1. Introduction
Aggressive prolactinoma (APRL) is a subgroup of
aggressive pituitary tumors (APTs) which represent a relatively new
concept defined by three main features: tumor invasion based on
radiological and/or histological features; a higher proliferation
profile than usual pituitary adenomas (also called typical
adenomas) and aggressive behavior such as rapidly developing
resistance to standard medication/protocols and increased risk of
early recurrence after surgical removal or after achieving
transitory control under traditional medical therapy (1). APTs, representing around 10% of all
pituitary tumors, require a complex multidisciplinary multimodal
approach that may include: neurosurgery (numerous repeated
procedures), radiotherapy, peptide receptor radionuclide therapy,
combined medical therapy depending on secretory profile (such as
cabergoline, pasireotide), but also uncommon medication as
temozolomide (an alkylating agent) if standard regimes are
inefficient (2).
Diagnosis of pituitary carcinoma (PC) is positive
only if distant or cerebrospinal fluid metastases are identified
(3). APTs and PC represents an
atypical subcategory of pituitary tumors which otherwise usually
display a more favorable profile, thus requiring a particular
approach as pointed by 2018 ESE (European Society of Endocrinology)
guideline (4).
Generally, prolactinoma is a subgroup of functioning
pituitary adenomas (a category that also includes corticotropinoma,
somatotropinoma, gonadotropinoma, and thyrotropinoma), knowing that
the most frequent abnormal pituitary secretion is prolactin
(between 47 and 66% of all secretor pituitary adenomas, depending
on the studies) (5). Functioning
and non-functioning hypophyseal adenomas represent 15% of all
intra-cranial tumors, being considered one of the most frequent
intra-cranial neoplasia (6). Their
associated co-morbidities due to the tumor itself and due to
hypersecretion/hyposecretion involve a relatively high rate of
morbidity and mortality (7).
The clinical spectrum due to hyperprolactinemia of
tumor origin varies from different manifestations of male/female
hypogonadism, mammary anomalies such as galactorrhea up to
osteoporosis and associated fractures and cardiovascular risk
(8-10).
In addition, prolactinomas of more than 1 cm in diameter up to
giant prolactin-secreting tumors are associated with local mass
effects as seen in other pituitary tumors, regardless of their
secretor features (11). Some
syndromic prolactinomas are synchronous or asynchronous with other
non-pituitary tumors as seen in adrenal glands or parathyroids
(12,13).
A total of 95% of all pituitary adenomas are
sporadic, while germline mutations have been reported, for
instance, multiple endocrine neoplasia type 1 (MEN1), aryl
hydrocarbon receptor-interacting protein (AIP), and
succinate dehydrogenase (SDHx) (14). AIP and MEN1 mutations
are correlated with a diagnosis of patients younger than 30 years
of age (or even 20 years); AIP mutation is mostly associated
with gigantism and large tumors; Xq26.3 mutations are
associated with pituitary tumors in very young children (15). Prolactinomas with underlying
AIP mutations represent a small subset of tumors with a
familial pattern which otherwise are typically growth
hormone-producing tumors or non-functioning adenomas (16). Both AIP- and
MEN1-related prolactinomas may be large and usually therapy
resistant, especially AIP-associated prolactinomas, as
similarly seen in somatotropinomas (17,18).
A complex multidisciplinary management may be
require for pituitary adenomas since their first line of approach
is surgical removal, except for prolactinomas which traditionally
respond to medical therapy such as dopamine agonists (DAs) and also
except for non-functioning microadenomas (usually named
incidentalomas) that do not require a specific approach, only
imaging follow-up (19-21).
