Introduction
Obesity is a metabolic disorder that remains a major
global public health threat, with affected individuals frequently
suffering from associated conditions, including type 2 diabetes and
nonalcoholic fatty liver disease (NAFLD) (1). The incidence rate of fatty liver was
50% in patients with type 2 diabetes patients and incidence rate of
fatty liver in alcoholics was 58% (1). In NAFLD, lipids accumulate at abnormal
levels within hepatic cells and drive significant inflammatory
activity, oxidative stress, fibrosis, metabolic dysregulation and
liver cancer development (1,2).
Although a wide range of metabolic and genetic factors have been
revealed to contribute to the development and progression of NAFLD,
the etiology of this disease remains incompletely understood
(2).
A previous study has suggested that the
NAD-dependent deacetylase sirtuin 1 (SIRT1), which is a metabolic
homeostasis regulator, may be associated with the development of
NAFLD (3). High levels of SIRT1
expression were found in the NALF liver, which has been indicated
to result in a notable upregulation of genes associated with
lipogenesis and gluconeogenesis compared to that in the
normal/healthy liver, thereby leading to an important increase in
intracellular glucose and lipid concentrations (3,4).
Consistent with this model, individuals with obesity suffering from
diabetes and NAFLD have been indicated to express lower SIRT1
levels compared with those in healthy controls (5,6).
The serine threonine kinase liver kinase B1 (LKB1)
has been demonstrated to phosphorylate and activate AMP-activated
protein kinase (AMPK), which regulates the metabolism of lipids and
carbohydrates within cells and detects their energy status
(7). Importantly, SIRT1 is known to
activate LKB1 by deacetylation, leading to AMPK activation
(8). The activation of AMPK alters
the NAD+/NADH ratio in cells, resulting in increased
SIRT1 activity in a metabolic regulatory feedback mechanism
(8). Short non-coding microRNA
(miRNA/miR) molecules have been indicated to be highly conserved
molecular entities capable of binding to the 3'-untranslated region
(3'-UTR) of specific target mRNA molecules, resulting in a
significant suppression of target gene expression either via
translational repression or mRNA cleavage (9,10).
Since miRNAs are important in all physiological contexts, the
relevance of miRNA dysregulation in metabolic disease contexts has
been specifically highlighted (11-14).
A previous study has suggested that miR-506-3p dysregulation may be
associated with the onset and progression of multiple forms of
cancer (15). Previous studies have
indicated that miR-506-3p may suppress hepatocellular carcinoma
(HCC) metastasis (16,17). Given that NAFLD is an important
cause of HCC (18) and considering
the close association between miR-506-3p and HCC, the present study
aimed to uncover the role of miR-506-3p in NAFLD. Subsequently, the
association of miR-506-3p with the pathogenesis of NAFLD was
explored, in order to study the underlying molecular mechanisms of
this condition.
Materials and methods
Cell culture and treatment
HepG2 and Huh7 cell lines were obtained from the
American Type Culture Collection and analyzed with short tandem
repeat profiling semiannually after the first recovery. HepG2 and
Huh7 cells were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS (HyClone; Cytiva), D-glucose (5.5 mM) and
1% penicillin/streptomycin at 37˚C in a 5% CO2 incubator. To model
hepatic steatosis, the cells were plated in 6-well plates
overnight, followed by a 4-h treatment in serum-free DMEM that was
subsequently supplemented with 100 mM D-glucose for an additional
48 h at 37˚C and the control group was treated with DMSO.
Oil Red O staining
HepG2 and Huh7 cells (1x106 cells/per
well) were seeded into 24-well plates, 4% formalin-fixed at room
temperature for 1 h and subsequently stained for 15 min at room
temperature with Oil Red O solution (cat. no. ab223796; Abcam) at
room temperature to detect intracellular lipids upon microscopic
analysis. In addition, Oil Red O staining quantification was
performed by treating samples with isopropanol to solubilize the
dye, and the absorbance of the isopropanol solution was
subsequently quantified at 510 nm with a microplate reader.
Intracellular triglyceride (TG)
measurements
TG levels were measured as in a previous study
(19). HepG2 and Huh7 cells
(1x106 cells/per well) were seeded into 24-well plates
and were lysed in NP-40 for 1 h at room temperature. Subsequently,
lysates were warmed for 5 min to 100˚C and cooled to room
temperature. This procedure was repeated one additional time to
fully solubilize lipids within these samples. Samples were then
centrifuged, and TG levels were measured using an enzymatic TG
assay kit (cat. no. K614-100; Biovision, Inc.) according to the
manufacturer's protocols, with total protein levels in each sample
used to normalize the TG content.
miRNA transfection
miR-506-3p mimics (50 nM), inhibitor (50 nM) and
corresponding controls (50 nM) were purchased from Shanghai
GenePharma Co., Ltd. Polyethylenimine (PEI; Sigma-Aldrich; Merck
KGaA) was used to transfect all cells in the present study. For
transfection, 1x105 HepG2 and Huh7 cells were plated for
24 h at 37˚C in serum- and antibiotic-free DMEM, following which
PEI-miRNA complexes in Opti-MEM (Gibco; Thermo Fisher Scientific,
Inc.) were added. After 4 h, fresh DMEM containing 10% FBS was
added, and the cells were incubated for an additional 48 h.
miR-505-3p mimics (5'-GGGAGCCAGGAACGAUUGAUGU-3'), inhibitor
(5'-ACUACUGAGCCGCAGUAGA-3'), mimics-NC
(5'-UUCUCCGAACGUGUCACGUTT-3') and inhibitor-NC
(5'-UUCUCCGAACGUGUCACGUTT-3') were used.
