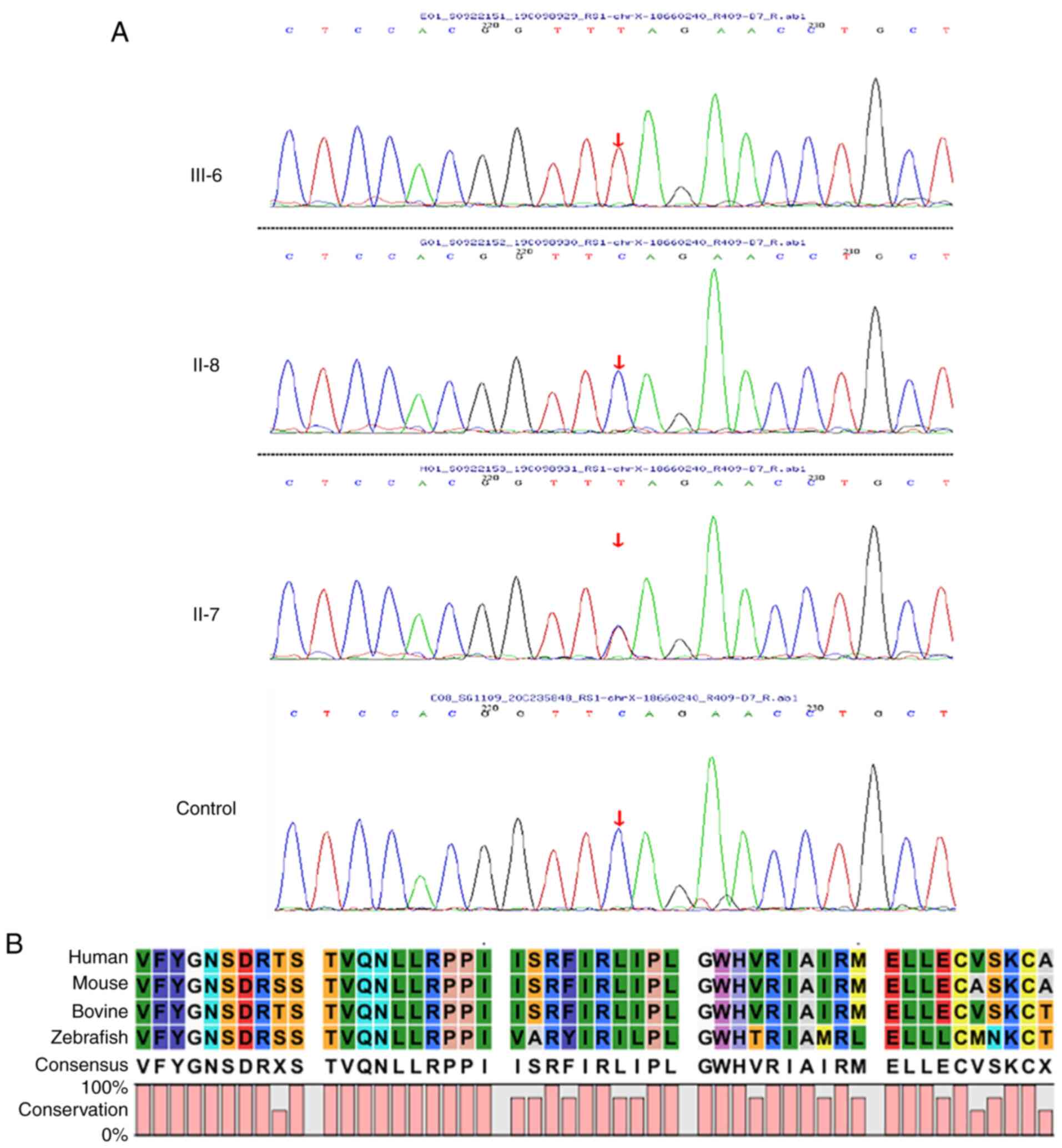 |
Figure 8
Sequencing analysis results. (A) Proband III-6 has an RS1:c.559C>T(p.Q187X) hemizygous mutation, the proband's mother (II-7) is a heterozygous mutation carrier and the proband's father (II-8) does not harbour the mutation. A normal control sequence with no mutation was provided for comparison. (B) Multispecies conserved sequence analysis revealed that this segment is highly conserved.
