Introduction
Acute respiratory distress syndrome (ARDS) is a
severe condition that may cause acute lung injury. ARDS is a
multifactorial syndrome that leads to significant morbidity and
mortality in infants and children, with a fatality rate ≤40%
worldwide (1,2). ARDS usually results from trauma,
hemorrhagic shock or toxic inhalation. However, the most common
cause of this syndrome is bacterial sepsis (3). The development of severe inflammation
involving pro-inflammatory cytokine production and neutrophil
integration is the main feature of ARDS, which may account for its
high mortality rate (4). Previous
studies conducted on ARDS have not been successful in developing
specific biomarkers and pharmacological targets for patients with
ARDS. Therefore, the identification of novel targets for the
treatment of ARDS is crucial.
At present, the pathogenesis of ARDS remains
unclear. It has been recognized that ARDS is primarily induced by
pathogenic inflammation. Lipopolysaccharide (LPS) is a main
component of the cell wall of Gram-negative bacteria and has been
reported to cause lung injury, which can subsequently progress into
ARDS (5,6). Chitinase-3-like-1 protein (CHI3L1 or
YKL-40) is also termed human cartilage glycoprotein-39. This
protein is a member of the mammalian chitinase-like protein family
and can be secreted by numerous cells, including macrophages,
vascular smooth muscle cells, endothelial cells, chondrocytes and
neutrophils (7,8). YKL-40 is considered as a
pro-inflammatory cytokine and its circulating levels have been
shown to be abnormally elevated in a wide range of inflammatory
disorder-associated diseases, including purulent meningitis,
rheumatoid arthritis and community-acquired pneumonia (9-12).
In addition, accumulating evidence has shown that YKL-40 might play
a crucial pathogenic role in chronic obstructive pulmonary disease
and hyperoxia-induced acute lung injury (13,14).
Interestingly, from the JASPAR database (http://jaspar.genereg.net/), a potential binding
relationship was found between YKL-40 promoter and Fos-related
antigen 1 (Fra-1). Fra-1 is a member of the Fos family of proteins
and a component of the activator protein-1 transcription factor
complex (15). Fra-1 is a
transcription factor involved in various pathological processes,
including cell proliferation and cell death, extracellular
remodeling, inflammation and immune response (16).
To the best of our knowledge, whether YKL-40 or
Fra-1 is involved in ARDS progression remains unknown. The present
study investigated therefore the role of YKL-40 in LPS-induced ARDS
and its potential underlying mechanisms.
Materials and methods
Cell culture and treatment
The human type II lung epithelial A549 cell line was
obtained from the American Type Culture Collection and cultured in
RPMI-1640 medium (Thermo Fisher Scientific, Inc.) supplemented with
10% FBS (Thermo Fisher Scientific, Inc.). The cells were maintained
at 37˚C in a humidified incubator containing 5% CO2.
A549 cells were treated with increasing concentrations of LPS (100,
500, 1,000 and 1,500 ng/ml) for 12 h for stimulation (17).
Cell transfection
Short hairpin (sh) RNA targeting Fra-1
(shRNA-Fra-1-1/2), sh-RNA targeting YKL-40 (sh-YKL-40-1/2) and
negative control shRNA (sh-NC) were obtained from Shanghai
GenePharma Co., Ltd. A YKL-40 overexpression plasmid
(pcDNA3.1-YKL-40) and a Fra-1 overexpression plasmid
(pcDNA3.1-Fra-1) were commercially constructed by Shanghai
GenePharma Co., Ltd. and the empty pcDNA 3.1 vector (pcDNA 3.1) was
used as the negative control. A549 cells were seeded in 6-well
plates (2x105 cells/well) in a humidified incubator
containing 5% CO2 at 37˚C. When cells reached 70-80%
confluence, they were transfected with sh-NC (500 ng/µl),
sh-YKL-40-1/2 (500 ng/µl), sh-Fra-1-1/2 (500 ng/µl), pcDNA3.1-Fra-1
(15 nM), pcDNA3.1-YKL-40 (15 nM) or pcDNA 3.1 (15 nM) using
Lipofetamine® 2000 reagent (Thermo Fisher Scientific,
Inc.) for 48 h according to the manufacturer's instructions.
Following transfection, cells were cultured at 37˚C in a humidified
incubator containing 5% CO2 for 48 h and the
transfection efficiency was determined using reverse
transcription-quantitative (RT-q) PCR and western blotting. After
48 h transfection, cells were harvested for subsequent
experiments.
Cell viability assay
Cell viability was determined using the Cell
Counting Kit-8 (CCK-8) assay. Cells were seeded in 96-well plates
at the density of 1x103 cells/well and treated with LPS
(0, 100, 500, 1,000 and 1,500 ng/ml) for 12 h. CCK-8 solution (10
µl; Dojindo Molecular Technologies, Inc.) was added to the cells
that were incubated for an additional 3 h. The absorbance was read
at 450 nm on a microplate reader (Bio-Rad Laboratories, Inc.).
RT-qPCR
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse-transcribed into cDNA using
PrimeScript™ RT Master Mix kit (Takara Bio, Inc.)
according to the manufacturer's instructions. The mRNA levels of
the genes were assessed by RT-qPCR according to the SYBR Premix
Ex-Taq Kit (Takara Bio, Inc.). The primers used were as follows:
YKL-40, forward, 5'-CCTGCTCAGCGCAGCACTGT-3' and reverse,
5'-GCTTTTGACGCTTTCCTGGTC-3'; Fra-1, forward,
5'-AGGAACTGACCGACTTCCTG-3' and reverse, 5'-CAGCTCTAGGCGCTCCTTC-3',
and GAPDH, forward, 5'-AGCCACATCGCTCAGACA-3' and reverse,
5'-GCCCAATACGACCAAATCC-3'. The qPCR thermocycling conditions were
as follows: 5 min at 95˚C, 40 cycles of 10 sec at 95˚C, 20 sec at
59˚C and 30 sec at 72˚C. The relative expression levels were
normalized to endogenous control and were expressed as
2-ΔΔCq (18).
