Introduction
5-Fluorouracil (5-FU) is an antimetabolic
chemotherapeutic drug that is widely used in the clinic (1). 5-FU is widely used in the treatment of
tumors in various parts of the body, such as breast, liver and
ovarian cancer, and has also been used for the treatment of
gastrointestinal tumors such as gastric cancer (2). 5-FU, as a commonly used chemotherapy
drug for digestive tract tumors, is very important for the
treatment of both elderly patients who cannot tolerate surgery and
need chemotherapy, and patients who have undergone surgery
(3,4). However, in addition to its antitumor
effects, patients may experience adverse reactions in various
organs, including tumor-associated cardiovascular disease (5,6).
Clinical data on the cardiotoxicity of antimetabolic
chemotherapeutic drugs has revealed that elderly patients,
particularly those with cardiovascular disease, exhibit a higher
incidence of cardiotoxicity and experience more adverse reactions
than younger patients without cardiovascular disease (7,8).
Therefore, the role of cardiotoxicity following the use of
chemotherapeutic drugs requires further investigation, particularly
in elderly patients with cancer. Current investigations aim to
minimize the heart damage experienced due to chemotherapy in
patients who undergo tumor-suppressing treatments (9,10).
The results of previous studies have demonstrated
the pathological mechanisms underlying 5-FU-induced cardiotoxicity,
including oxidative stress, cytotoxic effects, arterial vasospasm
outside the cardiac vasculature, apoptosis, autoimmune reactions
and changes in vascular endothelial function (11-13).
Lemaire et al (14) revealed
that 5-FU is converted to a number of metabolites, which leads to
mitochondrial swelling and a decrease in mitochondrial membrane
permeability (14). Eskandari et
al (12) demonstrated that 5-FU
induced oxidative stress and decreased mitochondrial function in
the myocardium, leading to apoptosis of cardiomyocytes and
cardiotoxicity in rabbits (12).
The results of a previous study indicated that
mitochondrial damage in cardiomyocytes is the basis of 5-FU
cardiotoxicity (15). Damaged
mitochondria are degraded or eliminated by autophagy to meet the
metabolic needs of the cells, for organelle regeneration and to
maintain cell homeostasis (16).
Insufficient or excessive mitochondrial autophagy causes myocardial
damage (17,18). However, the mechanisms underlying
5-FU-induced myocardial mitochondrial damage and mitochondrial
autophagy in aging organisms remain to be elucidated. Thus, the
dynamic changes induced by 5-FU on myocardial mitochondrial damage
were investigated in the aging rat model in the present study, and
the molecular mechanisms underlying cardiotoxicity were assessed
from the perspective of mitochondrial autophagy imbalance.
Materials and methods
Animal model and treatment
protocols
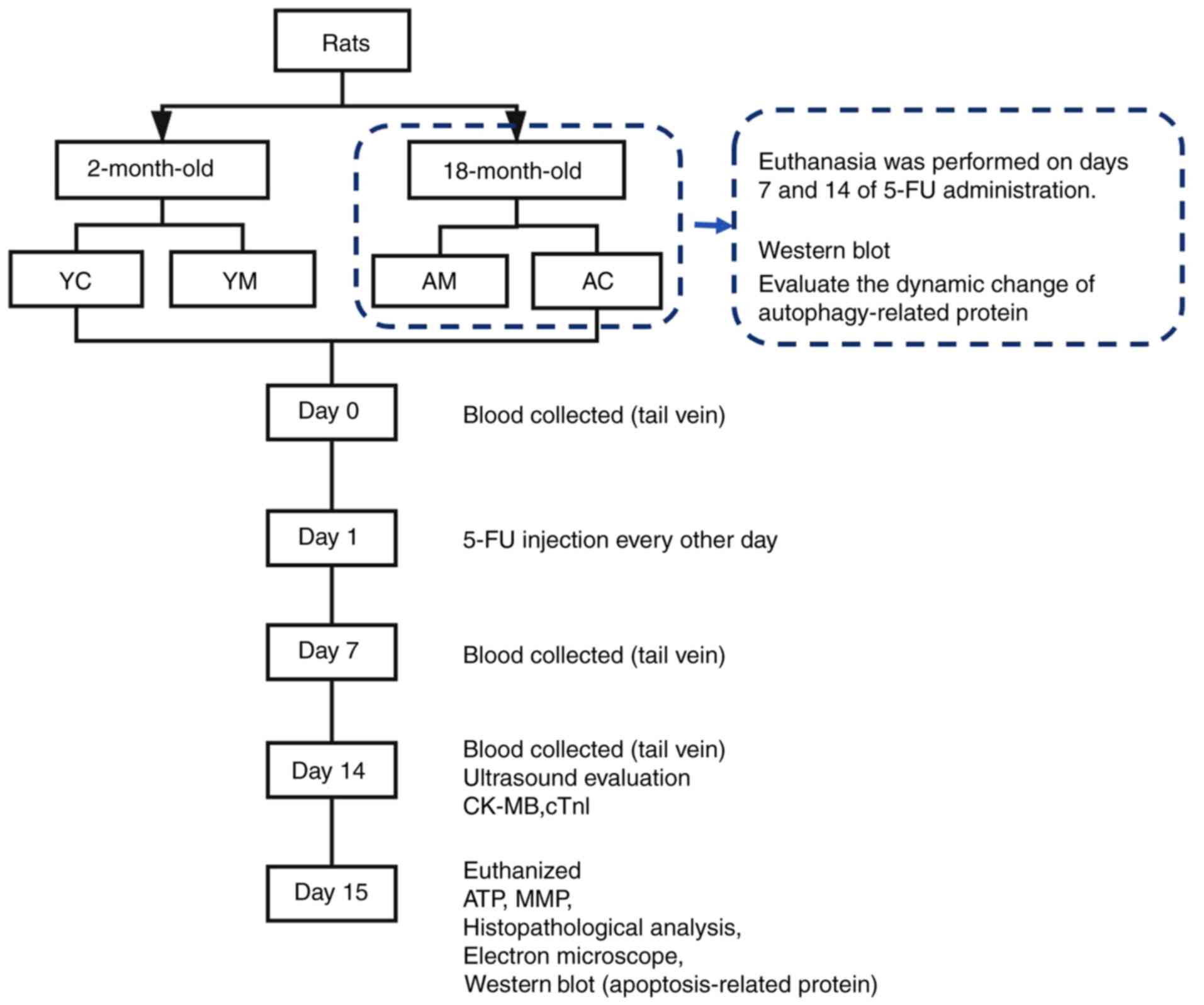
A total of 60 male adult Sprague-Dawley rats aged 2
(weight, 200±20 g) and 18 (weight, 600±50 g) months were purchased
from Beijing Vital River Laboratory Animal Technology Co., Ltd.;
Charles River Laboratories, Inc. and used in the present study. The
animals were kept under a 12-h light/dark cycle in an environment
with a temperature of 20˚C and a humidity of 50%, with free access
to food and water, and experiments were performed after 1 week of
acclimatization. All animals were used in accordance with ethics
guidelines, respecting animal welfare and minimizing discomfort.
The experimental protocol was approved by the Animal Care and Use
Committee of Tianjin Union Medical Center (approval no. 2020-B03;
Tianjin, China).
The 2-month-old rats were randomly divided into a
young age control (YC; n=10) and young age model (YM; n=10) groups.
The 18-month-old rats were randomly divided into an aging control
(AC; n=20) and aging model (AM; n=20) groups. The 5-FU-induced
cardiotoxicity model groups were administered an intraperitoneal
(i.p.) injection of 25 mg/kg 5-FU (Shanghai Xudong Haipu
Pharmaceutical Co., Ltd.) every other day for 1 or 2 consecutive
weeks. The control groups received the same volume of saline at the
same time points as the 5-FU group.
A total of 6 rats were randomly selected from each
group and 600-800 µl blood was collected from the tail vein of
these rats on days 0, 7 and 14. The blood was used for the
detection of creatine kinase isoenzyme (CK-MB) and cardiac troponin
I (cTnI). On day 14, all remaining rats underwent small animal
cardiac ultrasound evaluation. On day 15, the rats were
anesthetized following an i.p. injection with 3% pentobarbital
sodium solution at an anesthetic dose of 40 mg/kg (19,20),
blood was collected from the abdominal aorta, and rats were
sacrificed by cervical dislocation. Fresh myocardial tissues were
collected and washed with cold saline. A portion of each tissue was
used for mitochondrial extraction to determine the levels of ATP,
mitochondrial membrane potential (MMP) and specific mitochondrial
proteins. The remaining tissues were stored at -80˚C or fixed with
4% paraformaldehyde for 48 h and embedded in paraffin.
In order to further evaluate the degree of
myocardial mitochondrial autophagy following different 5-FU
treatment durations, the aged rats were divided into two subgroups.
Following the first week of 5-FU administration, 8 rats were
randomly selected from the aged group and euthanized, and the
remaining rats were euthanized after the second week of 5-FU
administration. Heart tissues were collected, and mitochondrial
proteins were extracted for the subsequent analysis of
autophagy-related protein expression. The overall experimental
process is displayed in Fig. 1.
Measurement of serum CK-MB and cTnI
levels
A total of 6 rats were randomly selected from each
group and blood was collected from the tail vein on days 0, 7 and
14. Following incubation at room temperature for 30 min, all blood
samples were centrifuged at 4˚C and 3,000 x g for 15 min to
separate the serum. The serum levels of CK-MB (cat. no.