RNA isolation and reverse
transcription quantitative-PCR (RT-qPCR)
Total RNA was extracted from HepG2 and Huh7 cells
using TRIzol® Reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Total RNA was reverse
transcribed into cDNA using the HiFiScipt cDNA Synthesis kit (CoWin
Biosciences) at 16˚C for 30 min, 42˚C for 30 min and 85˚C for 5
min. qPCR was performed using SYBR® Premix Ex Taq™
(Takara Biotechnology Co., Ltd.) and the ABI PRISM 7500 real-time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. qPCR reaction was
performed using the following conditions: Pre-denaturation at 95˚C
for 1 min, 40 cycles of denaturation at 95˚C for 30 sec, annealing
at 67˚C for 30 sec and extension at 72˚C for 30 sec, followed by a
final extension step at 72˚C for 5 min. The sequences of the
RT-qPCR primers were listed in Table
I. miRNA and mRNA expression levels were quantified using the
2-∆∆Cq method (20) and
normalized to the internal reference genes U6 and GAPDH,
respectively.
 | Table ISequences of primers used for reverse
transcription-quantitative PCR. |
Table I
Sequences of primers used for reverse
transcription-quantitative PCR.
| Primer | Sequence
(5'-3') |
|---|
| SIRT1 | F:
TGCGGGAATCCAAAGGATAA |
| | R:
CAGGCAAGATGCTGTTGCA |
| miR-506-3p | F:
TAAGGCACCCTTCTGAGTAGA |
| | R:
GCGAGCACAGAATTAATACGAC |
| SREBP1 | F:
CCACAATGCCATTGAGAAGCG |
| | R:
CTGACACCAGGTCCTTCAGTG |
| FASN | F:
TTGCTGGCACTACAGAATGC |
| | R:
AACAGCCTCAGAGCGACAAT |
| SCD1 | F:
CACACCTTCCCCTTCGACTA |
| | R:
TGACTCCCGTCTCCAGTTCT |
| ACC1 | F:
CTTGGAGCAGAGAACCTTCG |
| | R:
ACTTCCCGACCAAGGACTTT |
| U6 | F:
CTCGCTTCGGCAGCACA |
| | R:
AACGCTTCACGAATTTGCGT |
| GAPDH | F:
TGTGGGCATCAATGGATTTGG |
| | R:
ACACCATGTATTCCGGGTCAAT |
Western blotting
Huh7 and HepG2 cells were harvested and lysed in
RIPA lysis buffer supplemented with protease inhibitors (Bio Basic
Inc.). A bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.) was used to quantify protein levels in each
sample, following which 30 µg protein/sample were separated via 10%
SDS-PAGE prior to transfer onto PVDF membranes (Millipore Sigma).
The blots were then blocked using either 5% BSA (Gibco; Thermo
Fisher Scientific, Inc.) for AMKP and phosphorylated (p)-AMPK or
non-fat milk in TBST (for other proteins), followed by overnight
incubation with antibodies specific to SIRT1 (1:1,000; cat. no.
8469), AMPK (1:1,000; cat. no. 5831), p-AMPK (Threonine 172;
1:1,000; cat. no. 8208) and GAPDH (1:1,000; cat. no. 5174; all from
Cell Signaling Technology, Inc.) at 4˚C. The blots were
subsequently probed for 1 h at 4˚C with an appropriate horseradish
peroxidase-linked rabbit (1:10,000; cat. no. 7074) and anti-mouse
IgG (1:10,000; cat. no. 7076; Cell Signaling Technology, Inc.)
secondary antibodies followed by enhanced chemiluminescent
substrate visualization (GE Healthcare). ImageJ Software version
1.46 (National Institutes of Health) was used for densitometric
analyses.
SIRT1 activity measurement
SIRT1 activity in HepG2 and Huh7 cells transfected
with either miR-506-3p mimics or inhibitor was quantified with a
SIRT1 fluorometric assay kit (cat. no. CS1040; Sigma-Aldrich; Merck
KGaA) using manufacturer's protocols. For this assay, a substrate
that contained both a fluorophore and a quencher was used, so that
following SIRT1-mediated deacetylation in the presence of NAD, this
substrate was cleaved by a peptidase, leading to a fluorescent
signal proportional to the degree of SIRT1 activity. Excitation and
emission wavelengths at 350 and 460 nm, respectively, were applied.
Recombinant SIRT1 and fluoro-deacetylated peptides were used as
positive controls. NAD, samples or enzymes were omitted from the
assays as negative controls.
Luciferase reporter assay
The wild-type or mutated versions of SIRT1 3'-UTR
were cloned downstream of the firefly gene in the psiCHECK2 plasmid
(Promega Corporation), with the mutant construct containing
mutations designed to disrupt miR-506-3p binding to its cognate
target sequence. PEI transfection reagent (Sigma-Aldrich; Merck
KGaA) was used to transfect HepG2 and Huh7 cells with either
miR-506-3p mimics or inhibitor together with the vectors
encompassing the wild-type or mutated versions of SIRT1 3'-UTR.
Luciferase activity in these cells was analyzed at 48 h
post-transfection using a Dual-Luciferase Reporter Assay System
(Promega Corporation) based on the manufacturer's instructions,
with Renilla activity used for normalization purposes.