|
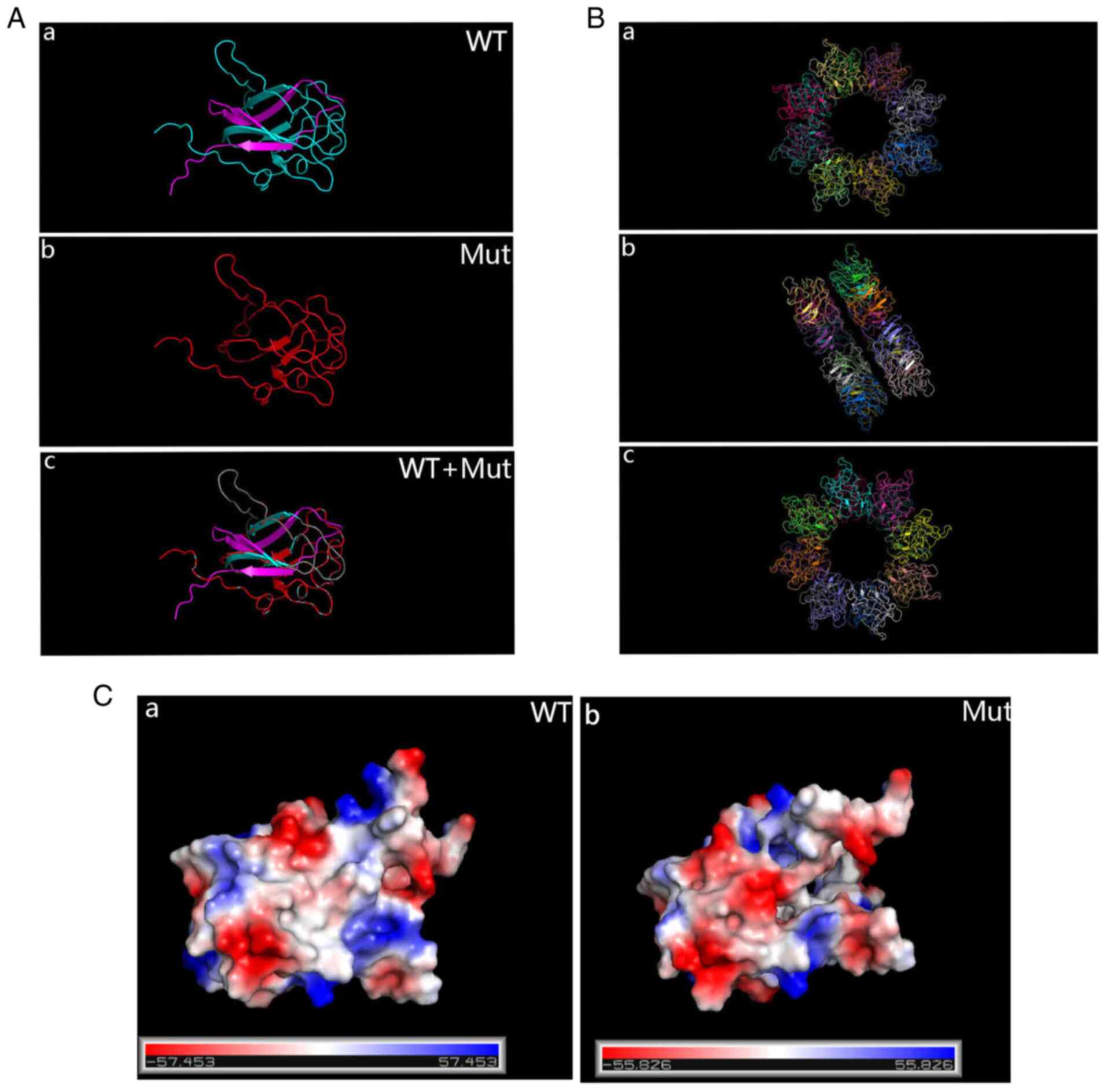 |
Figure 9
(A) PyMOL prediction of the protein structure of WT and Mut RS1. (a) WT; (b) Mut; (c) WT + Mut. For the Mut protein, translation is terminated early and the product is significantly shorter than the WT protein. (B) The functional RS1 forms a dimer of octamer rings comprising a hexadecamer. (a) Front; (b) flank; (c) back. (C) Surface electrostatics of the Mut protein changed drastically compared with the WT protein. (a) WT; (b) Mut. WT, wild-type; Mut, mutant; RS1, retinoschisin 1.