Western blotting
Cells were lysed in RIPA buffer (Wuhan Servicebio
Technology Co., Ltd.) on ice for 30 min. Following centrifugation
at 4˚C, 12,000 x g for 15 min, the supernatant was collected and
the protein concentration was determined using the BCA method.
Proteins (30 µg/lane) were separated by 10% SDS-PAGE and
subsequently transferred onto PVDF membranes. Membranes were
blocked with 5% skimmed milk for 1 h at room temperature and
incubated with primary antibodies against YKL-40 (1:1,000; cat. no.
ab180569; Abcam), Fra-1 (1:1,000; cat. no. ab124722; Abcam), Bcl-2
(1:2,000; cat. no. ab182858; Abcam), Bax (1:1,000; cat. no.
ab32503; Abcam), cleaved caspase 3 (1:500; cat. no. ab2302; Abcam),
caspase 3 (1:500; cat. no. ab13847; Abcam), cleaved caspase 9
(1:500; cat. no. ab2324; Abcam), caspase 9 (1:1,000; cat. no.
ab32539; Abcam) and GAPDH (1:2,500; cat. no. ab9485; Abcam) at 4˚C
overnight. Membranes were that incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(1:2,000; cat. no. ab6721; Abcam) at room temperature for 2 h.
Enhanced chemiluminescence reagent (Cytiva) was used to detect the
signal on the membrane. The data were analyzed via densitometry
using ImageJ software version 1.46 (National Institutes of Health)
and normalized to expression of the internal control GAPDH.
ELISA
Transfected cells were treated with 500 ng/ml LPS
for 12 h. Subsequently, the cell supernatant was collected. The
levels of tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and
IL-1β in the cell supernatant were measured using ELISA kits (cat.
no. 555220 for IL-6; cat. no. 557953 for IL-1β; and cat. no. 555212
for TNF-α; BD Biosciences) according to the manufacturer's
instructions.
TUNEL assay
Apoptosis was detected in vitro using TUNEL
staining. Following the cell treatment, cells were fixed with 4%
formaldehyde for 10 min at room temperature and permeabilized with
0.1% Triton X-100 for 2 min at room temperature. TUNEL assay Kit
(Roche Diagnostics) was used to detect the percentage of apoptotic
cells according to the instructions provided by the manufacturer.
The fluorescent images were captured using an inverted fluorescence
microscope (magnification, x200).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using the EpiQuik Chromatin
Immunoprecipitation Assay Kit (EpiGentek) according to the
manufacturer's instructions. An antibody against Fra-1 (1:30; cat.
no. ab252421; Abcam) or IgG (1:100; cat. no. ab172730; Abcam) was
used for immunoprecipitation. Subsequently, gel electrophoresis and
RT-qPCR were performed to detect the DNA fragments at the predicted
YKL-40 promoter binding sites.
Luciferase reporter assay
According to the JASPAR database (http://jaspar.genereg.net/), a potential binding
relationship between YKL-40 promoter and Fra-1 was predicted. Then,
this binding relationship was verified by Luciferase reporter
assay. In brief, the pcDNA 3.1-Fra-1 or pcDNA 3.1 vector was
co-transfected into A549 cells together with a luciferase reporter
plasmid driven by the full length (FL) of YKL-40 promoter, or
mutated YKL-40 promoter (targeting 2 E-Box motifs, named E1 Del and
E2 Del, respectively) using Lipofetamine® 2000 reagent
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. After 48 h of transfection, the Dual Reporter Assay
System (Promega Corporation) was used to measure luciferase
activity. The relative luciferase activity was normalized to that
of Renilla luciferase.
Statistical analysis
GraphPad Prism 5 software (GraphPad Software, Inc.)
was used for statistical analysis. The experimental data were
presented as the means ± standard deviation of three independent
experiments. Data were compared using one-way ANOVA followed by
Tukey's post hoc test when appropriate. P<0.05 was considered to
indicate a statistically significant difference.