Statistical analysis
GraphPad Prism 6 (GraphPad Software, Inc.) was used
for all statistical analyses in the present study. Data are
presented as the mean ± standard deviation from three experimental
repeats and one-way ANOVA followed by Tukey's post hoc test was
used to compare different groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
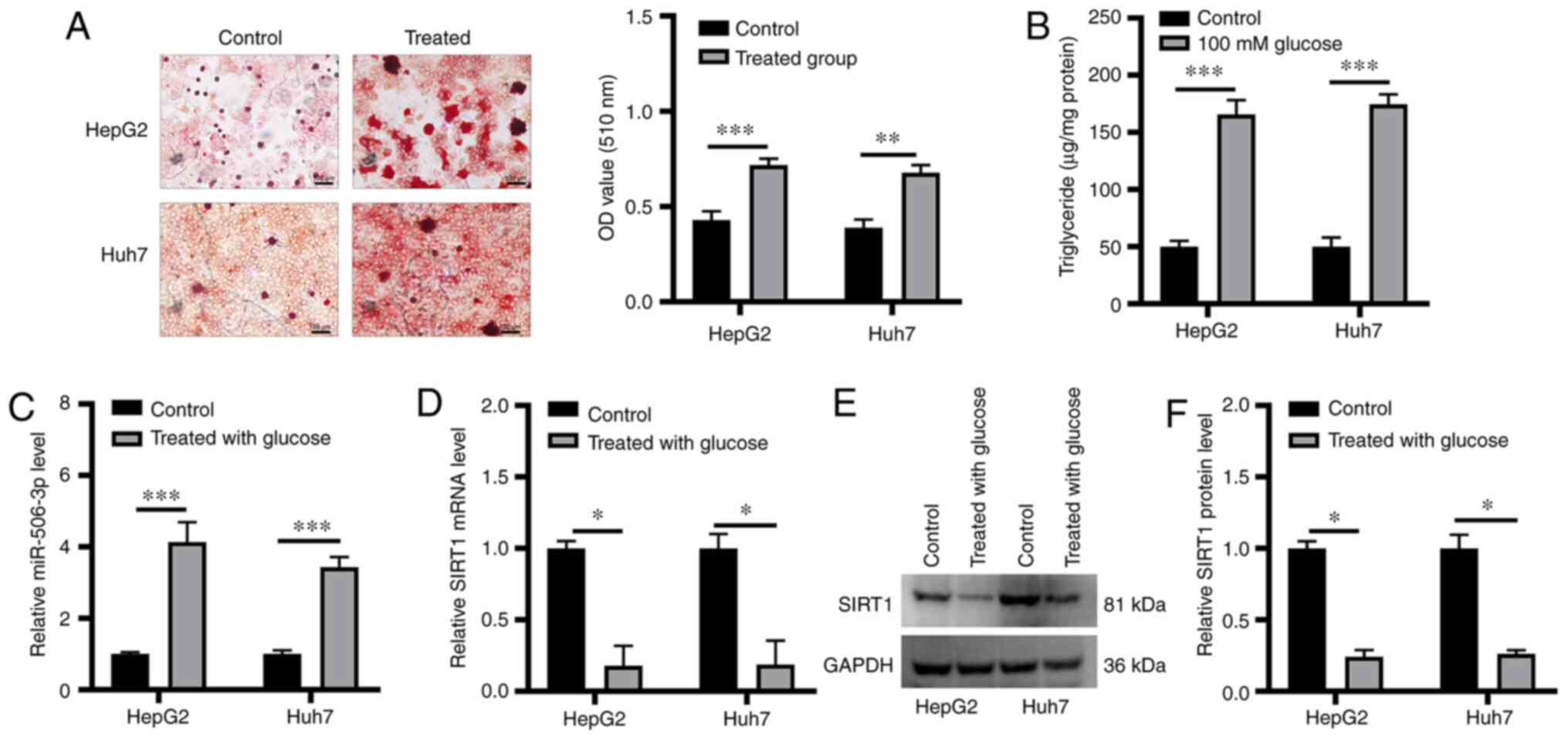
miR-506-3p and SIRT1 expression is
altered in a model of hepatic steatosis
Huh7 and HepG2 cells treated with high glucose
concentration were used to model hepatic steatosis. Lipid
accumulation within these cells was confirmed via Oil Red O
staining (Fig. 1A). TG content was
significantly increased in glucose-treated cells compared with
control cells (Fig. 1B). miR-506-3p
and SIRT1 expression level was examined in these cells, revealing
that miR-506-3p expression level was significantly increased in
glucose-treated cells (Fig. 1C),
whereas SIRT1 expression was markedly decreased at the RNA and
protein level in both cell lines following high glucose treatment
(Fig. 1D-F).
miR-506-3p induces lipid accumulation
in HepG2 and Huh7 cells
To explore the functional relevance of miR-506-3p in
the context of hepatic steatosis, HepG2 and Huh7 cells were
transfected with miR-506-3p mimics or inhibitor (Fig. 2A and B). The analysis of TG levels in these
cells following high glucose pretreatment revealed that
transfection with miR-506-3p mimics led to a significant
enhancement of glucose-induced TG accumulation compared with the
control group, whereas miR-506-3p inhibitor transfection exhibited
the opposite effect (Fig. 2C and
D). miR-506-3p mimics also resulted
in significant lipid accumulation within these cells compared with
the control group, demonstrated via Oil Red O staining, whereas
miR-506-3p inhibitor transfection had the opposite effect (Fig. 2E and F). Subsequently, the expression levels of
major lipogenic genes, such as sterol regulatory element-binding
protein 1 (SREBP1), fatty acid synthase (FASN), stearoyl-CoA
desaturase-1 (SCD1) and acetyl-CoA carboxylase 1 (ACC1) were
examined (21,22) in cells treated with high glucose
concentration with or without transfection with miR-506-3p
inhibitor or mimics. Treatment with high glucose increased the
expression level of these genes, as indicated by RT-qPCR; however,
their expression levels were decreased by miR-506-3p mimics
(Fig. 3G) and increased by
miR-506-3p inhibitor compared with non-transfected cells treated
with high-glucose (Fig. 3H).