|
Discussion
WES technology has been widely used in the diagnosis of clinically genetically heterogeneous diseases. In this context, WES was performed on a proband. Genes related to hereditary eye diseases were analysed via comparisons with the human reference genome sequence (hg38) and bioinformatics analysis using the dbSNP, the 1000 Genomes Project, ExAC, OMIM, HGMD, ClinVar and gnomAD, and sites with frequency not higher than 0.01 were selected. All candidate variants were evaluated according to ACMG standards and guidelines for further screening and detailed interpretation of variant sites combined with clinical phenotype and family verification. Candidate variations that were significantly inconsistent with the clinical phenotype were eliminated and conformation to the laws of heredity was determined. The robust results revealed that RS1:c.559C>T (p.Q187X) is a likely pathogenic variant, consistent with the patients' clinical phenotype and genetic pattern of the family. The proband was diagnosed with binocular XLRS. A linear OCT scan indicated bilateral foveal retinoschisis accompanying peripheral retinoschisis. Patient III-7 is a hemizygous mutation carrier; although he had not yet developed retinoschisis, he experienced abnormal blue-purple colour vision. Patient III-4 displayed a retinal split in the fovea of both eyes with macular holes. This patient also had abnormal blue-purple colour vision in the early stage, but did not pay any attention to it until he developed a macular hole in both eyes and underwent vitrectomy. Combined with the family's medical history, auxiliary examinations and WES, XLRS due to RS1:c.559C>T (p.Q187X) was diagnosed in I-1 and III-4. In addition, the proband's mother (II-7) and other female family members (II-3, II-5 and III-5) were heterozygous carriers, but they all had a normal fundus. A total of 4 patients in this family had a slow onset and variable fundus presentation. Numerous studies have demonstrated that this is a disease that slowly progresses with age (37,38). Furthermore, individuals harbouring the RS1 mutation display various clinical symptoms, including central retinal split, with or without a peripheral retinal split, macular hole, macular edema, retinal degeneration, retinal hemorrhage, retinal detachment and high myopia (39). Genetic modification and environmental factors that may be involved and the specific mechanisms require to be further explored. To date, >230 distinct RS1 gene mutations have been acknowledged to cause this particular disease (40,41). In the early stages of the disease, blue-purple colour vision abnormalities appear to be an important indicator and clinicians should consider the possibility of retinoschisis when patients complain of unexplained colour vision abnormalities. The early symptoms of retinoschisis may originate from disorganization of the inner retinal layers disrupting synaptic signal transmission from photoreceptors to outer nuclear layer (ON)-bipolar cells and lead to slowly progressive loss of vision and abnormal color vision.