Results
YKL-40 expression is upregulated in
LPS-treated A549 cells
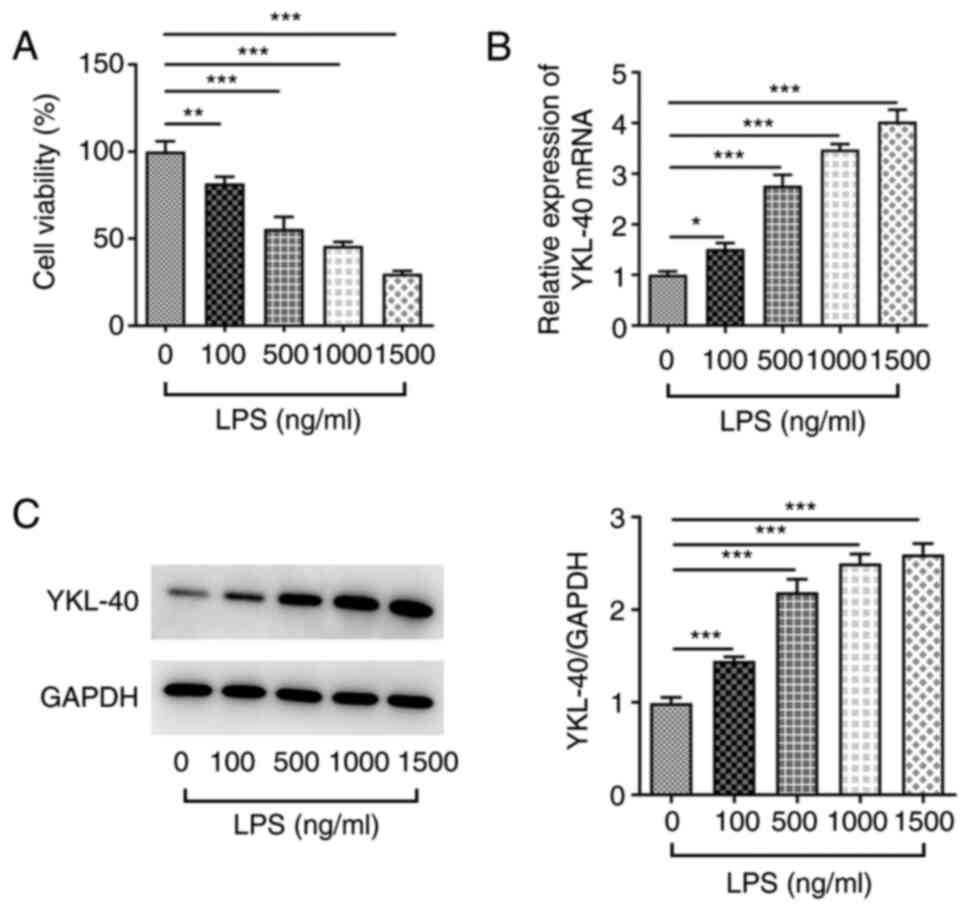
LPS was used to mimic the development of ARDS in
A549 cells. Cell viability was decreased following treatment with
increasing concentrations of LPS (0, 100, 500, 1,000 and 1,500
ng/ml; Fig. 1A). Furthermore, the
mRNA and protein expression of YKL-40 was significantly upregulated
(Fig. 1B and C) following LPS treatment. To retain a
relative cell viability >50% after LPS stimulation, a
concentration of 500 ng/ml LPS was used for induction of ARDS in
subsequent experiments.
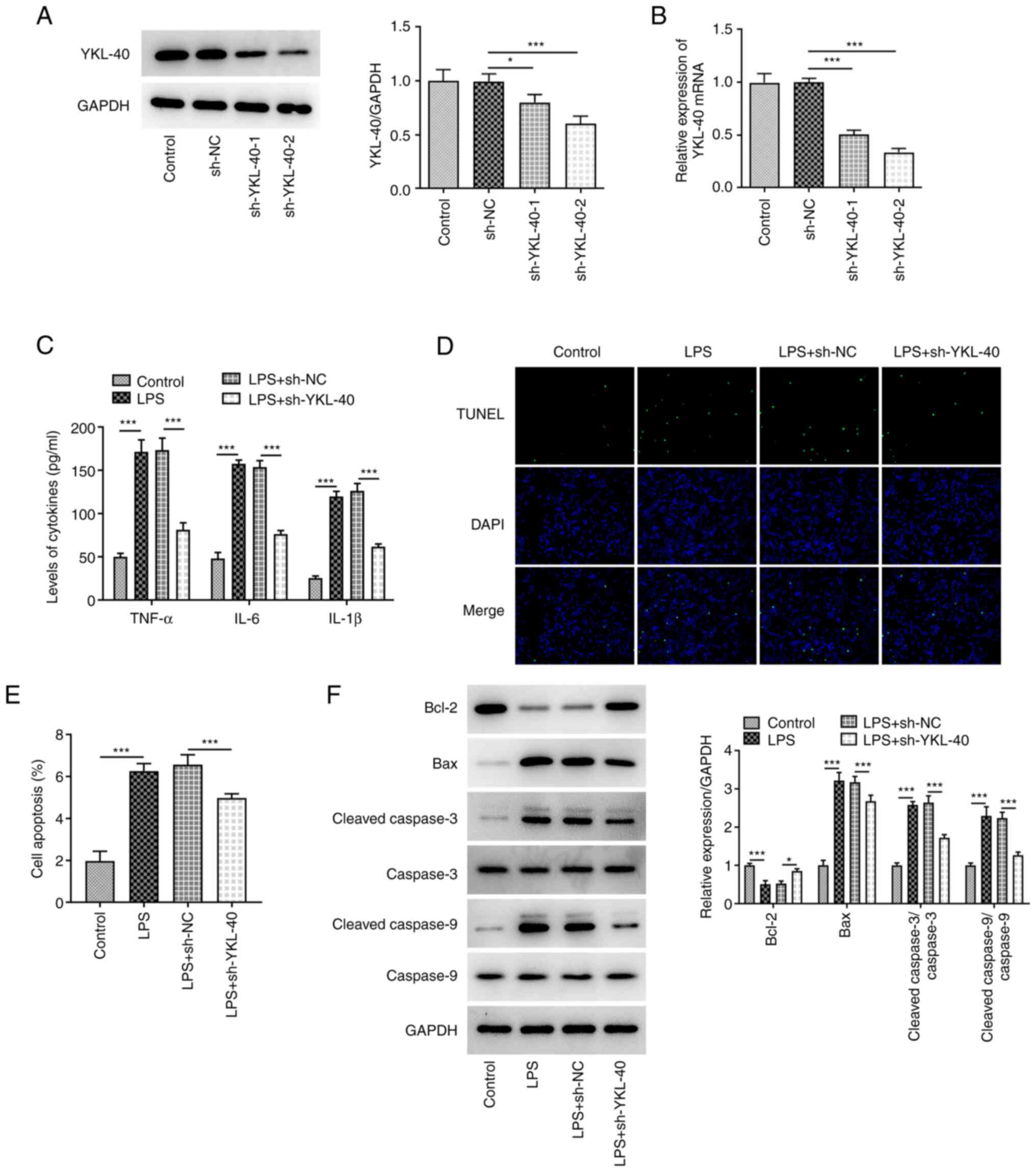
YKL-40 knockdown inhibits the
inflammatory response and apoptosis in LPS-treated A549 cells
To explore the role of YKL-40 in LPS-treated A549
cells, cells were transfected with sh-YKL-40-1/2. The results
indicated that the mRNA and protein expression of YKL-40 was
significantly downregulated following transfection, notably with
sh-YKL-40-2, which was used in subsequent experiments (Fig. 2A and B). A549 cells were treated with LPS and
transfected or not with sh-YKL-40. The concentration of certain
inflammatory cytokines was detected to assess the effect of YKL-40
on the induction of inflammation in LPS-treated A549 cells. The
results demonstrated that LPS treatment induced a severe
inflammatory response, with significantly increased levels of
TNF-α, IL-6 and IL-1β (Fig. 2C).