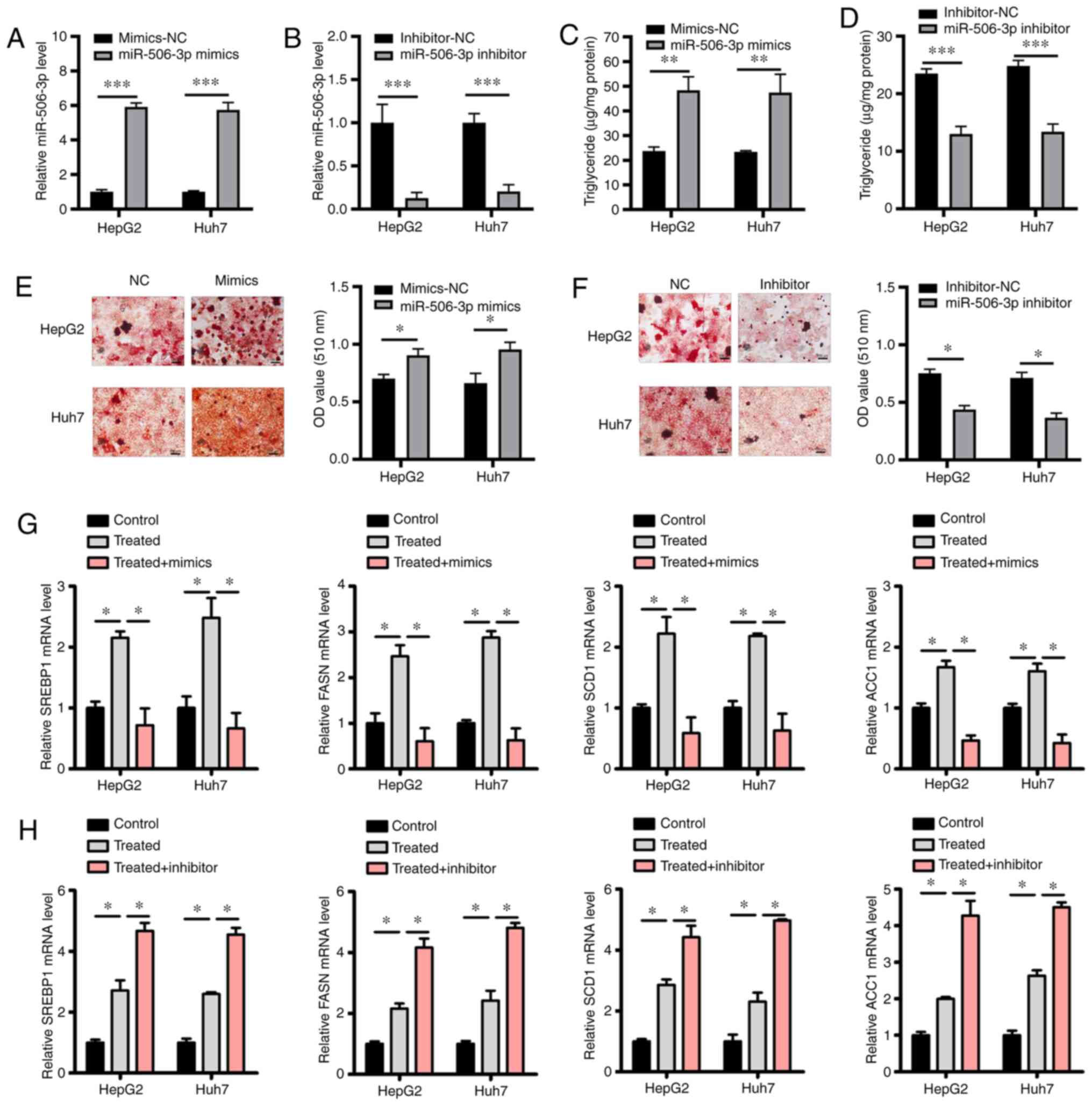 | Figure 2Overexpression of miR-506-3p induces
lipid accumulation in HepG2 and Huh7 cells. (A and B) Following
transfection with miR-506-3p mimics or inhibitor, miR-506-3p
expression in HepG2 and Huh7 cells was quantified via RT-qPCR. (C
and D) HepG2 and Huh7 cells were treated with high glucose prior to
transfection with miR-506-3p mimics or inhibitor or with
corresponding NC constructs, and an enzymatic method was used to
measure intracellular triglyceride levels. (E and F) HepG2 and Huh7
cells were treated with high glucose prior to transfection with
miR-506-3p mimics or inhibitor or with corresponding NC constructs.