In humans, the RS1 gene is expressed primarily in bipolar cells and photoreceptor cells of the retina. The gene is located on chromosome Xp22.1 and encodes the complex protein retinoschisin, which contains four important subdomains: Exons 1 and 2 encode an internal signal sequence of 23 amino acids for secretion, exon 3 encodes a 39-amino-acid retinoblastoma protein domain and exons 4 and 6 encode a highly conserved discoidin domain of 157 amino acids and a carbon terminal region (17). The highly conserved discoidin domain sequence (DS) motif is a transmembrane receptor that contributes to cell adhesion and signal transduction, usually mediating intercellular adhesion, managing extracellular mechanisms and communicating collagen. DS domain-containing proteins are signaling peptides that are cleaved by peptidase and involved in cell adhesion, migration, cell proliferation and blood coagulation (coagulation factors V and VIII) (7). Cleavage produces the mature retinoschisin protein, which has an important role in bipolar cell connections and synaptic connections of photoreceptors. RS1:c.559C>T (p.Q187X) located in exon 6 disrupts the discoidin domain. This mutation produces a nonsense mutation with early termination, resulting in a smaller version of the full-length protein, which may lead to loss of function. Software predicting protein structure changes indicated that the WT protein structure is significantly altered by mutations. Employing cryo-electron microscopy, it has been discovered that functional retinoschisin forms a dimer of octamer rings comprising a hexadecamer. Given this intricate structure, a mutation that affects the folded structure of the RS1 protein monomer is likely to inhibit the proper assembly of the 16-subunit oligomer (42,43), as evidenced by the large number of reported pathogenic mutations. The surface electrostatic potential of the variant protein is also markedly changed, which may influence normal polymerization of the RS1 protein and interaction with various other proteins. It is hypothesized that this mutation disrupts cell-to-cell communication and adhesion between retinal layers, leading to retinal interlayer division. At the early stage of retinoschisis, the condition may not be observed morphologically, but the abnormal protein caused may not be able to perform the normal adhesion function, leading to disruption of intercellular adhesion and signal transduction, starting with photoreceptor cell abnormality. Previous research revealed that RS1 mutations trigger severe dysontogenesis of the eye (44). Sauer et al (7) suggested that RS1 is a noteworthy pathogenic gene and Hu et al (15) identified 28 variants connected with congenital XLRS, 8 of which were novel. Chen et al (9) reported on six pedigrees with the disease in China, with certain sporadic cases and novel variants. In eukaryotes, if mRNA contains a premature termination codon, it is detected and destroyed by the detection mechanism of mRNA monitoring-nonsense mediated decay (NMD). However, this situation must satisfy that the sense codon is located >50 nucleotides upstream of the last exon node. RS1:c.559C>T (p.Q187X) is located in the last exon of RS1 and it is expected that RS1:c.559C>T escapes NMD, resulting in early termination of translation (45).
At present, there is no acceptable approach to halt progressive maculopathy due to XLRS. Gene therapy is considered a potential mainstay of treatment and there have been numerous reports on gene therapy for retinal cleavage. For instance, Bashar et al (46) reported that RS1 gene delivery avoids structural and functional defects in the RS1 knockout mouse model, inhibiting disease progression in this system. Cukras et al (47) injected RS1 complementary DNA into the eyeballs of mature RS1 knockout mice, eliminating the abnormal damage waveform of ERG and reconstructing the favourable alpha stage, contributing to the entire outermost bronchial rupture protein. It is thought that gene therapy may be an effective treatment for XLRS. WES may serve as an analytic method for the younger generation of patients in families with retinal splits, allowing them to select more optimized treatments prior to progression of the disease to the irreversible stage and laying the foundation for possible participation in gene therapy in the future. Therefore, it is significant to identify more mutated genes linked to retinoschisis and provide further evidence and benchmarks for genetic analysis and therapy.
In conclusion, the present analysis utilized WES to identify the RS1:c.559C>T (p.Q187X) mutation in a three-generation Chinese family with XLRS. The present study was the first to report on RS1:c.559C>T inducing XLRS in China. As previously described, impaired vision and colour vision are important features of disease progression and the early occurrence of blue-purple colour vision abnormalities in the patients of the present study is an important indication for clinicians to consider the possibility of retinal splitting when patients complain of unexplained colour vision abnormalities. WES has the ability to diagnose XLRS with characteristics of clinical and genetic heterogeneity.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 51573101), Beijing Natural Science Foundation (grant no. 7172056) and the Xinglin Scholar Scientific Research Promotion Program of Chengdu University of Traditional Chinese Medicine (grant no. BSH2019025).
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to local policy prohibiting data sharing but are available from the corresponding author on reasonable request.
Authors' contributions
DC performed the molecular genetic study, participated in the sequence alignment, drafted and edited the manuscript and prepared the figures. SZ conceived and designed the study and drafted the manuscript. Both authors read and approved the final manuscript. SZ and DC confirm the accuracy of all the raw data.
Ethics approval and consent to participate
The study protocol was approved by the Yin Hai Eye Hospital affiliated with Chengdu University of Traditional Chinese Medicine Ethics Committee (Chengdu, China; no. CMEC2010-22). Written informed consent was obtained from the participants or their guardians for genetic testing.