However, the increase in TNF-α, IL-6 and IL-1β release following
treatment with LPS was significantly inhibited in
sh-YKL-40-transfected cells, indicating that YKL-40 silencing
caused a significant inhibition in LPS-induced inflammation in A549
cells. Furthermore, cell apoptosis was also evaluated. The results
demonstrated that the number of apoptotic cells was elevated
following LPS treatment. This effect was partly abolished following
transfection with sh-YKL-40 (Fig.
2D and E). In addition, LPS
caused downregulation of the Bcl-2 protein and upregulation of Bax,
cleaved caspase-3 and cleaved caspase-9 proteins (Fig. 2F). These effects were partly
reversed following cell transfection with sh-YKL-40.
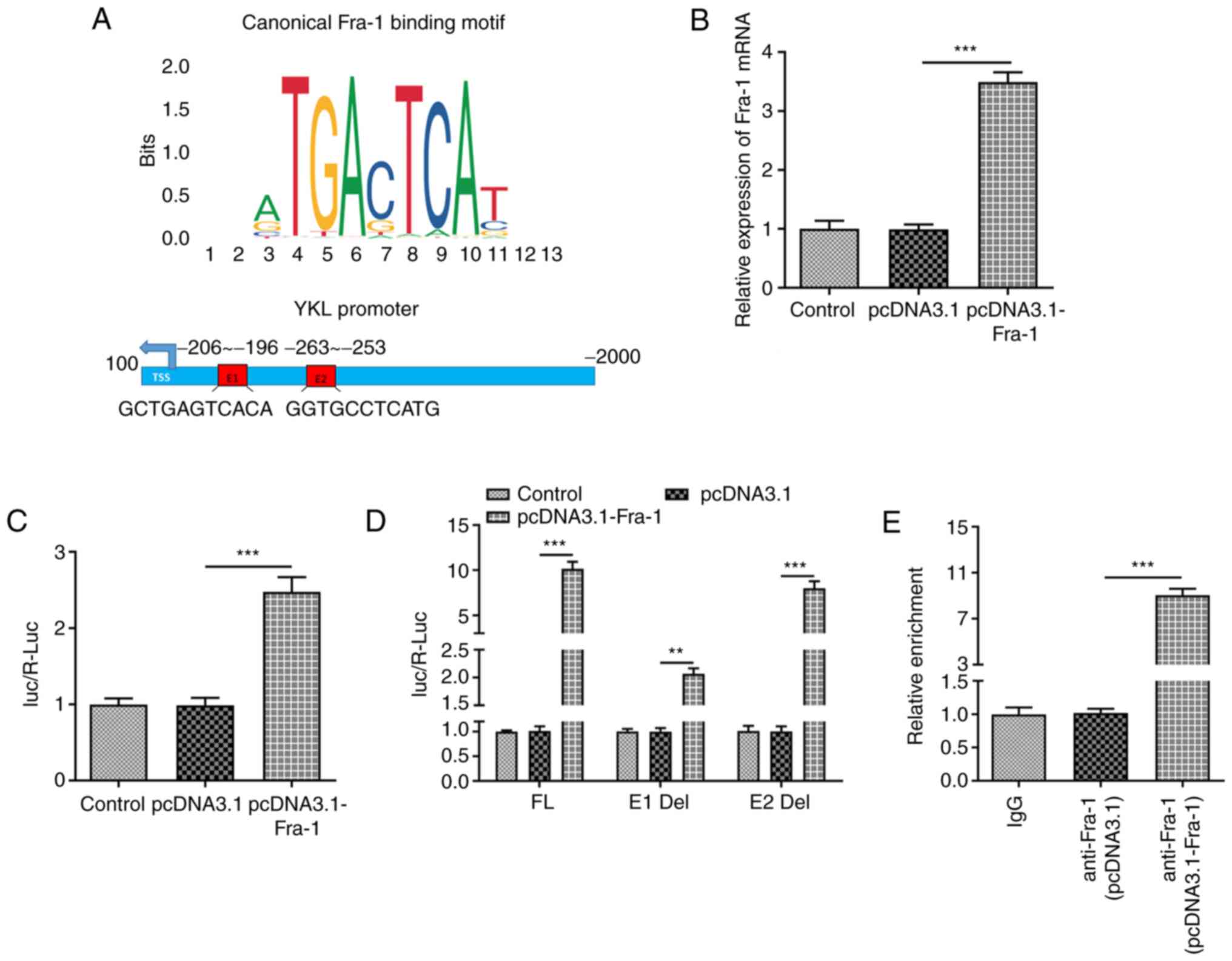
Fra-1 directly binds to YKL-40
promoter and regulates YKL-40 expression
The potential mechanism underlying the role of
YKL-40 in LPS-treated A549 cells was investigated. Firstly,
according to the JASPAR database (http://jaspar.genereg.net/), a potential binding
relationship between YKL-40 promoter and Fra-1 was predicted, and
the potential binding sites (named E1 and E2) between Fra-1 and the
YKL-40 promoter are displayed in Fig.