Oil Red O staining and quantification via spectrophotometry are
presented. Scale bars, 100 µm. (G and H) The relative expression
levels of SREBP1, FASN, SCD1 and ACC1 in high-glucose treated HepG2
and Huh7 cells were detected by RT-qPCR. Data are presented as the
mean ± SD (n=3). *P<0.05; **P<0.01;
***P<0.001. miR, microRNA; SIRT1, sirtuin 1; NC,
negative control; RT-qPCR, reverse transcription quantitative-PCR;
SREBP1, sterol regulatory element-binding protein 1; FASN, fatty
acid synthase; SCD1, stearoyl-CoA desaturase-1; ACC1, acetyl-CoA
carboxylase 1; OD, optical density. |
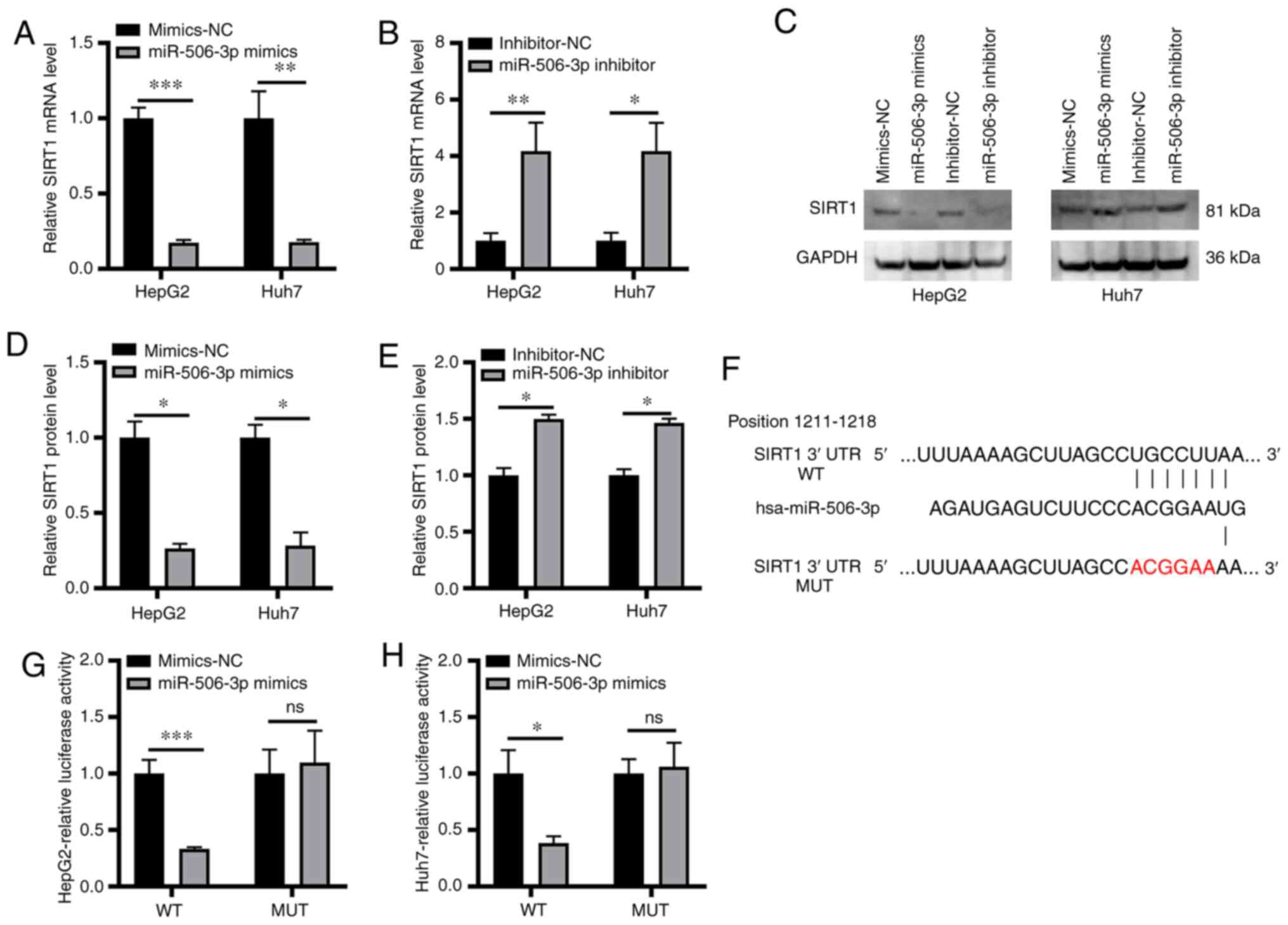
miR-506-3p binds to the 3'-UTR of
SIRT1 and suppresses its expression
The association between SIRT1 and miR-506-3p in the
cellular model of hepatic steatosis was further explored.
miR-506-3p mimics transfection in cells resulted in a significant
reduction in SIRT1 expression at the mRNA and protein level,
whereas the opposite effect was observed following miR-506-3p
inhibitor transfection (Fig. 3A-E).
These data suggested that miR-506-3p may directly or indirectly
regulate SIRT1 in this molecular context. To further explore this
regulatory association, the potential complementarity between SIRT1
mRNA and miR-506-3p was examined, revealing a putative miR-506-3p
binding site in the SIRT1 3'-UTR (Fig.
3F). Luciferase reporter assay revealed that miR-506-3p mimics
significantly suppressed the luciferase activity of reporter
constructs containing the wild-type but not a mutated version of
the SIRT1 3'-UTR sequence (Fig. 3G
and H). These results suggested
that miR-506-3p could directly bind to the 3'-UTR of SIRT1 and
suppress its expression.