Patient consent for publication
Written informed consent was obtained from the participants or their guardians for publication of clinical and genetic data and images.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Stephenson K, Dockery A, Wynne N, Carrigan M, Kenna P, Farrar GJ and Keegan D: Multimodal imaging in a pedigree of X-linked retinoschisis with a novel RS1 variant. BMC Med Genet. 19(195)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Grigg JR, Hooper CY, Fraser CL, Cornish EE, McCluskey PJ and Jamieson RV: Outcome measures in juvenile X-linked retinoschisis: A systematic review. Eye (Lond). 34:1760–1769. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lee Y and Oh BL: Retinal detachment in X-linked retinoschisis. N Engl J Med. 382(1149)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lezrek O, Matsanga OR, Htiti N, Tachfouti S, Laghmari M and Lezrek M: Vitreous veils in X-linked retinoschisis. J Fr Ophtalmol. 41:571–573. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fahim AT, Ali N, Blachley T and Michaelides M: Peripheral fundus findings in X-linked retinoschisis. Br J Ophthalmol. 101:1555–1559. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tsang SH and Sharma T: X-linked juvenile retinoschisis. Adv Exp Med Biol. 1085:43–48. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sauer CG, Gehrig A, Warneke-Wittstock R, Marquardt A, Ewing CC, Gibson A, Lorenz B, Jurklies B and Weber BH: Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 17:164–170. 1997.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sudha D, Patric IR, Ganapathy A, Agarwal S, Krishna S, Neriyanuri S, Sripriya S, Sen P, Chidambaram S and Arunachalam JP: Genetic studies in a patient with X-linked retinoschisis coexisting with developmental delay and sensorineural hearing loss. Ophthalmic Genet. 38:260–266. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chen C, Xie Y, Sun T, Tian L, Xu K, Zhang X and Li Y: Clinical findings and RS1 genotype in 90 Chinese families with X-linked retinoschisis. Mol Vis. 26:291–298. 2020.PubMed/NCBI
|
|
10
|
Huang KC, Wang ML, Chen SJ, Kuo JC, Wang WJ, Nguyen PN, Wahlin KJ, Lu JF, Tran AA, Shi M, et al: Morphological and molecular defects in human three-dimensional retinal organoid model of X-linked juvenile retinoschisis. Stem Cell Reports. 13:906–923. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Alfonso-Muñoz EA, Català-Mora J and Díaz-Cascajosa J: X-linked retinoschisis without macular retinoschisis: A new RS1 mutation. Ophthalmol Retina. 4(719)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Huang L, Sun L, Wang Z, Chen C, Wang P, Sun W, Luo X and Ding X: Clinical manifestation and genetic analysis in Chinese early onset X-linked retinoschisis. Mol Genet Genomic Med. 8(e1421)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Plössl K, Schmid V, Straub K, Schmid C, Ammon M, Merkl R, Weber BH and Friedrich U: Pathomechanism of mutated and secreted retinoschisin in X-linked juvenile retinoschisis. Exp Eye Res. 177:23–34. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Huang Y, Mei L, Gui B, Su W, Liang D, Wu L and Pan Q: A novel deletion mutation in RS1 gene caused X-linked juvenile retinoschisis in a Chinese family. Eye (Lond). 28:1364–1369. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hu QR, Huang LZ, Chen XL, Xia HK, Li TQ and Li XX: Genetic analysis and clinical features of X-linked retinoschisis in Chinese patients. Sci Rep. 7(44060)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Staffieri SE, Rose L, Chang A, De Roach JN, McLaren TL, Mackey DA, Hewitt AW and Lamey TM: Clinical and molecular characterization of females affected by X-linked retinoschisis. Clin Exp Ophthalmol. 43:643–647. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Vijayasarathy C, Sui R, Zeng Y, Yang G, Xu F, Caruso RC, Lewis RA, Ziccardi L and Sieving PA: Molecular mechanisms leading to null-protein product from retinoschisin (RS1) signal-sequence mutants in X-linked retinoschisis (XLRS) disease. Hum Mutat. 31:1251–1260. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Han P, Wei G, Cai K, Xiang X, Deng WP, Li YB, Kuang S, Dong Z, Zheng T, Luo Y, et al: Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J Cell Mol Med. 24:1286–1299. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zheng Y, Xu J, Liang S, Lin D and Banerjee S: Whole exome sequencing identified a novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front Genet. 9(129)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang R, Chen S, Han P, Chen F, Kuang S, Meng Z, Liu J, Sun R, Wang Z, He X, et al: Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes meckel syndrome. J Cell Mol Med. 24:1906–1916. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Dai Y, Liang S, Dong X, Zhao Y, Ren H, Guan Y, Yin H, Li C, Chen L, Cui L and Banerjee S: Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy-dystroglycanopathy. J Cell Mol Med. 23:811–818. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gao FJ, Dong JH, Wang DD, Chen F, Hu FY, Chang Q, Xu P, Liu W, Li JK, Huang Y, et al: Comprehensive analysis of genetic and clinical characteristics of 30 patients with X-linked juvenile retinoschisis in China. Acta Ophthalmol. 99:e470–e479. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Renner AB, Kellner U, Fiebig B, Cropp E, Foerster MH and Weber BH: ERG variability in X-linked congenital retinoschisis patients with mutations in the RS1 gene and the diagnostic importance of fundus autofluorescence and OCT. Doc Ophthalmol. 116:97–109. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Martin M: Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17(14806)2011.