3A. The expression level of Fra-1 was significantly increased
following cell transfection with pcDNA3.1-Fra-1. The results from
luciferase reporter assay demonstrated that the transcriptional
activity of YKL-40 was increased following Fra-1 overexpression
(Fig. 3B and C). To clarify which binding site was
mainly responsible for the increase in transcriptional activity, E1
and E2 were deleted. The results indicated that the change in the
transcriptional activity was more apparent when E1 was deleted,
indicating that this region was mainly responsible for binding to
Fra-1 (Fig. 3D). Furthermore, the
results from ChIP assay revealed that Fra-1 was enriched at the
YKL-40 promoter within the E1 region, demonstrating the binding
association between the YKL-40 promoter and Fra-1 (Fig. 3E).
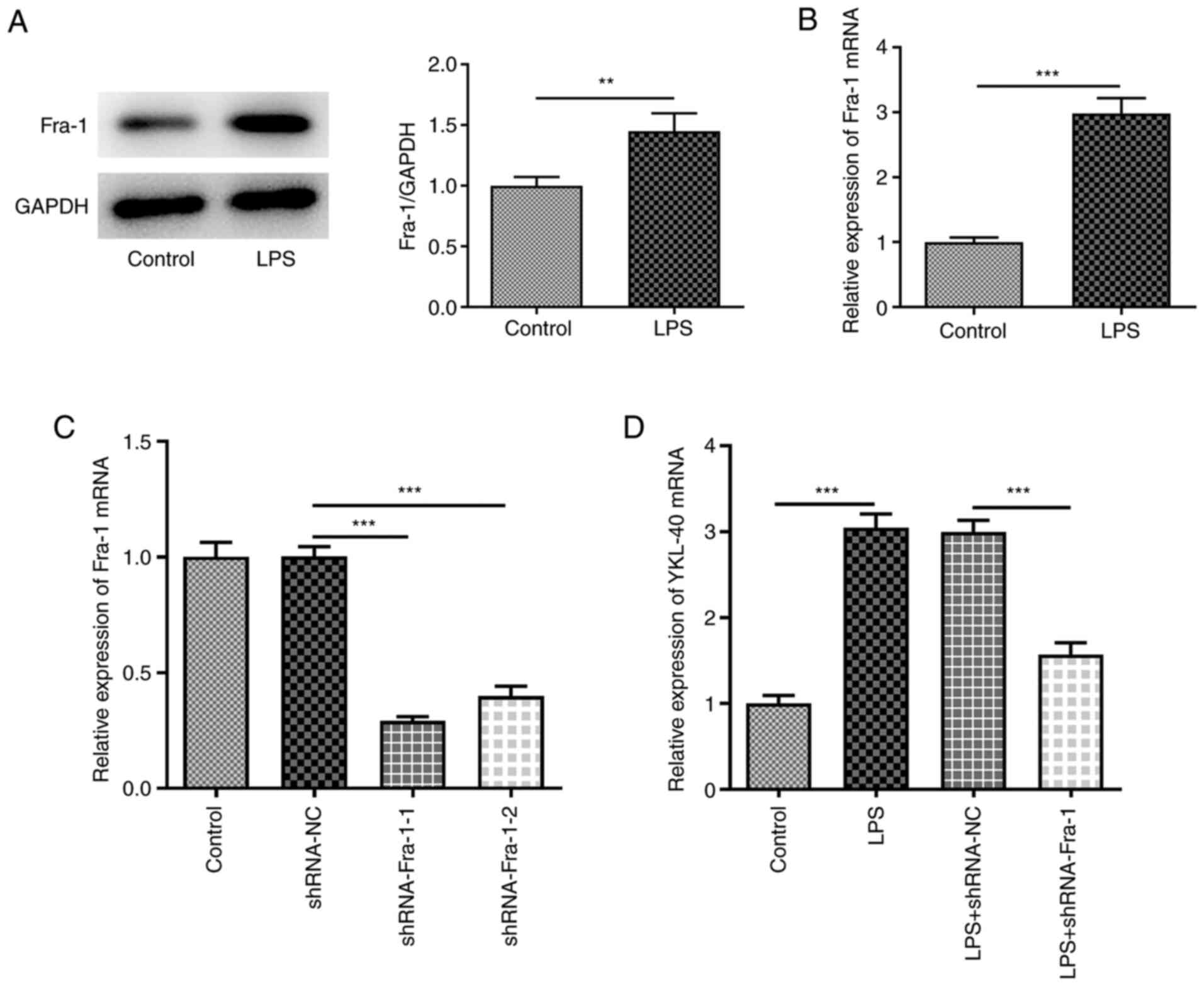
Based on these findings, the role of Fra-1 was
investigated in LPS-treated A549 cells. Both the protein and mRNA
expression of Fra-1 was significantly increased following treatment
of A549 cells with LPS (Fig. 4A
and B). Cell transfection with
sh-Fra-1-1/2 was successful (Fig.