2. Aim of the review
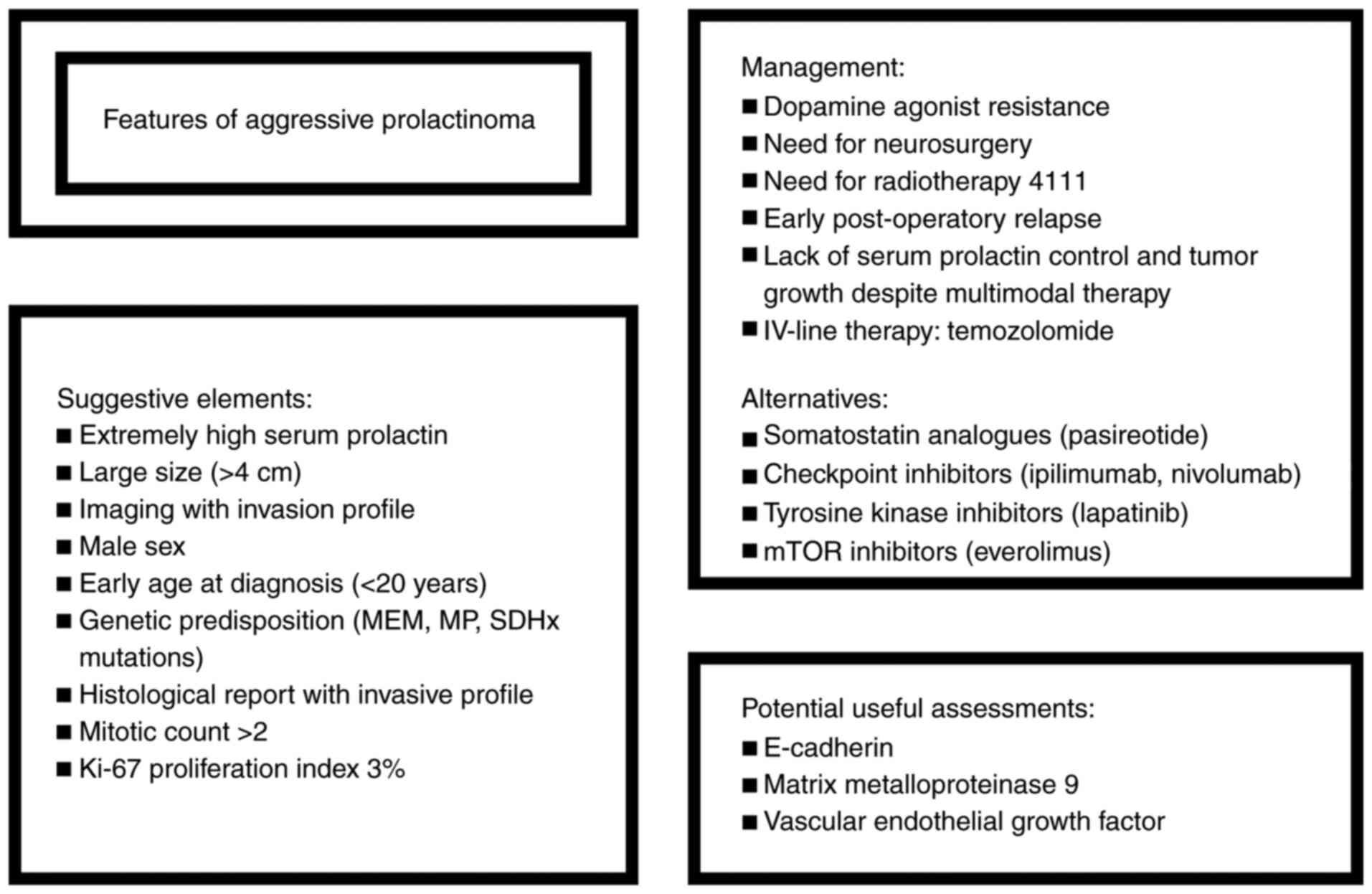
We aimed to focus on APRL in terms of both
presentation, but also management and associated prognostic factors
(Fig. 1). This is a narrative
review based on a PubMed search using key words, ‘aggressive
prolactinoma’, ‘aggressive pituitary tumor’, and ‘pituitary
carcinoma’. A number of 81 references are included from 2009 to
2021. Inclusion criteria included English language and full-length
articles. Exclusion criteria included papers published before
2009.
3. Presentation
Prolactinoma affects both females and males, having
an incidence of 3-5 cases/100,000 individuals/year and a general
prevalence of 50 cases/100,000 individuals which is higher in
subjects with hypogonadism since increased prolactin levels induce
central inhibition of gonadotropes through kisspeptin neurons
(22). The detection of APRLs may
not start from the clinical presentation itself, but the imaging
detection of a large pituitary mass in addition to large
mass-associated symptoms and signs and extremely elevated prolactin
levels are important clues, further followed by a poor response to
DA medication and need for neurosurgery/early relapse after
hypophysectomy due to the unusual rapid growth (23). Generally, a patient with
prolactinoma becomes a neurosurgery candidate in the event apoplexy
or cystic transformation is present, neurological/ophthalmic
deficit has been identified, and the patient develops DA resistance
or intolerance (24).
Post-operative histological and immunohistochemistry
reports provide additional elements of the aggressive profile in
association with clinical outcome after removal. This can sometimes
occur from the beginning even when the surgical procedure is for
debulking purposes only (25).
Additional poor prognostic elements are represented
by epigenetic data regarding APTs that demonstrate a high DNA
methyltransferase overexpression, p53-related histone anomalies,
and upregulation of citrullinating enzymes. Yet, currently research
involving new predictive factors of aggressive behavior is still in
progress (26). Sometimes the term
‘refractory’ pituitary adenoma is used as an open category of APT
with high Ki-67, rapid growth and lack of response to a traditional
guideline approach (25).