E-EL-R1327c) and cTnI (cat. no. E-EL-R1253c) were subsequently
detected by ELISA (Elabscience Biotechnology, Inc.). The optical
density was determined using a microplate reader at 450 nm, a
standard curve was plotted and the sample index concentration was
calculated (CK-MB, y=135.64x2+700.01x-40.521,
R2=0.999; cTnI, y=3984.1x2-330.74x+27.441,
R2=0.996).
Echocardiography
On day 14 of 5-FU administration, rats were
anesthetized by an i.p. injection with 3% pentobarbital sodium
solution at a dose of 40 mg/kg. The fur was removed from the chest
and the rats were fixed on a platform with a horizontal tilt of
15˚. Short-axis m-type echocardiography was used to determine left
ventricular internal dimension in diastole (LVID-d), left
ventricular internal dimension in systole (LVID-s), left
ventricular ejection fraction (LVEF; %) and left ventricular
shortening fraction (LVFS; %) using a Vevo2100 high-resolution
imaging system (VisualSonics, Inc.).
Heart weight index (HWI)
assessment
Total heart tissue was obtained and washed with cold
PBS, and excess water was removed using an absorbent paper towel.
The adipose tissue was removed, the aorta was separated from the
atrium and the weight of each heart was determined in mg. HWI was
defined as the ratio of heart weight to body weight (mg/g).
Measurement of ATP content
Fresh myocardial tissue (20 mg) was obtained, added
to pyrolysis liquid (cat. no. S0027-4; Beyotime Institute of
Biotechnology), homogenized on ice and centrifuged at 4˚C for 5 min
at 12,000 x g to obtain the supernatant. Standard curves were
prepared by diluting the standard ATP solution to different
concentrations (0.01, 0.03, 0.1, 0.3, 1, 3 and 10 µM) using an
enhanced ATP assay kit (cat. no. S0027; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. A
working concentration of ATP detection solution (1X) was produced,
and 100 µl was added to an opaque 96-well plate. In order to
consume the ATP in the detection solution, the 96-well plate was
placed at room temperature for 5 min, thus reducing the background
fluorescence. After the ATP background was eliminated, the samples
were added to the 96-well plate. The relative light unit value was
determined using a luminometer (Multifunctional Microplate
Absorbance Reader; Bio-Rad Laboratories, Inc.). The protein
concentration of the homogenates was determined using a BCA protein
kit and the final ATP concentration was converted to nmol/mg
protein for subsequent quantification.
MMP assay
Fresh myocardial tissue (20 mg) was obtained and
minced with scissors, prechilled for 3 min using cold PBS, and
centrifuged at 4˚C for 20 sec at 600 x g to obtain the precipitate.
Tissues were subsequently prechilled for 20 min using Trypsin-EDTA
Solution (cat. no. C3606-3; Beyotime Institute of Biotechnology)
and centrifuged at 4˚C for 20 sec at 600 x g to obtain the
precipitate. The mitochondria were extracted using a mitochondrial
separation reagent (cat. no. C3606-2; Beyotime Institute of
Biotechnology), homogenized on ice and centrifuged at 4˚C at 12,000
x g. This treatment was repeated twice. JC-1 buffer (1X) was
prepared using an MMP assay kit with JC-1 (cat. no. C2006; Beyotime
Institute of Biotechnology), and 10 mM carbonyl cyanide
m-chlorophenylhydrazone was diluted to 10 µM for use as a positive
control. JC-1 detection fluid (1X) and the samples were added to a
96-well light-proof plate at a ratio of 9:1 to load the purified
mitochondria with JC-1. A fluorescence microplate reader was
preheated, and the excitation and emission values were set to 490
and 530 nm, respectively, to detect the JC-1 monomer and 525 and
590 nm, respectively, for the polymer. The relative ratio of red
and green fluorescence was used to determine the proportion of
mitochondrial depolarization.
Histopathological analysis
Left ventricular tissues were fixed with 4%
paraformaldehyde at room temperature for 48 h. Following
dehydration with 95 and 100% ethanol, tissues were incubated in
xylene, embedded in paraffin and cut into 4-µm sections. Sections
were stained with hematoxylin for 5 min, followed by eosin for 5
min at room temperature. H&E trichrome staining was
subsequently performed. Sections were stained with Weigert iron
hematoxylin solution (cat. no. G1340; Beijing Solarbio Science and
Technology Co., Ltd.) for 5 min, then differentiated with 1%
hydrochloric acid for 1 min and rinsed for 1.5 h. Ponceau stain was
added for 7 min, and distilled water was used for washing. The
sections were treated with a 1% aqueous solution of phosphomolybdic
acid for ~5 min and counterstained with an aniline blue solution
for 5 min. Finally, the sections were treated with 1% glacial
acetic acid for 1 min. After staining, the slices were dehydrated
with different concentrations of ethanol and cleared with 100%
xylene. Masson's trichrome staining were performed. All sections
were imaged using a light microscope (magnification, x200 and
x400). Histopathological analysis was performed in six randomly
selected regions from each section. ImageJ software (version 1.8.0;
National Institutes of Health) was used for image analysis, and
collagen volume fraction (CVF) was calculated as the left
ventricular collagen area/field area.
TUNEL apoptosis analysis
Apoptosis analysis was conducted using the TUNEL
apoptosis detection kit (cat. no. C1091; Beyotime Institute of
Biotechnology). Sections were dewaxed for 40 min at 70˚C, and
incubated in xylene for 10 min, 100% ethanol for 5 min, 100%
ethanol for 2 min, 90% ethanol for 2 min, 80% ethanol for 2 min,
70% ethanol for 2 min and washed in distilled water. Following
dewaxing and rehydration, the paraffin sections were treated with
proteinase K (cat. no. ST352; Beyotime Institute of Biotechnology)
and incubated at room temperature for 15 min. Sections were
subsequently incubated in Enhanced Endogenous Peroxidase Blocking
Buffer (cat. no. P0100B; Beyotime Institute of Biotechnology) at
room temperature for 15 min, followed by washing three times for 10
min with PBS. The negative control samples were treated with 1X
Reaction Buffer (cat. no. C-1082-2; Beyotime Institute of
Biotechnology) instead of TUNEL Reaction mixture, and the positive
control sections were treated with DNase I (cat. no. C-1082-1;
Beyotime Institute of Biotechnology), and incubated at 25˚C for 15
min to generate double-stranded DNA breaks. Following washing with
PBS three times at room temperature for 5 min each,
3,3'-diaminobenzidine substrate solution was added, and the
sections were incubated at 37˚C for 5 min prior to signal
detection. The samples were counterstained with hematoxylin at room
temperature for 5 min. The plates were sealed using gelatin and
glycerinum, and imaged under a light microscope (magnification,
x400), followed by analysis using ImageJ software (version, 1.8.0;
National Institutes of Health). A total of six random regions were
selected from each section for counting and immunohistochemical
scoring, according to the following equation: IHS=A x B, where A is
the score of positive cells (0, 0-1; 1, 1-10; 2, 10-50; 3, 50-80;
and 4, 80-100%) and B is the color intensity score (0, negative; 1,
weakly positive; 2, positive; and 3, strongly positive) (21).
Electron microscopic observation of
the mitochondrial ultrastructure
Fresh myocardial tissues were collected (volume,
<1 mm3) and fixed in 2.5% glutaraldehyde fixation
fluid (in 0.1 mol/l phosphate buffer) at 4˚C for 24 h. Following
washing three times for 15 min with PBS, the samples were fixed in
1% osmium fixative solution at 4˚C for 2 h, dehydrated two times
for 15 min in a graded ethanol series (50, 70, 80, 90 and 100%),
embedded in epoxy resin at 37˚C overnight under a vented hood and
subsequently cut into 70-nm sections. The ultrathin sections were
stained with lead citrate and uranium acetate at room temperature
for 15 min, and observed and photographed under a JEM-1200
transmission electron microscope (JEOL Ltd.; magnification, x10,000
and x20,000). Subsequently, a total of three samples were collected
from each group, six regions were selected from each section for
histopathological analysis, and semi-quantitative analysis was
conducted according to the Flameng classification system using
ImageJ (version, 1.8.0; National Institutes of Health) (22).
Protein extraction, quantification and
western blotting
Samples of fresh myocardial tissue (30 mg/sample)
were collected, and the mitochondrial protein was extracted using
the Tissue Mitochondrial Isolation kit (cat. no. C3606; Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. The protein concentrations were determined using a
BCA protein assay kit (cat. no. P0010S; Beyotime Institute of
Biotechnology), and the samples were mixed with 5X loading buffer
for denaturation at 95˚C for 10 min. A total of 30 µg protein per
sample was isolated by SDS-PAGE on 10 or 15% gels and transferred
to a PVDF membrane. The membrane was subsequently blocked with
blocking solution (PBST; 1X; cat. no. P0226; Beyotime Institute of
Biotechnology) for 15 min at room temperature and incubated with
the appropriate primary antibody diluted in TBS-Tween-20 (1%
Tween-20) at 4˚C overnight. Following washing three times for 10
min with TBS-T, the membrane was incubated with a horseradish
peroxidase (HRP)-conjugated secondary antibody, (1:10,000; cat. no.
2722564; ProteinTech Group, Inc.) at room temperature for 90 min,
and visualization was performed using luminol reagent (cat. no.