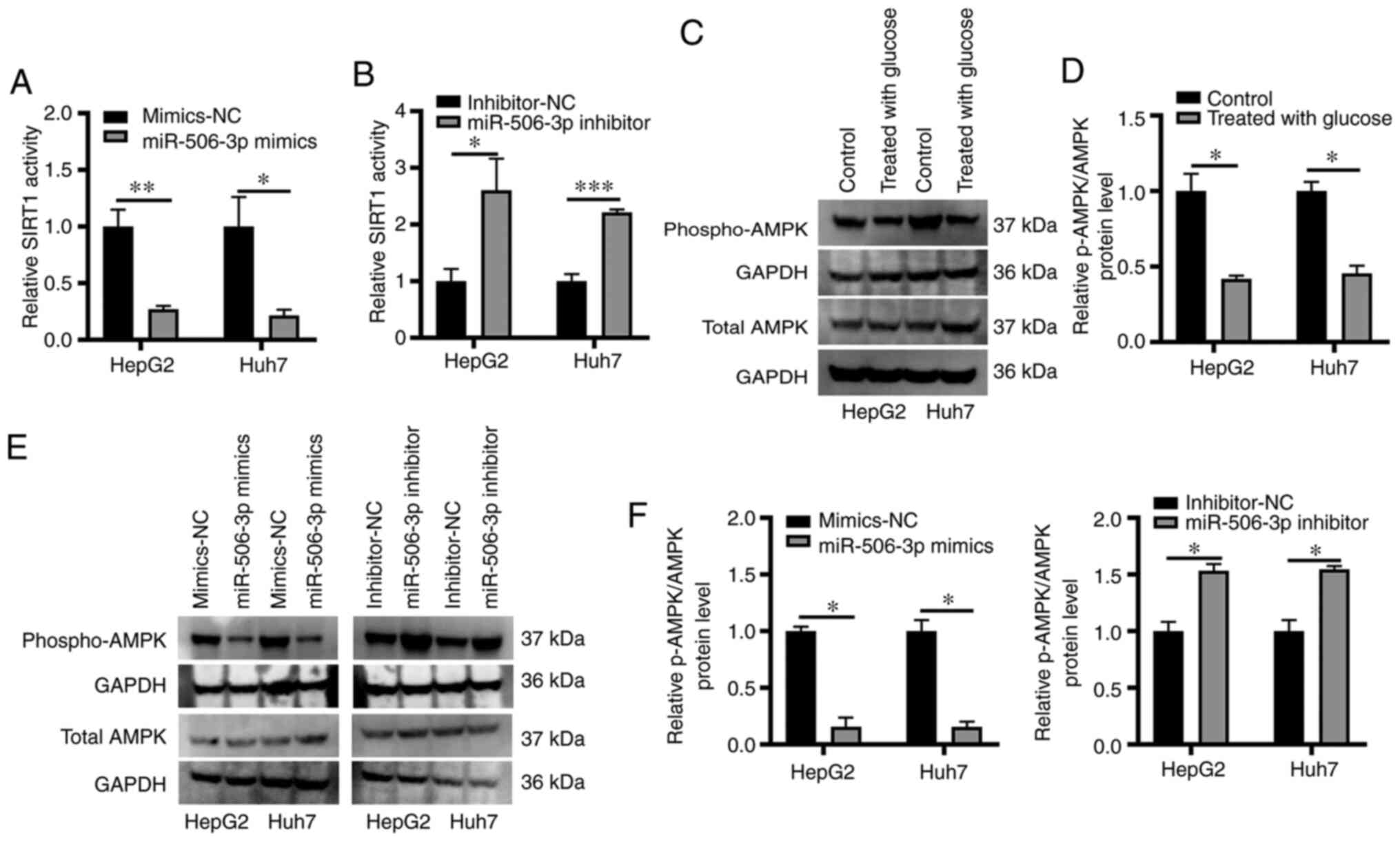
miR-506-3p modulates AMPK
phosphorylation in an in vitro model of hepatic steatosis
The AMPK pathway has been indicated to be associated
with the onset of NAFLD (23), and
enhance intracellular NAD+ levels, thereby leading to
enhanced SIRT1 activation (24).
Similarly, SIRT1 overexpression has been demonstrated to result in
increased AMPK activation, highlighting a regulatory feedback
mechanism in this metabolic context (25). In the present study, SIRT1
deacetylase activity was measured in samples obtained from HepG2
and Huh7 cells that were not treated with high glucose transfected
with miR-506-3p mimics or inhibitor using a fluorometric assay,
revealing that miR-506-3p mimics transfection resulted in markedly
reduced SIRT1 activity, whereas miR-506-3p inhibition exhibited the
opposite effect (Fig. 4A and
B). The role of the AMPK pathway as
a mediator of SIRT1 activity in this cellular model of hepatic
steatosis was subsequently examined by detecting AMPK
phosphorylation via western blotting. The results revealed that
lipid accumulation in high glucose-treated HepG2 and Huh7 cells was
associated with a significant reduction in AMPK phosphorylation
(Fig. 4C and D). In addition, miR-506-3p mimics
transfection into these cells significantly decreased the level of
AMPK phosphorylation, whereas miR-506-3p inhibitor transfection led
to significantly enhanced AMPK phosphorylation compared with the
control group (Fig. 4E and F).
Discussion
NAFLD is a disease that arises from hepatic
steatosis, which is itself associated with other metabolic
disorders, including insulin resistance, hyperglycemia and elevated
fatty acid levels (26). Consistent
with these associations, the present study indicated that treatment
of Huh7 and HepG2 cells with high glucose levels led to increased
uptake of lipids by these hepatocytes, thus generating an in
vitro model of hepatic steatosis.
SIRT1 is a key regulator of lipid metabolism within
cells that improves tissue insulin sensitivity and suppresses the
onset of hepatic steatosis (27).
Significant SIRT1 downregulation was observed in HepG2 and Huh7
cells following high glucose treatment and lipid accumulation. A
previous study has demonstrated that SIRT1 activity could prevent
the onset of hepatic steatosis, and that patients with NAFLD
present lower SIRT1 expression compared with healthy individuals
(3). Knockdown of SIRT1 in
hepatocytes has been indicated to result in decreased fatty acid
oxidation, whereas lipid metabolism was enhanced via peroxisome
proliferator-activated receptor-α signaling upon SIRT1
overexpression (28). Consistent
with these findings, mice with a hepatocyte-specific SIRT1 knockout
exhibited increased steatosis and inflammation (28). However, the mechanistic basis of
SIRT1 effect on hepatic steatosis remains poorly elucidated.
A previous study has highlighted the ability of
miRNAs to modulate a wide range of physiological and pathological
conditions owing to their ability to post-transcriptionally
regulate mRNA translation and degradation (29,30).