4C), and sh-Fra-1-1 was used for subsequent experiments due to
its higher transfection efficacy. In addition, Fra-1 knockdown
decreased LPS-induced increase in YKL-40 expression level (Fig. 4D). Fra-1 expression was upregulated
in LPS-treated A549 cells, and the data indicated that Fra-1 could
directly bind to the YKL-40 promoter and positively regulate its
expression.
YKL-40 overexpression partly abolishes
the inhibitory effects of Fra-1 knockdown on LPS-induced
inflammation and apoptosis in A549 cells
YKL-40 overexpression was established following cell
transfection with pcDNA3.1-YKL-40 (Fig. 5A). Fra-1 knockdown decreased
LPS-induced elevated production of TNF-α, IL-6 and IL-1β and
reduced apoptotic cell numbers in LPS-induced A549 cells (Fig. 5B and C, respectively). This reduction in
apoptosis was accompanied by increased expression of Bcl-2 and
decreased expression of Bax, cleaved caspase 3 and cleaved caspase
9 (Fig. 5D). These alterations
were partly abolished by YKL-40 overexpression (Fig. 5B-D).
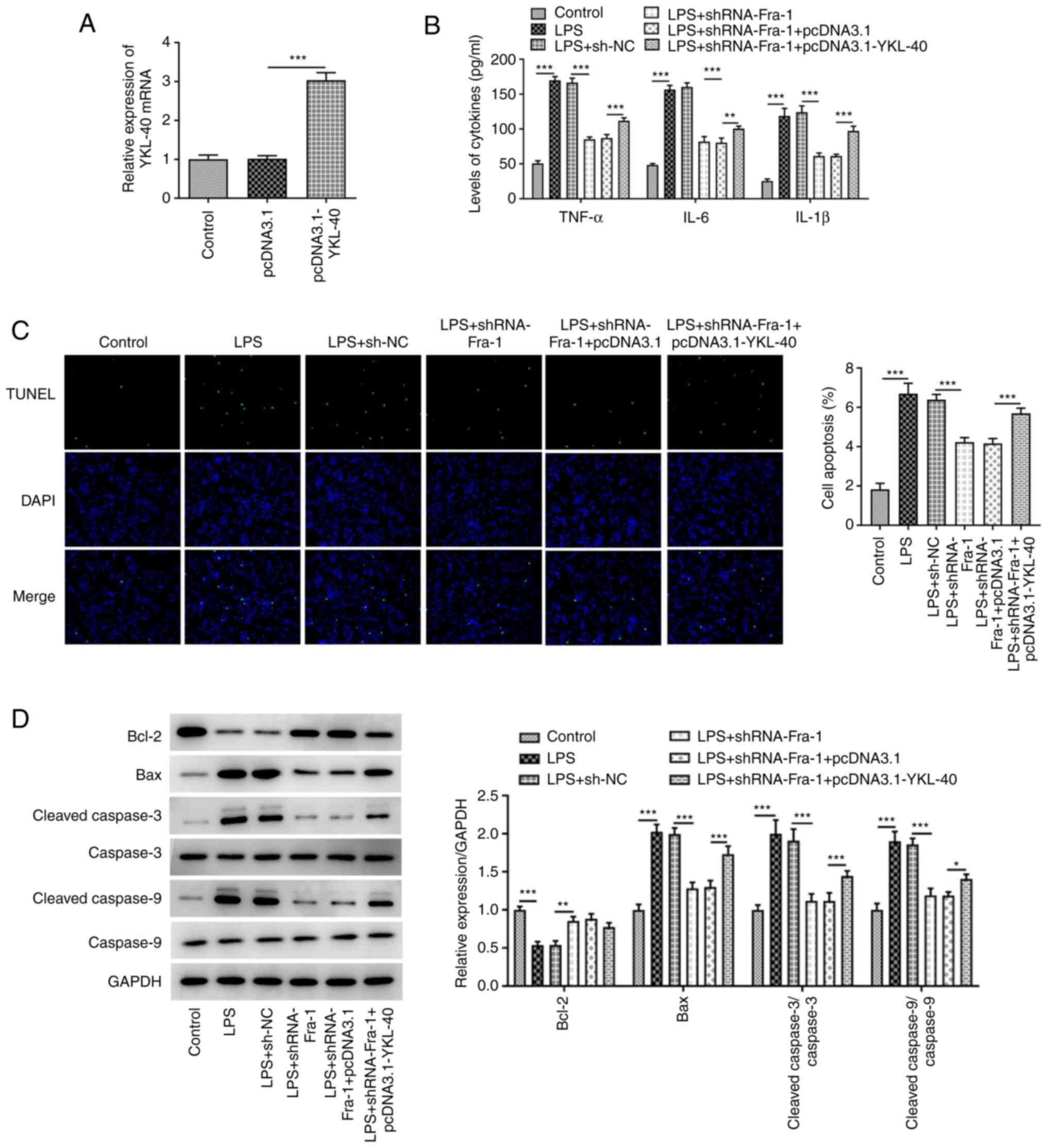 | Figure 5YKL-40 overexpression partly abolishes
the inhibitory effects of Fra-1 knockdown on LPS-induced
inflammation and apoptosis in A549 cells. A549 cells were
transfected with pcDNA3.1 or pcDNA3.1-YKL-40 and the mRNA
expression of YKL-40 was evaluated. (A) LPS-treated A549 cells were
transfected with sh-NC or sh-Fra-1 or co-transfected with sh-Fra-1
and pcDNA3.1 or pcDNA3.1-YKL-40. (B) Production of inflammatory
cytokines, including TNF-α, IL-6 and IL-1β, was measured using
ELISA kits. (C) TUNEL assay was used to detect apoptotic cells, and
the apoptotic cells were quantified. (D) Western blotting was used
to evaluate the protein expression of apoptosis-related proteins.