P0018AM; Beyotime Institute of Biotechnology). β-actin was used as
a loading control. The primary antibodies used in the present study
were as follows: Anti-β-actin (1:1,000; 4970s; Cell Signaling
Technology, Inc.), Anti-Bax (1:10,000; cat. no. 50599-2-lg;
ProteinTech Group, Inc.), anti-Bcl-2 (1:3,000; cat. no. 26593-1-AP;
ProteinTech Group, Inc.), anti-beclin-1 (1:1,000; #3738 Cell
Signaling Technology, Inc.), anti-Parkin (1:1,000; #2132 Cell
Signaling Technology, Inc.), anti-microtubule-associated protein
1A/1B-light chain 3 (LC3) A/B (1:1,000; #12741 Cell Signaling
Technology, Inc.), anti-PTEN-induced putative kinase protein 1
(PINK-1; 1:500; cat. no. 23274-1-AP; ProteinTech Group, Inc.) and
anti-cytochrome c oxidase subunit 4 (COX-IV; 1:1,000; #4844 Cell
Signaling Technology, Inc.).
Statistical analysis
The data were statistically analyzed using SPSS 23.0
(IBM Corp.), and the figures were generated using GraphPad 5.0
(GraphPad Software, Inc.). Normally distributed data are presented
as the mean ± standard deviation. Comparisons among groups were
analyzed by one-way ANOVA, and Tukey's post hoc test was used for
multiple comparisons. In cases of non-normality, the non-parametric
Kruskal-Wallis test followed by Bonferroni post hoc analysis was
used for multiple testing. Repeated measurement data were analyzed
by repeated measures multi-factor ANOVA and means by LSD test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
5-FU induces weight loss and
myocardial injury in rats
To evaluate the extent of myocardial damage induced
by 5-FU, a myocardial toxicity model was established in rats aged 2
and 18 months. Notably, the baseline body weight was inconsistent
between rats of different ages; thus, the difference between weight
gain and loss was used as the evaluation index. The results of the
present study demonstrated that 5-FU stimulation significantly
reduced the rat body weight compared with that in the model groups.
The AM group experienced weight loss from day 3 of 5-FU
administration, whereas the YM group exhibited weight loss from day
9, with the difference in weight loss significantly lower than that
of the YM group (Fig. 2A). In
addition, HWI evaluation was conducted, but due to 5-FU-induced
gastrointestinal toxicity, the weight of the rats in the model
group was significantly reduced compared with the control groups,
which may have led to inaccurate results; therefore, the heart
weight comparisons were combined to facilitate a comprehensive
assessment. The results indicated that the heart weight and HWI of
AM groups demonstrated an increasing trend compared with the
control groups, although the increase in heart weight was not
significant in the YM group. 5-FU may have induced structural
changes, such as hypertrophy and dilatation of the myocardium, and
induced more serious myocardial injury in aging rats (Fig. 2B and C).
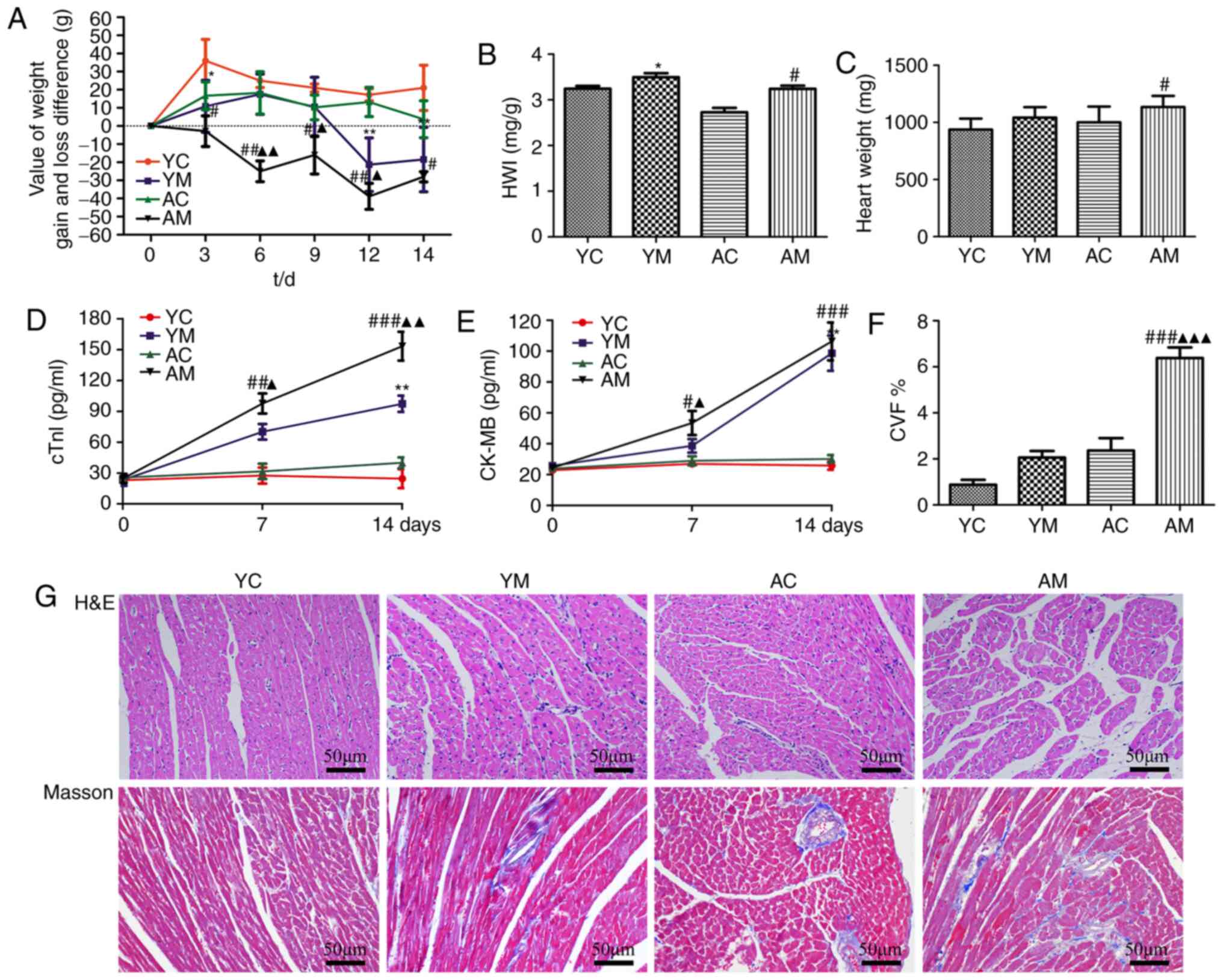 | Figure 25-FU induces weight loss and
myocardial injury in rats. (A) Variation in weight gain and loss in
each group at different time points during 5-FU administration
(n=8). Comparison of (B) HWI and (C) heart weight in each group
(n=6). ELISA was used to examine the levels of myocardial enzymes
in rat myocardial tissues, (D) cTnI and (E) CK-MB (n=6). (F) CVF of
myocardial tissues was determined by quantification of the left
ventricular collagen area/field area (n=6). (G) H&E and
Masson's trichrome staining of cardiac tissues (magnification,
x200). Blue staining represents fibrosis. *P<0.05 and
**P<0.01 vs. YC; #P<0.05,
##P<0.01 and ###P<0.001 vs. AC;
▲P<0.05, ▲▲P<0.01 and
▲▲▲P<0.001 vs. YM. 5-FU, 5-fluorouracil; CK-MB,
creatine kinase isoenzyme; cTnI, cardiac troponin I; CVF, collagen
volume fraction; YC, young age control group; YM, young age model
group; AC, aging control group; AM, aging model group; HWI, heart
weight index. |
In order to observe whether 5-FU-induced
cardiotoxicity was time-dependent, blood was collected from the
tail vein on days 0, 7 and 14 post-5-FU administration, and CK-MB
and cTnI detection was conducted (Fig.
2D and E). Serum cTnI values
increased on days 7 and 14 following 5-FU treatment in both the YM
and AM groups compared with those in the corresponding control
groups; this increase was more significant in the AM group
(Fig. 2D). Furthermore, the CK-MB
level trend was consistent with that of cTnI, although the YM group
only demonstrated an increasing trend on day 7 of 5-FU
administration, and there was no significant difference compared
with the YC group (Fig. 2E). The
index of CK-MB growth demonstrated a significant upward trend with
prolonged 5-FU administration. However, by day 14, the CK-MB values
of the AM and YM groups were no longer significantly different. In
conclusion, following prolonged administration of 5-FU, both the
serum CK-MB levels and cTnI of rats in the 5-FU model group
demonstrated an increasing trend compared with those in the
corresponding control groups, and the corresponding detection
indices of the AM group were all greater compared with those of the
YM group.
The H&E staining results revealed enlargement of
the myofibrillar interstitium and the muscle fiber gap of the YM
and AM groups compared with the corresponding control groups, and
inflammatory cell infiltration was observed in the myocardial
interstitium (Fig. 2G). These
pathomorphological effects were the most prominent in the AM group.
Masson's staining further demonstrated that the myocardial tissues
of the YM and AM groups were irregularly arranged, and that the
myocardial interstitial tissue was slightly fibrotic in the YM
group. By contrast, in the AM group, the myocardial fibers were
thickened with increased spacing between fibers, and the
blue-stained interstitial area was increased compared with YM
group, which was consistent with the H&E staining results
(Fig. 2G). The CV results indicated
that the CVF of the YM and AM groups were significantly increased,
compared with the corresponding control groups (Fig. 2F).