The present study indicated SIRT1 to be a predicted target of
miR-506-3p, with a notable complementarity between miR-506-3p and
SIRT1 3'-UTR. A significant increase in miR-506-3p expression level
was observed in high glucose-treated HepG2 and Huh7 cells that
coincided with intracellular lipid accumulation. Interestingly,
transfection of these hepatocytes with miR-506-3p mimics resulted
in a significant enhancement in lipid accumulation within these
cells upon glucose exposure, whilst transfection of these
hepatocytes with miR-506-3p inhibitor resulted in a significant
decrease in lipid accumulation within these cells upon glucose
exposure. This suggests that miR-506-3p serves a direct role in
regulating the development of hepatic steatosis.
To explore the mechanisms by which miR-506-3p
regulated the present model of hepatic steatosis, SIRT1 expression
was examined in particular, owing to SIRT1 downregulation in NAFLD
and its high complementarity with miR-506-3p (31). Previous studies have suggested that
SIRT1 upregulation may be of therapeutic value in NAFLD (32), resulting in altered regulation of a
number of key metabolism-related genes, including SREBP1, FASN and
SCD1 (33,34). Using a luciferase reporter assay, a
direct interaction between miR-506-3p and SIRT1 3'-UTR was
confirmed. Multiple miRNAs have previously been demonstrated to
promote lipogenesis and therefore drive the onset of NAFLD,
including miR-9(35) and miR-34a
(36). The present study, however,
is the first to similarly highlight miR-506-3p as a potential
regulator of NAFLD owing to its ability to inhibit SIRT1
expression, to the best of our knowledge.
AMPK activation has been indicated to decrease lipid
production and enhance fatty acid oxidation, ultimately inhibiting
hepatic TG accumulation and NAFLD development (37). In the present study, a significant
reduction in AMPK phosphorylation level following high glucose
treatment in HepG2 and Huh7 cells was observed, while miR-506-3p
inhibitor transfection significantly enhanced AMPK phosphorylation
and miR-506-3p mimic transfection significantly decreased AMPK
phosphorylation. These data suggested that the SIRT1/AMPK axis
mediated at least partially the metabolic effects of miR-506-3p in
the present in vitro model of hepatic steatosis.
In conclusion, the results of the present study
suggested that miR-506-3p suppressed SIRT1 expression, thereby
impairing AMPK signaling. These results further suggested that
miR-506-3p may be associated with the pathogenesis of hepatic
steatosis and may represent either a biomarker or a therapeutic
target in NAFLD.
Acknowledgements
Not applicable.
Funding
The present study was supported by Shanghai Science and
Technology Committee (grant no. 17ZR1427000), Shanghai Municipal
Health Committee (grant no. 201740156), National Natural Science
Foundation of China (grant no. 81772591), National Key Basic
Research Program of China (grant no. 2014CB542102), Shanghai Health
and Family Planning Commission Foundation (grant no. 20164Y0189),
National Human Genetic Resources Sharing Service Platform (grant
no. 2005DKA21300), Science Fund for Creative Research Groups, NSFC
(grant no. 81521091) and State Key Infectious Disease Project of
China (grant no. 2017ZX10203208).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XFX conceived the study and designed the
experiments. LKH, JQC, HZ, YPT, YY and XFX performed the
experimnts. LKH, JQC, HZ, YPT and XFX wrote and revised the
manuscript. All authors contributed to the interpretation and
discussion of the results and reviewed the manuscript. LKH, JQC and
XFX confirm the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Gui Y, Pan Q, Chen X, Xu S, Luo X and Chen
L: The association between obesity related adipokines and risk of
breast cancer: A meta-analysis. Oncotarget. 8:75389–75399.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Benedict M and Zhang X: Non-alcoholic
fatty liver disease: An expanded review. World J Hepatol.
9:715–732. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nassir F and Ibdah JA: Sirtuins and
nonalcoholic fatty liver disease. World J Gastroenterol.
22:10084–10092. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tobita T, Guzman-Lepe J, Takeishi K, Nakao
T, Wang Y, Meng F, Deng CX, Collin de l'Hortet A and Soto-Gutierrez
A: SIRT1 disruption in human fetal hepatocytes leads to increased
accumulation of glucose and lipids. PLoS One.
11(e0149344)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Arab Sadeghabadi Z, Nourbakhsh M, Pasalar
P, Emamgholipour S, Golestani A, Larijani B and Razzaghy-Azar M:
Reduced gene expression of sirtuins and active AMPK levels in
children and adolescents with obesity and insulin resistance. Obes
Res Clin Pract. 12:167–173. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kitada M and Koya D: SIRT1 in type 2
diabetes: mechanisms and therapeutic potential. Diabetes Metab J.
37:315–325. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hardie DG: Minireview: the AMP-activated
protein kinase cascade: the key sensor of cellular energy status.
Endocrinology. 144:5179–5183. 2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hou X, Xu S, Maitland-Toolan KA, Sato K,
Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, et al:
SIRT1 regulates hepatocyte lipid metabolism through activating
AMP-activated protein kinase. J Biol Chem. 283:20015–20026.
2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jinek M and Doudna JA: A three-dimensional
view of the molecular machinery of RNA interference. Nature.
457:405–412. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Rottiers V and Näär AM: MicroRNAs in
metabolism and metabolic disorders. Nat Rev Mol Cell Biol.
13:239–250. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Ceccarelli S, Panera N, Gnani D and Nobili
V: Dual role of microRNAs in NAFLD. Int J Mol Sci. 14:8437–8455.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kerr TA, Korenblat KM and Davidson NO:
MicroRNAs and liver disease. Transl Res. 157:241–252.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lakner AM, Bonkovsky HL and Schrum LW:
microRNAs: Fad or future of liver disease. World J Gastroenterol.