*P<0.05, **P<0.01 and
***P<0.001. YKL-40, chitinase-3-like-1 protein;
Fra-1, Fos-related antigen 1; LPS, lipopolysaccharide; sh, short
hairpin RNA; NC, negative control; TNF-α, tumor necrosis factor-α;
IL, interleukin. |
Discussion
ARDS is a life-threatening pulmonary inflammatory
disease characterized by pulmonary edema, refractory hypoxemia and
multiple organ failure (19).
Although YKL-40 is regarded as a pro-inflammatory cytokine and is
involved in various inflammatory diseases, its role in the
pathophysiology of ARDS remains unknown. In the present study,
LPS-treated A549 cells were used to establish an in vitro
model of ARDS. Upregulation of YKL-40 expression was observed in
LPS-treated A549 cells. Furthermore, YKL-40 knockdown inhibited the
inflammatory response and induction of cell apoptosis in
LPS-treated A549 cells. These phenomena were partly regulated by
the transcription factor Fra-1. These data suggested a potential
role of YKL-40 in the development of ARDS and may provide a
potential target for ARDS treatment.
Accumulating evidence has confirmed that excessive
production of pro-inflammatory cytokines and uncontrolled systemic
inflammatory response account for impaired lung function in ARDS
(20,21). Therefore, the reduction in the
production of pro-inflammatory cytokines and the maintenance of the
balance between the anti-inflammatory and the pro-inflammatory
response may be a promising therapeutic strategy for ARDS. It has
been previously reported that certain anti-inflammatory genes can
be used as drug targets for ARDS treatment. For example, Pooladanda
et al (6) reported that
nimbolide could ameliorate LPS-induced ARDS partly by inhibiting
inflammation. Zhou et al (22) demonstrated that the long non-coding
RNA NEAT1 is highly expressed following LPS-induced lung injury in
mice or A549 cells. In addition, suppression of NEAT1 expression
restrains LPS-induced production of the inflammatory cytokines
TNF-α, IL-6 and IL-1β via high mobility group box 1/receptor for
advanced glycation end products signaling. In addition, microRNA
(miR)-297, miR-150 and miR-216a are expressed at low levels in
ARDS. These miRs exert protective effects against LPS-induced
injury in A549 cells by reducing inflammatory cytokine secretion
(17,23,24).
The present study reported an excessive production of TNF-α, IL-6
and IL-1β in LPS-treated A549 cells, which was consistent with the
findings reported previously. Furthermore, YKL-40 expression was
upregulated following A549 cell exposure to LPS. YKL-40 knockdown
markedly decreased the production of TNF-α, IL-6 and IL-1β, which
partially alleviated the inflammatory response in LPS-treated A549
cells.
A previous study reported that Fra-1 could impair
inflammatory response and suppress inflammation-induced
chondrogenesis in fracture healing (25). In addition, Fra-1 can mediate some
anti-fibrotic effects in the lung via the regulation of
pro-inflammatory, pro-fibrotic and anti-fibrotic gene expression
(26). Fra-1 is notably activated
during inflammatory lung injury in vivo. Fra-1-null mice are
less susceptible to LPS-induced lung injury and mortality than
wild-type mice (27), indicating
that Fra-1 serves a key role in the lung inflammatory response. In
line with previous studies, the present study demonstrated that
Fra-1 expression was upregulated in LPS-treated A549 cells. It is
interesting to note that Fra-1 was predicted to bind to YKL-40
promoter from the JASPAR database. This prediction was subsequently
verified by ChIP and luciferase reporter assays, and Fra-1 could
positively regulate the expression of YKL-40. In addition, the
inhibitory effects of Fra-1 knockdown on LPS-induced inflammatory
response and cell apoptosis in A549 cells were partly abolished by
YKL-40 overexpression, indicating that the functional role of
YKL-40 in LPS-treated A549 cells may be partly mediated by
Fra-1.
This study presented some limitations. The present
study only performed in vitro experiments and lacked results
from in vivo and clinical research. In vivo
experiments and clinical research are of great significance to
verify the role of YKL-40 in ARDS and should be performed in our
future work. In addition, the transcription factors predicted to
bind to YKL-40 promoter are multiful. Whether the other
transcription factors can also directly bind to YKL-40 promoter and
regulate YKL-40 requires some clarification, and the potential
mechanism underlying the role of the other unknown potential
transcription factors and YKL-40 during ARDS progression should be
further investigated.
Taken together, the results from the present study
demonstrated a high expression of YKL-40 in LPS-induced ARDS. In
addition, YKL-40 knockdown exerted some protective effects against
LPS-induced inflammatory response and apoptosis in A549 cells. The
transcription factor Fra-1 could directly bind to the YKL-40
promoter and was partly responsible for mediating the function of
YKL-40. These findings indicated that YKL-40 may represent a
promising target for the development of therapeutic strategies for
ARDS.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DW made substantial contributions to the conception
and design of the study. FW and WL made substantial contributions
to data acquisition. FW, ZL and RY were responsible for the
development of the study methodology, analysis and interpretation
of the data. DW, FW and WL were involved in drafting the manuscript
or revising it critically for important intellectual content. DW
and FW confirm the authenticity of the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Amigoni A, Pettenazzo A, Stritoni V and
Circelli M: Surfactants in acute respiratory distress syndrome in
infants and children: Past, present and future. Clin Drug Investig.