5-FU induces ventricular enlargement,
decreases myocardial contractile function and decreases LVEF in
aged rats
Previous studies have demonstrated that 5-FU-induced
cardiac injury mainly occurs due to a decrease in myocardial
systolic function (23-25).
Left ventricular structure and function is a suitable indicator to
assess the systolic and diastolic function of the heart. In
addition, the low volume allows for adequate fixation, dehydration,
transparency and staining of the tissue, avoiding the creation of
slits in the sections and ensuring that pathological changes can be
assessed. Therefore, in the present study, only the left ventricle
of rats was selected for histopathological analysis.
Echocardiography was used to evaluate the changes in
cardiac structure and function. Compared with those in the control
group, LVID-d and LVID-s in the rat hearts demonstrated an
increasing trend following 5-FU treatment, whereas LVEF and LVFS
demonstrated a decreasing trend. However, in the YM group, there
were no significant differences in LVID-d, LVID-s and LVEF compared
with those in the YC group (Fig.
3). These results confirmed that 5-FU induced ventricular
enlargement, decreased myocardial contractile function and
decreased the LVEF in aged rats. Compared with AC and YM groups,
the induced cardiac structure and LVEF were more severely impaired
in aged rats.
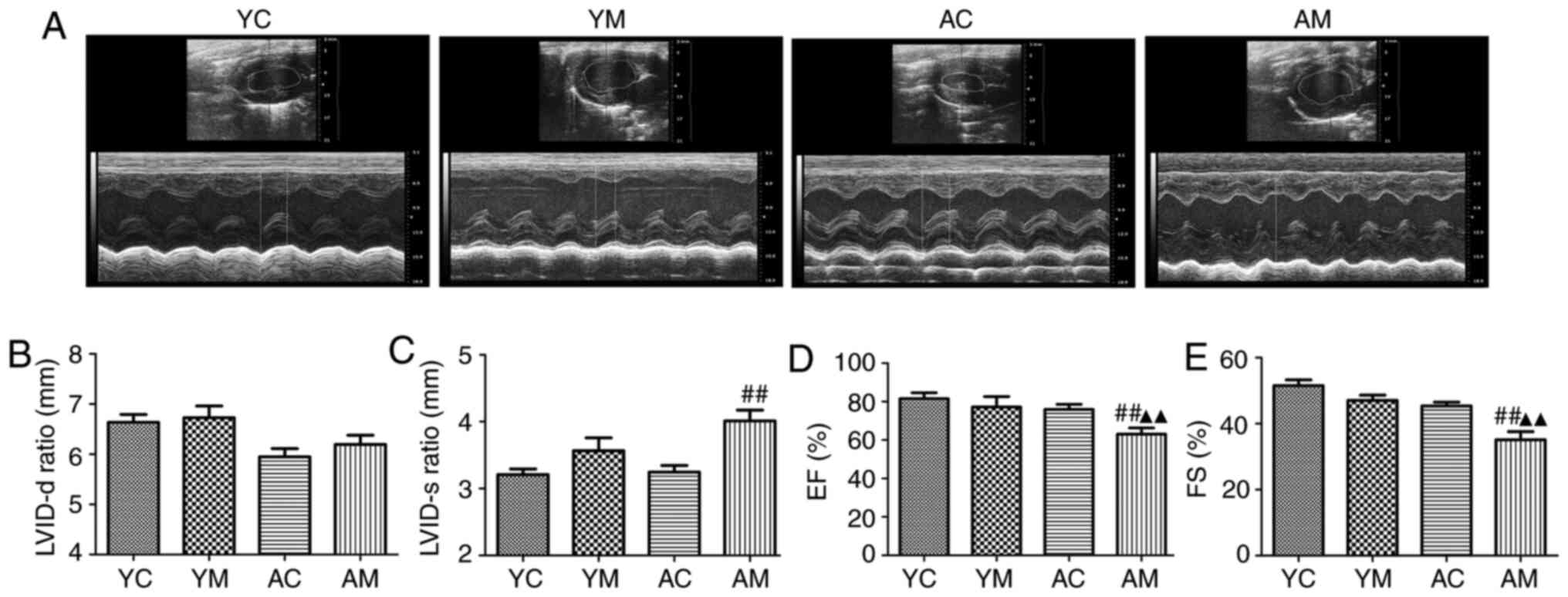 | Figure 35-Fluorouracil induces ventricular
enlargement and myocardial systolic dysfunction in rats. (A)
Representative m-mode echocardiograms for each group of rats. Bar
charts demonstrating (B) the LVID-d ratio, (C) the LVID-s ratio,
(D) LVEF and (E) LVFS (n=6). ##P<0.01 vs. AC;
▲▲P<0.01 vs. YM. LVID-d, left ventricular internal
dimension in diastole; LVID-s, left ventricular internal dimension
in systole; LVEF, left ventricular ejection fraction; LVFS, left
ventricular shortening fraction; YC, young age control group; YM,
young age model group; AC, aging control group; AM, aging model
group. |
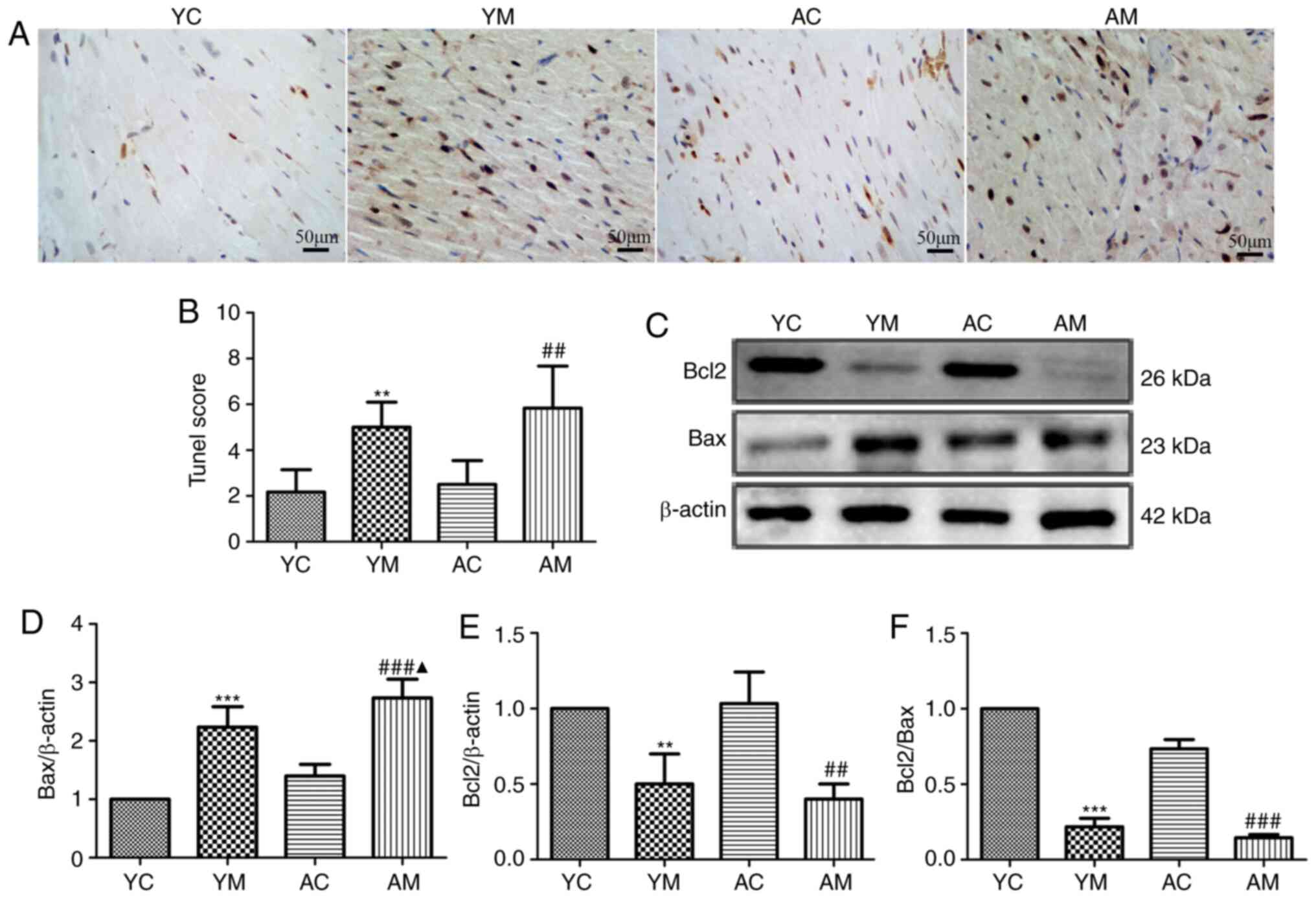
5-FU induces cardiomyocyte apoptosis
in rats
Apoptosis and autophagy share a number of common
signaling pathways and regulatory proteins, such as the bcl-2
family of proteins, caspases, ATG proteins and P53 (26,27).
Bcl-2 proteins serve a key dual regulatory role between apoptosis
and autophagy (28). In the present
study, the levels of apoptosis and the expression levels of
apoptosis-related proteins were determined in order to analyze the
effects of 5-FU on myocardial cell apoptosis. The results of the
TUNEL assay demonstrated that cells in the YM, AC and AM groups
exhibited varying degrees of cell hyperchromemia and nuclear
fragmentation compared with those in the YC group (Fig. 4A), and nuclear staining of the YM
and AM group samples was increased. The apoptotic index of the YM
and AM groups was significantly higher compared with that of the YC
and AC groups, and was higher in the AM compared with the YM group.