17:2536–2542. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hu CY, You P, Zhang J, Zhang H and Jiang
N: MiR-506-3p acts as a novel tumor suppressor in prostate cancer
through targeting GALNT4. Eur Rev Med Pharmacol Sci. 23:5133–5138.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Deng Q, Xie L and Li H: MiR-506 suppresses
cell proliferation and tumor growth by targeting Rho-associated
protein kinase 1 in hepatocellular carcinoma. Biochem Biophys Res
Commun. 467:921–927. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang Z, Si M, Yang N, Zhang H, Fu Y, Yan
K, Zong Y, Zhu N and Wei Y: MicroRNA-506 suppresses invasiveness
and metastasis of human hepatocellular carcinoma cells by targeting
IL8. Am J Cancer Res. 8:1586–1594. 2018.PubMed/NCBI
|
|
18
|
Debes JD, Boonstra A and de Knegt RJ:
NAFLD-related hepatocellular carcinoma and the four horsemen of the
apocalypse. Hepatology. 71:774–776. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Teras LR, DeSantis CE, Cerhan JR, Morton
LM, Jemal A and Flowers CR: 2016 US lymphoid malignancy statistics
by World Health Organization subtypes. CA Cancer J Clin.
66:443–459. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lin MJ, Dai W, Scott MJ, Li R, Zhang YQ,
Yang Y, Chen LZ and Huang XS: Metformin improves nonalcoholic fatty
liver disease in obese mice via down-regulation of apolipoprotein
A5 as part of the AMPK/LXRα signaling pathway. Oncotarget.
8:108802–108809. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
da Silva-Santi LG, Antunes MM,
Caparroz-Assef SM, Carbonera F, Masi LN, Curi R, Visentainer JV and
Bazotte RB: Liver fatty acid composition and inflammation in mice
fed with high-carbohydrate diet or high-fat diet. Nutrients.
8(8)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Santamarina AB, Oliveira JL, Silva FP,
Carnier J, Mennitti LV, Santana AA, de Souza GH, Ribeiro EB, Oller
do Nascimento CM, Lira FS, et al: Green tea extract rich in
epigallocatechin-3-gallate prevents fatty liver by AMPK activation
via LKB1 in mice fed a high-fat diet. PLoS One.
10(e0141227)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Cantó C, Gerhart-Hines Z, Feige JN,
Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P and Auwerx
J: AMPK regulates energy expenditure by modulating NAD+
metabolism and SIRT1 activity. Nature. 458:1056–1060.
2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lan F, Cacicedo JM, Ruderman N and Ido Y:
SIRT1 modulation of the acetylation status, cytosolic localization,
and activity of LKB1. Possible role in AMP-activated protein kinase
activation. J Biol Chem. 283:27628–27635. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kitade H, Chen G, Ni Y and Ota T:
Nonalcoholic fatty liver disease and insulin resistance: new
insights and potential new treatments. Nutrients.
9(9)2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cao Y, Jiang X, Ma H, Wang Y, Xue P and
Liu Y: SIRT1 and insulin resistance. J Diabetes Complications.
30:178–183. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Purushotham A, Schug TT, Xu Q, Surapureddi
S, Guo X and Li X: Hepatocyte-specific deletion of SIRT1 alters
fatty acid metabolism and results in hepatic steatosis and
inflammation. Cell Metab. 9:327–338. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ,
Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al: A long noncoding RNA
activated by TGF-β promotes the invasion-metastasis cascade in
hepatocellular carcinoma. Cancer Cell. 25:666–681. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297.
2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Castro RE, Ferreira DM, Afonso MB,
Borralho PM, Machado MV, Cortez-Pinto H and Rodrigues CM:
miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat
liver and activated by disease severity in human non-alcoholic
fatty liver disease. J Hepatol. 58:119–125. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Colak Y, Yesil A, Mutlu HH, Caklili OT,
Ulasoglu C, Senates E, Takir M, Kostek O, Yilmaz Y, Yilmaz Enc F,
et al: A potential treatment of non-alcoholic fatty liver disease
with SIRT1 activators. J Gastrointestin Liver Dis. 23:311–319.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wang LF, Wang XN, Huang CC, Hu L, Xiao YF,
Guan XH, Qian YS, Deng KY and Xin HB: Inhibition of NAMPT
aggravates high fat diet-induced hepatic steatosis in mice through
regulating Sirt1/AMPKα/SREBP1 signaling pathway. Lipids Health Dis.
16(82)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sun L, Wang Y, Song Y, Cheng XR, Xia S,
Rahman MR, Shi Y and Le G: Resveratrol restores the circadian
rhythmic disorder of lipid metabolism induced by high-fat diet in
mice. Biochem Biophys Res Commun. 458:86–91. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ao R, Wang Y, Tong J and Wang BF: Altered
microRNA-9 Expression Level is Directly Correlated with
Pathogenesis of Nonalcoholic Fatty Liver Disease by Targeting
Onecut2 and SIRT1. Med Sci Monit. 22:3804–3819. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kim HJ, Joe Y, Yu JK, Chen Y, Jeong SO,
Mani N, Cho GJ, Pae HO, Ryter SW and Chung HT: Carbon monoxide
protects against hepatic ischemia/reperfusion injury by modulating
the miR-34a/SIRT1 pathway. Biochim Biophys Acta. 1852:1550–1559.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Boudaba N, Marion A, Huet C, Pierre R,
Viollet B and Foretz M: AMPK re-activation suppresses hepatic
steatosis but its downregulation does not promote fatty liver
development. EBioMedicine. 28:194–209. 2018.PubMed/NCBI View Article : Google Scholar
|