|
|
25
|
Li H and Durbin R: Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al: From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang K, Li M and Hakonarson H: ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kumar P, Henikoff S and Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Schwarz JM, Cooper DN, Schuelke M and Seelow D: MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A and Batzoglou S: Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 6(e1001025)2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Riera M, Wert A, Nieto I and Pomares E: Panel-based whole exome sequencing identifies novel mutations in microphthalmia and anophthalmia patients showing complex Mendelian inheritance patterns. Mol Genet Genomic Med. 5:709–719. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
ACMG Board of Directors. ACMG policy statement: Updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 17:68–69. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, et al: ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 15:565–574. 2013.
|
|
36
|
Seeliger D and de Groot BL: Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 24:417–422. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ambrosio L, Hansen RM, Kimia R and Fulton AB: Retinal function in X-linked juvenile retinoschisis. Invest Ophthalmol Vis Sci. 60:4872–4881. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bowles K, Cukras C, Turriff A, Sergeev Y, Vitale S, Bush RA and Sieving PA: X-linked retinoschisis: RS1 mutation severity and age affect the ERG phenotype in a cohort of 68 affected male subjects. Invest Ophthalmol Vis Sci. 52:9250–9256. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hinds AM, Fahim A, Moore AT, Wong SC and Michaelides M: Bullous X linked retinoschisis: Clinical features and prognosis. Br J Ophthalmol. 102:622–624. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. The Retinoschisis Consortium. Hum Mol Genet. 7:1185–1192. 1998.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A and Cooper DN: The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 133:1–9. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tolun G, Vijayasarathy C, Huang R, Zeng Y, Li Y, Steven AC, Sieving PA and Heymann JB: Paired octamer rings of retinoschisin suggest a junctional model for cell-cell adhesion in the retina. Proc Natl Acad Sci USA. 113:5287–5292. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ramsay EP, Collins RF, Owens TW, Siebert CA, Jones RPO, Wang T, Roseman AM and Baldock C: Structural analysis of X-linked retinoschisis mutations reveals distinct classes which differentially effect retinoschisin function. Hum Mol Genet. 25:5311–5320. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yi J, Li S, Jia X, Xiao X, Wang P, Guo X and Zhang Q: Novel RS1 mutations associated with X-linked juvenile retinoschisis. Int J Mol Med. 29:644–648. 2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wilkinson MF: A new function for nonsense-mediated mRNA-decay factors. Trends Genet. 21:143–148. 2005.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bashar AE, Metcalfe AL, Viringipurampeer IA, Yanai A, Gregory-Evans CY and Gregory-Evans K: An ex vivo gene therapy approach in X-linked retinoschisis. Mol Vis. 22:718–733. 2016.PubMed/NCBI
|
|
47
|
Cukras C, Wiley HE, Jeffrey BG, Sen HN, Turriff A, Zeng Y, Vijayasarathy C, Marangoni D, Ziccardi L, Kjellstrom S, et al: Retinal AAV8-RS1 gene therapy for X-linked retinoschisis: Initial findings from a phase I/IIa trial by intravitreal delivery. Mol Ther. 26:2282–2294. 2018.PubMed/NCBI View Article : Google Scholar
|