The apoptotic index of the AC group appeared to be increased
compared with that of the YC group, although the difference was not
significant (Fig. 4B).
The results of the western blot analysis
demonstrated that the expression levels of the antiapoptotic
protein Bcl-2 were decreased in the YM and AM groups compared with
those in the YC and AC groups. The expression levels of the
proapoptotic protein Bax were increased in the YM and AM groups
compared with those in the YC and AC groups, respectively, and were
significantly increased in the AM group compared with those in the
YM group. Furthermore, compared with those in the AC and YM groups,
the Bcl-2/Bax ratio was the most notably reduced in the AM group
(Fig. 4C-F). The results of the
present study suggested that the 5-FU-induced cardiomyocyte
apoptosis may be accompanied by autophagy.
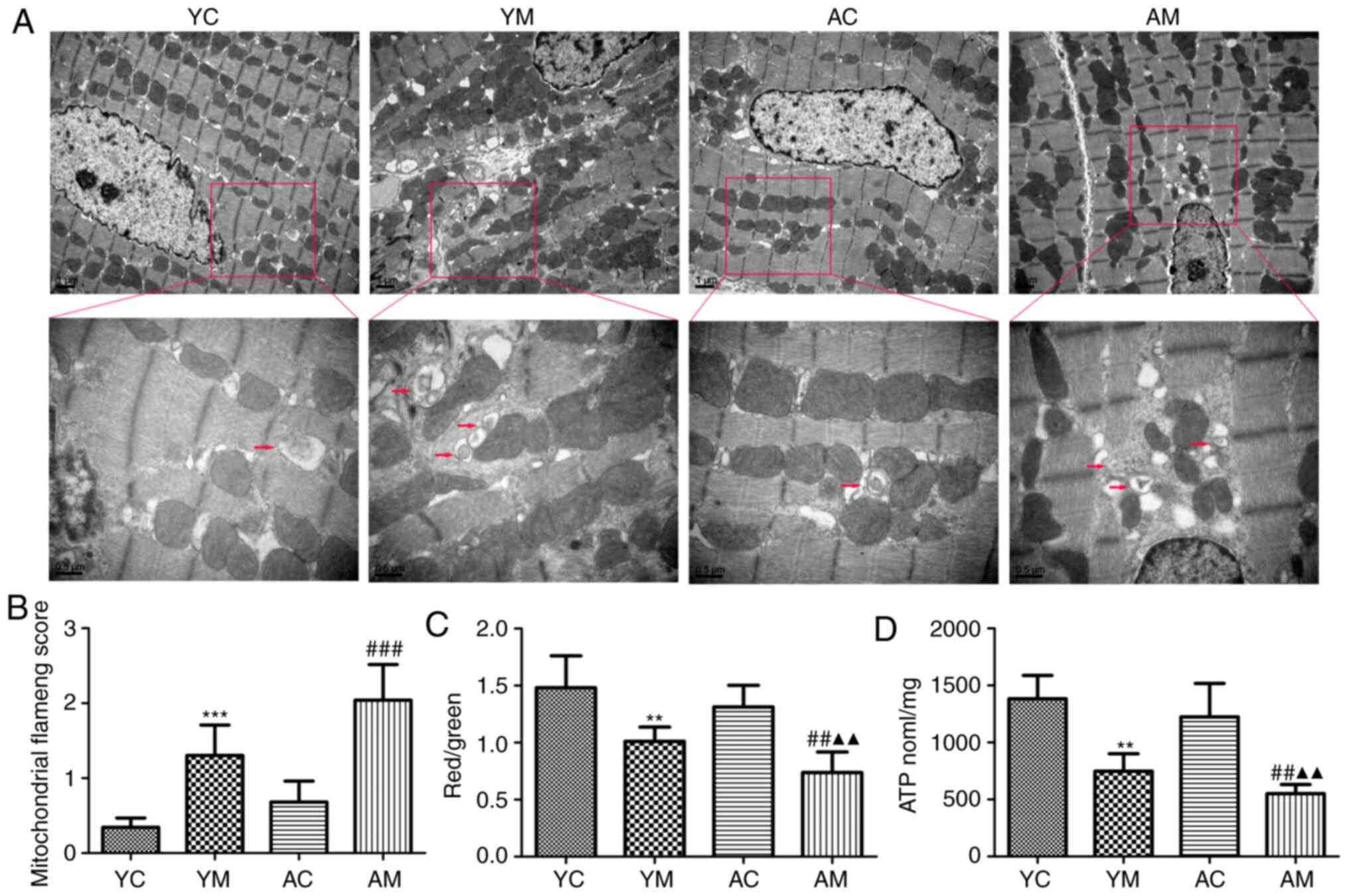
5-FU induces myocardial mitochondrial
damage in rats
In order to investigate whether mitochondrial
functional injury was involved in the mechanism underlying the
5-FU-induced myocardial toxicity, changes in MMP and ATP levels
were determined in rat myocardial tissues. The results demonstrated
that 5-FU stimulation significantly reduced mitochondrial MMP and
ATP levels in YM and AM groups rats compared with the corresponding
control groups, and the ATP level was significantly decreased in
the AM group compared with YM group (Fig. 5C and D). These results further confirmed that
5-FU induced mitochondrial energy metabolism disorder in the
myocardium, with more severe damage in the AM group compared with
the YM group.
Electron microscopy was subsequently used to observe
the mitochondrial structure in the rat myocardial tissue. In the YC
group, the mitochondrial structure was normal, with a single
autophagosome observed. In the AC group, the mitochondria were
sparse, however, the structure and arrangement were normal.
However, in the YM group, mitochondrial swelling, partial fusion,
ridge breakage and an increased number of autophagosomes were
observed. In the AM group, the mitochondria were sparse with a
condensed structure, ridge fracture, increased lipid droplet
deposition and a greater number of autophagosomes compared with the
AC and YM groups (Fig. 5A).
The semi-quantitative scoring of mitochondrial
damage revealed that following 5-FU administration, compared with
the corresponding control groups, myocardial mitochondrial damage
was more severe in the YM and AM groups, and compared with the YM
group, the degree of damage was the most severe in the AM group
(Fig. 5B). The results of the
present study suggested that 5-FU induced myocardial mitochondrial
injury and may enhance mitochondrial autophagy.
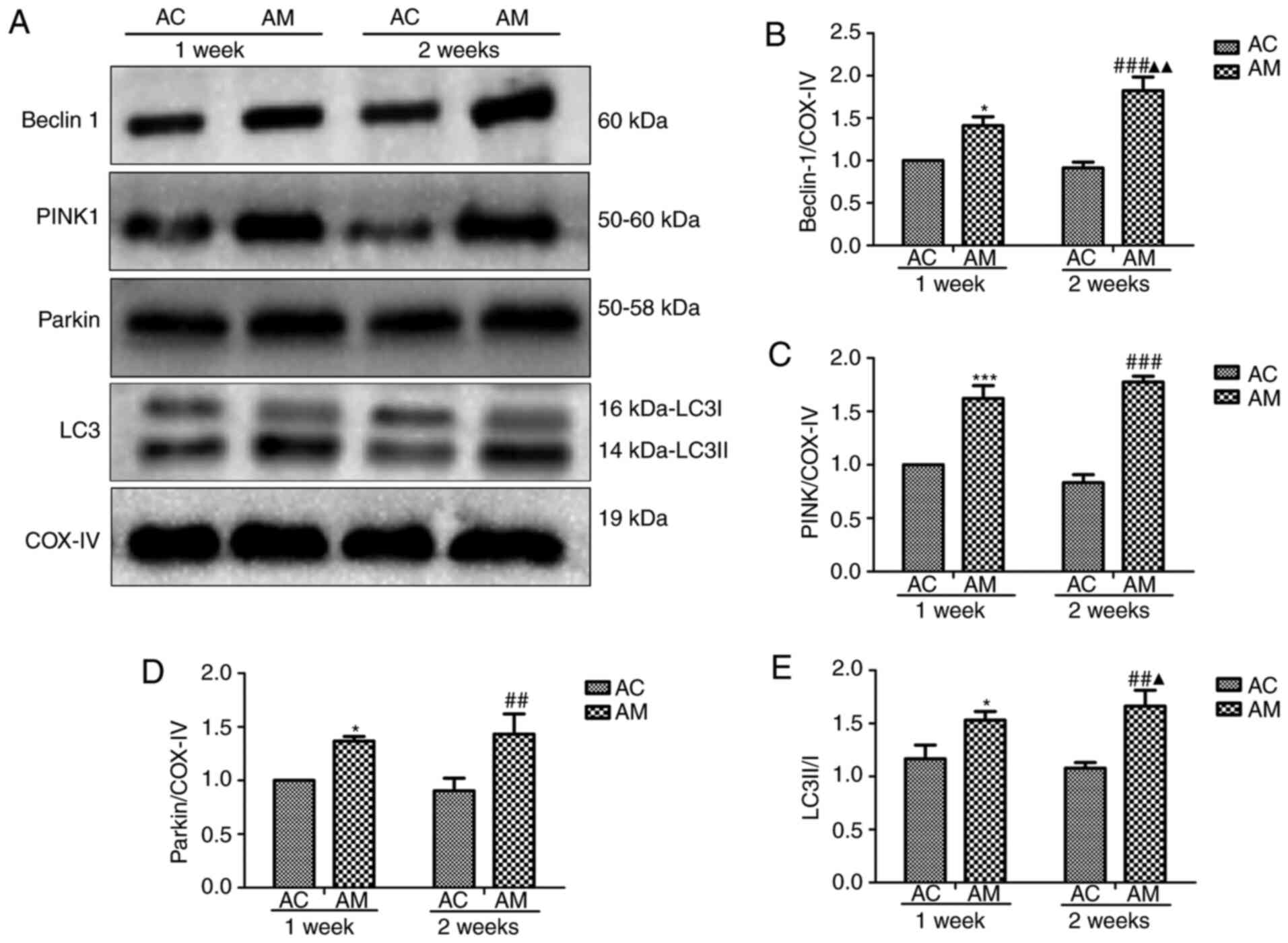
5-FU enhances mitochondrial autophagy
in rats
In the present study, mitochondrial damage was
revealed to induce mitochondrial autophagy and 5-FU caused more
serious myocardial damage in aging rats. Thus, 5-FU was predicted
to induce excessive mitochondrial autophagy in aging rats, and the
degree of autophagy may be associated with the duration of 5-FU
exposure. Therefore, the expression of autophagy-related proteins
on days 7 and 14 of 5-FU administration was quantitatively analyzed
by western blotting (Fig. 6A).