37:729–736. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349.
2000.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jung YJ, Park YY, Huh JW and Hong SB: The
effect of human adipose-derived stem cells on
lipopolysaccharide-induced acute respiratory distress syndrome in
mice. Ann Transl Med. 7(674)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li D, Sun T, Chi L, Zhao D and Li W:
Acupoint catgut embedding improves the lipopolysaccharide-induced
acute respiratory distress syndrome in rats. Biomed Res Int.
2020(2394734)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Pooladanda V, Thatikonda S, Bale S,
Pattnaik B, Sigalapalli DK, Bathini NB, Singh SB and Godugu C:
Nimbolide protects against endotoxin-induced acute respiratory
distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3
nuclear translocation. Cell Death Dis. 10(81)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lee CG, Hartl D, Lee GR, Koller B,
Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, et
al: Role of breast regression protein 39 (BRP-39)/chitinase
3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J
Exp Med. 206:1149–1166. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Johansen JS: Studies on serum YKL-40 as a
biomarker in diseases with inflammation, tissue remodelling,
fibroses and cancer. Dan Med Bull. 53:172–209. 2006.PubMed/NCBI
|
|
9
|
Kastrup J: Can YKL-40 be a new
inflammatory biomarker in cardiovascular disease? Immunobiology.
217:483–491. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jafari-Nakhjavani MR, Ghorbanihaghjo A,
Bagherzadeh-Nobari B, Malek-Mahdavi A and Rashtchizadeh N: Serum
YKL-40 levels and disease characteristics in patients with
rheumatoid arthritis. Caspian J Intern Med. 10:92–97.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ostergaard C, Johansen JS, Benfield T,
Price PA and Lundgren JD: YKL-40 is elevated in cerebrospinal fluid
from patients with purulent meningitis. Clin Diagn Lab Immunol.
9:598–604. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nordenbaek C, Johansen JS, Junker P,
Borregaard N, Sorensen O and Price PA: YKL-40, a matrix protein of
specific granules in neutrophils, is elevated in serum of patients
with community-acquired pneumonia requiring hospitalization. J
Infect Dis. 180:1722–1726. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Tong X, Wang D, Liu S, Ma Y, Li Z, Tian P
and Fan H: The YKL-40 protein is a potential biomarker for COPD: A
meta-analysis and systematic review. Int J Chron Obstruct Pulmon
Dis. 13:409–418. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sohn MH, Kang MJ, Matsuura H, Bhandari V,
Chen NY, Lee CG and Elias JA: The chitinase-like proteins breast
regression protein-39 and YKL-40 regulate hyperoxia-induced acute
lung injury. Am J Respir Crit Care Med. 182:918–928.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wagner EF and Eferl R: Fos/AP-1 proteins
in bone and the immune system. Immunol Rev. 208:126–140.
2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Burch PM, Yuan Z, Loonen A and Heintz NH:
An extracellular signal-regulated kinase 1- and 2-dependent program
of chromatin trafficking of c-Fos and Fra-1 is required for cyclin
D1 expression during cell cycle reentry. Mol Cell Biol.
24:4696–4709. 2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xi X, Yao Y, Liu N and Li P: MiR-297
alleviates LPS-induced A549 cell and mice lung injury via targeting
cyclin dependent kinase 8. Int Immunopharmacol.
80(106197)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bein T, Briegel J and Annane D: Steroids
are part of rescue therapy in ARDS patients with refractory
hypoxemia: Yes. Intensive Care Med. 42:918–920. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao D, Ding R, Mao Y, Wang L, Zhang Z and
Ma X: Heparin rescues sepsis-associated acute lung injury and
lethality through the suppression of inflammatory responses.
Inflammation. 35:1825–1832. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hu H, Shi D, Hu C, Yuan X, Zhang J and Sun
H: Dexmedetomidine mitigates CLP-stimulated acute lung injury via
restraining the RAGE pathway. Am J Transl Res. 9:5245–5258.
2017.PubMed/NCBI
|
|
22
|
Zhou H, Wang X and Zhang B: Depression of
lncRNA NEAT1 antagonizes LPS-evoked acute injury and inflammatory
response in alveolar epithelial cells via HMGB1-RAGE signaling.
Mediators Inflamm. 2020(8019467)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li P, Yao Y, Ma Y and Chen Y: MiR-150
attenuates LPS-induced acute lung injury via targeting AKT3. Int
Immunopharmacol. 75(105794)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li P, Yao Y, Ma Y and Chen Y: MiR-30a-5p
ameliorates LPS-induced inflammatory injury in human A549 cells and
mice via targeting RUNX2. Innate Immun. 27:41–49. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yamaguchi T, Takada Y, Maruyama K, Shimoda
K, Arai Y, Nango N, Kosaki N, Takaishi H, Toyama Y and Matsuo K:
Fra-1/AP-1 impairs inflammatory responses and chondrogenesis in
fracture healing. J Bone Miner Res. 24:2056–2065. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Rajasekaran S, Vaz M and Reddy SP:
Fra-1/AP-1 transcription factor negatively regulates pulmonary
fibrosis in vivo. PLoS One. 7(e41611)2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rajasekaran S, Tamatam CR, Potteti HR,
Raman V, Lee JW, Matthay MA, Mehta D, Reddy NM and Reddy SP:
Visualization of Fra-1/AP-1 activation during LPS-induced
inflammatory lung injury using fluorescence optical imaging. Am J
Physiol Lung Cell Mol Physiol. 309:L414–L424. 2015.PubMed/NCBI View Article : Google Scholar
|