COX-IV is a common internal reference gene located in the
mitochondria and is commonly used as a mitochondrial loading
control. Compared with the 1 week-AC group, 5-FU treatment
upregulated the levels of PINK-1, Parkin and beclin-1 expression in
the 2 weeks-AM groups, and increased the LC3II/I ratio. In
addition, the protein expression levels of beclin-1 and LC3II/I in
the 2 weeks group were significantly higher compared with those in
the 1-week group (Fig. 6B-E). These
results indicated that 5-FU induced myocardial mitochondrial
autophagy, and that autophagy was increased with prolonged 5-FU
administration. Therefore, the pathological mechanisms underlying
5-FU cardiotoxicity may involve the induction of myocardial damage
through the promotion of mitochondrial autophagy.
Discussion
Considerable cardiotoxicity may occur during 5-FU
chemotherapy, with high levels of toxicity observed in the elderly
and patients with prior cardiovascular disease (7,8).
Therefore, detailed investigation is required into the prevention
and treatment of cardiac toxicity in elderly patients, focusing on
the damaging effects of chemotherapeutic drugs.
The pathogenesis of 5-FU-induced cardiotoxicity is
multifactorial, although mitochondrial injury is a potential
initiating mechanism (29,30). According to the pharmacological
mechanism of 5-FU, F-citrate, a metabolite of 5-FU, acts on the
mitochondria to block the tricarboxylic acid cycle, reduce the
production of ATP and alter mitochondrial membrane permeability,
resulting in mitochondrial dysfunction (14). Mitochondrial damage leads to the
progressive accumulation of defective organelles, which further
induces cell and tissue damage, resulting in the initiation of
mitochondrial autophagy (31).
Autophagy is a programmed intracellular degradation process that
initiates autophagosome formation by encapsulating degraded
macromolecules (32). These
autophagosomes fuse with lysosomes for digestion to meet cell
metabolic needs, promote organelle renewal and cell homeostasis
(32). Although it remains
controversial whether the induction of autophagy may be detrimental
to myocardial tissue, the activation of autophagy is generally
considered to be cardioprotective. However, excessive autophagy
results in cell death and myocardial damage (33,34).
Therefore, in the present study, the 5-FU-induced dynamic changes
to myocardial and mitochondrial injury were investigated in aging
rats, and the varying trends of mitochondrial autophagy in
5-FU-induced myocardial injury were discussed.
Cardiotoxic drugs act directly on cardiomyocytes,
and myocardial cTnI is a protein only expressed by atrial and
ventricular myocytes (35,36). When cardiomyocytes are damaged,
cardiac troponin (cTn) is released into the blood, and its levels
are proportional to the area and degree of myocardial cell injury
(37). In the present study,
dynamic changes in myocardial enzyme levels, namely CK-MB and cTnI,
were detected in young and aged rats prior to 5-FU treatment as
well as on days 7 and 14 of 5-FU administration. The results of the
present study demonstrated increasing trends in CK-MB and cTnI
levels on day 7 of 5-FU administration in the YM and AM groups, and
the levels of these two indicators continued to increase with
prolonged 5-FU administration. However, cTnI exhibited a rapid rise
within 7 days of 5-FU administration, compared with the rising
trend of CK-MB occurring later in the 5-FU administration period.
These results suggested that 5-FU induced myocardial injury in
rats, and the degree of injury increased with the accumulation of
the drug. Furthermore, the degree of damage was more severe in
aging rats.
Previous studies have reported that 5-FU-induced
cardiac damage results in decreased myocardial contractility and,
in severe cases, congestive heart failure (23). In the present study, 5-FU was also
demonstrated to induce the expansion of the rat ventricle, and
decrease myocardial contractility and LVEF. However, no significant
changes in the cardiac structure and function were observed. This
may indicate that the 5-FU administration time was too short, or
that the dosage was too low to cause cardiac dysfunction. However,
5-FU also induces intestinal mucosal damage (38,39).
Preliminary experiments demonstrated that following prolonged
duration of 5-FU administration or increasing the administered
dosage, there were significant gastrointestinal events, which
resulted in increased mortality. Therefore, previously used doses
and methods (intraperitoneal (i.p.) injection of 25 mg/kg 5-FU
every other day for 1 or 2 consecutive weeks of 5-FU administration
were used to investigate cardiac injury (30).
In order to verify the 5-FU-induced myocardial
mitochondrial damage, changes in myocardial mitochondrial ATP
levels and MMP were assessed in rats after 14 days of 5-FU
administration in the present study. The results demonstrated that
in the YM and AM groups, the myocardial MMP was significantly
decreased compared with that in the respective control groups, with
more notable effects in the AM group. MMP is a sensitive indicator
of the integrity of mitochondrial function, and the dissipation of
MMP prevents cells from synthesizing sufficient ATP to complete
normal physiological activities (40,41).
In the present study, the ATP levels in each group followed the
same trend as the levels of MMP. Eskandari et al (12) have reported that a high
concentration of 5-FU results in damage beyond the compensation
function of the mitochondria, resulting in a reduction in ATP
production. These results were consistent with those of the current
study. In addition, the ultrastructure of the myocardial
mitochondria was observed using electron microscopy. The
mitochondria in the YM group exhibited myofibril rupture, swelling
and crest fracture or fusion, as well as a high number of
autophagosomes. The AM group exhibited mitochondrial sparseness,
shrinking mitochondrial structure and a breaking crest, as well as
high lipid droplet deposition and autophagosome formation. The
results of the present study revealed abnormal morphological
changes in the mitochondria and confirmed that 5-FU induced
abnormal mitochondrial ultrastructure in rats, resulting in a
decline or loss of mitochondrial function.
Mitochondrial damage is associated with
cardiomyocyte apoptosis, necrosis and autophagy (28). Autophagy, also termed type II
programmed cell death, is involved in the pathogenesis of many
diseases, such as heart-related disease (42), cancer (43), vascular dementia (44) and chronic respiratory disease
(45). The complex interaction
between autophagy and apoptosis cooperatively regulates cell death,
and studies have indicated that both may be induced by the same
external stimuli (46,47). A study by Pattingre et al
(48) has revealed that in the
treatment of breast and colon tumors, apoptosis and autophagy are
concurrently upregulated, and the use of 3-methyladenine to inhibit
autophagy also inhibits caspase activation and reduces apoptosis.
In these cases, interaction was apparent between apoptosis and
autophagy, and both processes exhibited complementary cooperation
(48).
A number of common signaling pathways and regulatory
proteins exist between apoptosis and autophagy (28,49).
These include the Bcl-2 family of proteins, which play a key dual
regulatory role between both processes (48). Beclin-1 directly regulates autophagy
and apoptosis by binding to Bcl-2 anti-apoptotic proteins. The
Bcl-2/Bcl-XL complex inhibits the activation of autophagy by
beclin-1. However, when Bcl-2 competitively binds Bcl-2/Bcl-XL,
beclin-1 is released, which induces autophagy (49). Furthermore, Lindqvist et al
(50) have determined that the
antiapoptotic proteins Bcl-2 and Bcl-XL indirectly inhibit Bax and
Bcl-2 homologous antagonist/killer. In summary, apoptosis and
autophagy are complex, closely associated processes, and Bcl-2
proteins serve an important role in dynamically regulating and
maintaining the balance between them (48). In the present study, the expression
levels of Bax and Bcl-2 were detected in the myocardial tissues of
model rats following 5-FU administration, and the results
demonstrated that the expression levels of Bax were decreased in
the YM and AM groups, while the expression levels of Bcl-2 were
increased compared with those in the corresponding control groups.
Thus, we hypothesized that 5-FU-induced myocardial cell apoptosis
may be accompanied by autophagy.
Mitochondrial autophagic homeostasis is considered
to be an effective and indispensable factor for the mitochondrial
maintenance of cellular homeostasis (51). To a certain extent, the maintenance
of mitochondrial health depends on mitochondrial dynamics, which
involves sufficient, but not excessive autophagy (18,52).
When this balance is disrupted, it induces excessive autophagy and
aggravate damage. Through animal and clinical experimentation,
Eisenberg et al (53)
demonstrated that spermidine may serve a protective role in the
heart by inducing stable autophagy. However, other studies have
reported that excessive mitochondrial autophagy results in impaired
autophagosome clearance, which directly promotes cardiac cell death
(54,55).
Mitochondrial quality control is necessary to
maintain normal cellular activity, for which mitochondrial
autophagy is crucial (56).
PINK-1/Parkin is a regulatory signaling pathway involved in
mitochondrial autophagy (57,58).
Previous studies have confirmed that in a healthy state, PINK-1
degrades through the actions of presenilins-associated
rhomboid-like protein (59,60). However, during mitochondrial damage,
hypoxia and external stimulation, PINK-1 is stabilized and recruits
Parkin ligase to initiate autophagy (59,60).
Mitochondrial membrane proteins induce autophagy junction protein
aggregation through the ubiquitination of Parkin (59,60).
Autophagic junction protein binds to LC3 through the
LC3-interacting region, and forms LC3II through further lipidation,
participating in the completion stage of phagocytic vesicle
membrane formation (61,62). Therefore, the conversion from LC3I
to LC3II suggests an increase in mitochondrial autophagy flux
(62). The increased expression of
LC3II may indicate an increase in mitochondrial autophagy; however,
autophagosome clearance may be impaired (63). In order to determine the effects of
5-FU-induced mitochondrial autophagy, mitochondrial
autophagy-related protein expression changes were examined in the
present study 7 and 14 days after 5-FU administration. The
expression levels of mitochondrial autophagy-related proteins
PINK-1, Parkin, beclin-1 and LC3 were increased on days 7 and 14 of
5-FU administration. In addition, the expression levels of beclin-1
and LC3II were significantly higher in the 2-weeks group compared
with those in the 1-week group. Thus, the results of the present
study demonstrated that 5-FU induced excessive mitochondrial
autophagy and decreased the scavenging ability of damaged
mitochondria, which induced myocardial injury. These pathological
observations were more prominent in aging rats.
The results of previous studies have suggested that
activation of autophagy rescues myocardial injury (64,65).
However, autophagy needs to be maintained within a specific level
to be beneficial, as aforementioned. The results of the present
analysis were inconsistent with the results of previous studies. We
hypothesized that the effects of moderate and excessive autophagy
may be different within the organism; for example, an appropriate
level of autophagy may facilitate the survival of cells, whereas
excessive autophagy may promote cell death. Additionally, autophagy
is a dynamic process, and moderate autophagy of the myocardial
mitochondria may serve a protective role in the early stage of 5-FU
intervention. In the present study, with prolonged administration
of 5-FU, mitochondrial autophagy was increased, and excessive
autophagy induced or exacerbated myocardial injury. Further studies
should focus on observing the extent of mitochondrial autophagy on
a daily basis, either by electron microscopy or by detection of
autophagy-related proteins. However, frequent sampling and testing
is not possible in animal experiments; thus, in vitro
experiments are required to complement the animal experiments.
Mitochondrial division inhibitor 1 (Mdivi-1)
inhibits mitochondrial autophagy, which reduces mitochondrial and
myocardial damage (65). Further
in vitro experiments, Mdivi-1 and autophagy activator
ras-related protein will be used to reveal the dynamic changes in
autophagic processes in 5-FU-induced myocardial injury and the
potential signaling pathways involved. Future studies will also
further investigate whether inhibiting mitochondrial autophagy
reduces 5-FU-induced mitochondrial damage, which may promote the
development of novel therapeutic strategies for the prevention and
reduction of 5-FU-induced myocardial injury.
In conclusion, the results of the present study
demonstrated that autophagy was acutely activated following
5-FU-induced myocardial injury. Consistent 5-FU administration
induced excessive autophagy in damaged mitochondria, and an
impaired ability of autophagosome clearance resulted in direct
myocardial cell damage, exacerbating cardiac dysfunction over
time.
Acknowledgements
Not applicable.
Funding
This research was supported by the Graduate Students Innovation
Fund of Tianjin University of Traditional Chinese Medicine (grant
no. ZXYCXLX201801), Tianjin Binhai New Area Health Committee
Science and Technology Key Project (grant no. 2019BWKZ004), Tianjin
Union Medical Center Scientific Research Project (grant no.
2019YJZD001) and the Special Basic Research Project Cooperation in
Beijing, Tianjin and Hebei (grant no. 19JCZDJC63900).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW supervised and designed the study, and revised
and approved the manuscript. YL conceived and designed the study,
performed the experiments and data analysis, and wrote the
manuscript. XLi and YZ performed the experiments and translated the
manuscript. XZ co-designed the study and acquired funding. XLe
analyzed the animal ultrasound results. LW and YL confirm the
authenticity of all the raw data. All authors have read the final
manuscript.
Ethics approval and consent to
participate
The animal study was reviewed and approved by The
Animal Care and Use Committee of Tianjin Union Medical Center,
Tianjin, China (2020-B03).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McQuade RM, Stojanovska V, Bornstein JC
and Nurgali K: Colorectal cancer chemotherapy: The evolution of
treatment and new approaches. Curr Med Chem. 24:1537–1557.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li C, Ngorsuraches S, Chou C, Chen L and
Qian J: Risk factors of fluoropyrimidine induced cardiotoxicity
among cancer patients: A systematic review and meta-analysis. Crit
Rev Oncol Hematol. 162(103346)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kastl S, Brunner T, Herrmann O, Riepl M,
Fietkau R, Grabenbauer G, Sauer R, Hohenberger W and Klein P:
Neoadjuvant radio-chemotherapy in advanced primarilynon-resectable
carcinomas of the pancreas. Eur J Surg Oncol. 26:578–582.
2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Abbruzzese JL and Levin B: Treatment of
advanced colorectal cancer. Hematol Oncol Clin North Am. 3:135–153.
1989.PubMed/NCBI
|
|
5
|
Chang HM, Moudgil R, Scarabelli T, Okwuosa
TM and Yeh ETH: Cardiovascular complications of cancer therapy:
Best practices in diagnosis, prevention, and management: Part 1. J
Am Coll Cardiol. 70:2536–2551. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Raber I, Warack S, Kanduri J, Pribish A,
Godishala A, Abovich A, Orbite A, Dommaraju S, Frazer M, Peters ML
and Asnani A: Fluoropyrimidine-associated cardiotoxicity: A
retrospective case-control study. Oncologist. 25:e606–e609.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Polk A, Vaage-Nilsen M, Vistisen K and
Nielsen DL: Cardiotoxicity in cancer patients treated with
5-fluorouracil or capecitabine: A systematic review of incidence,
manifestations and predisposing factors. Cancer Treat Rev.
39:974–984. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kanduri J, More LA, Godishala A and Asnani
A: Fluoropyrimidine-associated cardiotoxicity. Cardiol Clin.
37:339–405. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Abdel-Rahman O: 5-Fluorouracil-related
cardiotoxicity; findings from five randomized studies of
5-fluorouracil-based regimens in metastatic colorectal cancer. Clin
Colorectal Cancer. 18:58–63. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sorrentino MF, Kim J, Foderaro AE and
Truesdell AG: 5-Fluorouracil induced cardiotoxicity: Review of the
literature. Cardiol J. 19:453–458. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lamberti M, Porto S, Marra M, Zappavigna
S, Grimaldi A, Feola D, Pesce D, Naviglio S, Spina A, Sannolo N and
Caraglia M: 5-Fluorouracil induces apoptosis in rat cardiocytes
through intracellular oxidative stress. J Exp Clin Cancer Res.
31(60)2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Eskandari MR, Moghaddam F, Shahraki J and
Pourahmad J: A comparison of cardiomyocyte cytotoxic mechanisms for
5-fluorouracil and its pro-drug capecitabine. Xenobiotica.
45:79–87. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Focaccetti C, Bruno A, Magnani E,
Bartolini D, Principi E, Dallaglio K, Bucci EO, Finzi G, Sessa F,
Noonan DM and Albini A: Effects of 5-fluorouracil on morphology,
cell cycle, proliferation, apoptosis, autophagy and ROS production
in endothelial cells and cardiomyocytes. PLoS One.
10(e0115686)2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lemaire L, Malet-Martino MC, Forni M,
Martino R and Lasserre B: Cardiotoxicity of commercial
5-fluorouracil vials stems from the alkaline hydrolysis of this
drug. Br Cancer. 66:119–127. 1992.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Polk A, Vistisen K, Vaage-Nilsen M and
Nielsen DL: A systematic review of the pathophysiology of
5-fluorouracil-induced cardiotoxicity. BMC Pharmacol Toxicol.
15(47)2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Mizushima N: A brief history of autophagy
from cell biology to physiology and disease. Nat Cell Biol.
20:521–527. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ong SB, Kalkhoran SB, Cabrera-FUentes HA
and Hausenloy DJ: Mitochondrial fusion and fission proteins as
novel therapeutic targets for treating cardiovascular disease. Eur
J Pharmacol. 763:104–114. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Dombi E, Mortiboys H and Poulton J:
Modulating mitophagy in mitochondrial disease. Curr Med Chem.
25:5597–5612. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yu YL, Wang MS, Wang ZS, Yu JQ, Song YY,
Li BZ, Hong ZZ, Wen ZN, Xu GS, Liang SJ, et al: A guidebook for the
care and use of laboratory animals. In: Chinese society for the
laboratory animal science Publ. Taiwan, pp64-75, 2004.
|
|
20
|
Zou YH, Xu ZW, Huang R, Chen LM, Yu L,
Wang X, Wang SC, Guo XJ, Zhang W, Cai ZZ, et al: Laboratory animal
science. In: Science Press Publ. Beijing, pp200-203, 2016.
|
|
21
|
Soslow RA, Dannenberg AJ, Rush D, Woerner
BM, Khan KN, Masferrer J and Koki AT: COX-2 is expressed in human
pulmonary, colonic, and mammary tumors. Cancer. 89:2637–2645.
2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Flameng W, Borgers M, Daenen W and
Stalpaert G: Ultrastructural and cytochemical correlates of
myocardial protection by cardiac hypothermia in man. J Thora
Cardiovasc Surg. 79:413–424. 1980.PubMed/NCBI
|
|
23
|
Mishra T, Shokr M, Ahmed A and Afonso L:
Acute reversible left ventricular systolic dysfunction associated
with 5-fluorouracil therapy: A rare and increasingly recognised
cardiotoxicity of a commonly used drug. BMJ Case Rep.
12(e230499)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Iskandar MZ, Quasem W and El-Omar M:
5-Fluorouracil cardiotoxicity: Reversible left ventricular systolic
dysfunction with early detection. BMJ Case Rep.
2015(bcr2015209347)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Depetris I, Marino D, Bonzano A, Cagnazzo
C, Filippi R, Aglietta M and Leone F: Fluoropyrimidine-induced
cardiotoxicity. Crit Rev Oncol Hematol. 124:1–10. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What is the connection? Trends Cell Biol. 21:387–392.
2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013(102735)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen Q, Kang J and Fu C: The independence
of and associations among apoptosis, autophagy, and necrosis.
Signal Transduct Target Ther. 3(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sara JD, Kaur J, Khodadadi R, Rehman M,
Lobo R, Chakrabarti S, Herrmann J, Lerman A and Grothey A:
5-Fluorouracil and cardiotoxicity: A review. Ther Adv Med Oncol.
10(1758835918780140)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang D and Ma J: Mitochondrial dynamics
in rat heart induced by 5-fluorouracil. Med Sci Monit.
24:6666–6672. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Roca-Agujetas V, de Dios C, Lestón L, Marí
M, Morales A and Colell A: Recent insights into the mitochondrial
role in autophagy and its regulation by oxidative stress. Oxid Med
Cell Longev. 2019(3809308)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Maejima Y: The critical role of autophagy
in heart failure. Nihon Yakurigaku Zasshi. 151:100–105.
2018.PubMed/NCBI View Article : Google Scholar : (In Japanese).
|
|
34
|
Sridhar S, Botbol Y, Macian F and Cuervo
AM: Autophagy and disease: Always two sides to a problem. J Pathol.
226:255–273. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Novo G, Cadeddu C, Sucato V, Pagliaro P,
Romano S, Tocchetti CG, Zito C, Longobardo L, Nodari S and Penco M:
Role of biomarkers in monitoring antiblastic cardiotoxicity. J
Cardiovasc Med (Hagerstown). 17 (Suppl 1):S27–S34. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Jones M, O'Gorman P, Kelly C, Mahon N and
Fitzgibbon MC: High-sensitive cardiac troponin-I facilitates timely
detection of subclinical anthracycline-mediated cardiac injury. Ann
Clin Biochem. 54:149–157. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mokhtar AT, Begum J, Buth KJ and Legare
JF: Cardiac troponin T is an important predictor of mortality after
cardiac surgery. J Crit Care. 38:41–46. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Rtibi K, Selmi S, Grami D, Amri M, Sebai H
and Marzouki L: Contribution of oxidative stress in acute
intestinal mucositis induced by 5 fluorouracil (5-FU) and its
pro-drug capecitabine in rats. Toxicol Mech Methods. 28:262–267.
2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
McQuade RM, Stojanovska V, Donald E, Abalo
R, Bornstein JC and Nurgali K: Gastrointestinal dysfunction and
enteric neurotoxicity following treatment with anticancer
chemotherapeutic agent 5-fluorouracil. Neurogastroenterol Motil.
28:1861–1875. 2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Murphy E, Ardehali H, Balaban RS, DiLisa
F, Dorn GW II, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, et
al: Mitochondrial function, biology, and role in disease: A
scientific statement from the American heart association. Circ Res.
118:1960–1991. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Basu Ball W, Neff JK and Gohil VM: The
role of nonbilayer phospholipids in mitochondrial structure and
function. FEBS Lett. 592:1273–1290. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Onorati AV, Dyczynski M, Ojha R and
Amaravadi RK: Targeting autophagy in cancer. Cancer. 124:3307–3318.
2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wang W, Qiao O, Ji H, Zhang X, Han X,
Zhang Y, Wang J, Li X and Gao W: Autophagy in vascular dementia and
natural products with autophagy regulating activity. Pharmacol Res.
170(105756)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Racanelli AC, Kikkers SA, Choi AMK and
Cloonan SM: Autophagy and inflammation in chronic respiratory
disease. Autophagy. 14:221–232. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mukhopadhyay S, Panda PK, Sinha N, Das DN
and Bhutia SK: Autophagy and apoptosis: Where do they meet?
Apoptosis. 19:555–566. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Young MM, Kester M and Wang HG:
Sphingolipids: Regulators of crosstalk between apoptosis and
autophagy. J Lipid Res. 54:5–19. 2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell.
122:927–939. 2005.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Nakajima S, Aikawa C, Nozawa T,
Minowa-Nozawa A, Toh H and Nakagawa I: Bcl-xL affects group a
streptococcus-induced autophagy directly, by inhibiting fusion
between autophagosomes and lysosomes, and indirectly, by inhibiting
bacterial internalization via interaction with beclin 1-UVRAG. PLoS
One. 13(e0170138)2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lindqvist LM, Heinlein M, Huang DC and
Vaux DL: Prosurvival Bcl-2 family members affect autophagy only
indirectly, by inhibiting Bax and Bak. Proc Nati Acad Sci USA.
111:8512–8517. 2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Haeussler S, Köhler F, Witting M, Premm
MF, Rolland SG, Fischer C, Chauve L, Casanueva O and Conradt B:
Autophagy compensates for defects in mitochondrial dynamics. PLoS
Genet. 16(e1008638)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yoo SM and Jung YK: A molecular approach
to mitophagy and mitochondrial dynamics. Mol Cells. 41:18–26.
2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Eisenberg T, Abdellatif M, Schroeder S,
Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann
A, Schmidt A, et al: Cardioprotection and lifespan extension by the
natural polyamine spermidine. Nat Med. 22:1428–1438.
2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lin XL, Xiao WJ, Xiao LL and Liu MH:
Molecular mechanisms of autophagy in cardiac ischemia/reperfusion
injury (review). Mol Med Rep. 18:675–683. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Liu CY, Zhang YH, Li RB, Zhou LY, An T,
Zhang RC, Zhai M, Huang Y, Yan YW, Dong YH, et al: LncRNA CAIF
inhibits autophagy and attenuates myocardial infarction by blocking
p53-mediated myocardin transcription. Nat Commun.
9(29)2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Ng MYW, Wai T and Simonsen A: Quality
control of the mitochondrion. Dev Cell. 56:881–905. 2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Hamacher-Brady A and Brady NR: Mitophagy
programs: Mechanisms and physiological implications of
mitochondrial targeting by autophagy. Cell Mol Life Sci.
73:775–795. 2016.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Tanaka K: The PINK1-Parkin axis: An
overview. Neurosci Res. 159:9–15. 2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chun Y and Kim J: Autophagy: An essential
degradation program for cellular homeostasis and life. Cells.
7(278)2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kawajiri S, Saiki S, Sato S, Sato F,
Hatano T, Eguchi H and Hattori N: PINK1 is recruited to
mitochondria with parkin and associates with LC3 in mitophagy. FEBS
Lett. 584:1073–1079. 2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Sciarretta S, Yee D, Nagarajan N, Bianchi
F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schirone L,
et al: Trehalose-induced activation of autophagy improves cardiac
remodeling after myocardial infarction. J Am Coll Cardiol.
71:1999–2010. 2018.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Kabeya Y, Mizushima N, Yamamoto A,
Oshitani-Okamoto S, Ohsumi Y and Yoshimori T: LC3, GABARAP and
GATE16 localize to autophagosomal membrane depending on form-II
formation. J Cell Sci. 117:2805–2812. 2004.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Wu X, Qin Y, Zhu X, Liu D, Chen F, Xu S,
Zheng D, Zhou Y and Luo J: Increased expression of DRAM1 confers
myocardial protection against ischemia via restoring autophagy
flux. J Mol Cell Cardiol. 124:70–82. 2018.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Liang H, Su X, Wu Q, Shan H, Lv L, Yu T,
Zhao X, Sun J, Yang R, Zhang L, et al: LncRNA 2810403D21Rik/Mirf
promotes ischemic myocardial injury by regulating autophagy through
targeting Mir26a. Autophagy. 16:1077–1691. 2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Dong Y, Undyala VVR and Przyklenk K:
Inhibition of mitochondrial fission as a molecular target for
cardioprotection: Critical importance of the timing of treatment.
Basic Res Cardiol. 111(59)2016.PubMed/NCBI View Article : Google Scholar
|