Overall, the cluster of elements used for evaluating
the potential aggressive behavior starts with high prolactin levels
at first admission and large diameter upon computed tomography or
magnetic resonance imaging; yet these themselves do not necessarily
indicate an APRL (27,28). If the patient is a male or there is
an early age at diagnosis or a genetic predisposition is
identified, the prognosis may be poor (27,29).
Furthermore, the development of DA (mostly cabergoline) resistance
represents another useful clue (27,30).
Since the subject is referred to neurosurgery, the
post-hypophysectomy histological report pointing out a mitotic
count higher than 2 and a Ki-67 at least 3% is an indicator of
increased proliferation risk. Yet, in the case that we analyze
these characteristics apart from all other elements, these do not
necessarily indicate an APRL (27,31).
The evolution of prolactin levels and tumor size under multimodal
therapy are also indicators of APRL (27,32).
Other indicators of poor prognosis may be provided
by the assessment of E-cadherin, matrix metalloproteinase (MMP)-9,
growth factors such as vascular endothelial growth factor (VEGF),
and abnormal gene expression (AIP, MEN1, p53) or even breast
cancer gene 1 (BRCA1) mutation has been described (27,33).
At first glance, no single element of the features mentioned above
is enough to reveal an APRL. This is a step by step process, while
the clear difference between an adenoma and a carcinoma comes only
with confirmation of metastases, not by just analyzing the
pituitary tumor histology and immunohistochemistry (19,27).
4. Management
Management of APRLs goes beyond the typical scenario
of most prolactinoma cases that are not candidate to non-medical
approach (34). In this particular
situation, a complex team is eventually expected to be necessary,
including approaches in neurosurgery, radiotherapy, and even
oncology since the APRL profile is contoured step by step starting
with the identification of a large invasive tumor and continuing to
small changes in serum prolactin levels under DA (35). A total of 1-5% of prolactinomas are
giant at presentation (a diameter larger than 4 cm), while most
macro-prolactinomas have a diameter between 1 and 4 cm (36). DA is expected to induce tumor
shrinkage and normalization of prolactin levels due to the abundant
expression of type 2 dopamine receptors; yet 10-20% of
prolactinomas are DA resistant (10% for cabergoline, respective
20-30% for bromocriptine; thus bromocriptine-resistance patients
should be first switched to cabergoline with an 80% expected rate
of response in most cases of prolactinomas) (37). APRLs are frequently resistant to
traditional therapy with DA (mainly cabergoline); DA resistance is
also seen in familial forms prolactinomas with underlying
AIP or MEN1 mutations (38). In patients younger than 20 years, DA
resistance is correlated with high serum levels of prolactin at
first presentation and increased tumor diameter in addition to
AIP/MEN1 mutations which represent an independent predictor
factor for the poor rate of DA response, accounting for 14% of all
cases with suboptimal evolution under DA (39). DA resistance is an essential feature
of APRLs, but, on the other hand, not all DA-resistant
prolactinomas are actually APRLs (40,41).
According to current opinions, DA resistance means the lack of
achieving normal prolactin levels and at least 30% reduction in the
maximum diameter under high cabergoline doses (at least 3.5
mg/week) (40,41). The most useful clinical element of
response is restoration of gonadal axe function. Usually lifelong
administration of cabergoline is necessary in responsive cases
since stopping the medication is associated with a 60-80% relapse
rate (42).
Subjects with DA resistance become candidates for
hypophysectomy and some experts consider that early recognition of
patients who are DA non-responsive improves the success of the
neurosurgical procedure due to tumor-associated fibrosis which is a
direct contributor to incomplete resection (43-45).
Retrospective post-surgery studies have shown that a value of Ki-67
of more than 3% is associated with a higher risk of resistance to
medication and post-operatory relapse; however, Ki-67 is not the
only useful indicator and this isolated value itself is subject to
controversy regarding APRL prognosis (44,45).
In cases with persistent or progressive disease after surgery or if
only debulking surgery is feasible, DA should be re-considered in
addition to irradiation therapy, preferable gamma knife (46).
Radiotherapy for pituitary tumors is useful if other
medical and surgical procedures do not control the condition; the
risk of hypopituitarism is high and in certain circumstances the
risk of a secondary brain tumor should be taken into account
(47). Some authors suggest that
radiation therapy applied for invasive prolactinomas that are
non-responsive to surgery and DA may be a promotor of distant
metastases, thus subscribing to a PC profile, but this aspect is
still a matter of debate (48).
Temozolomide, an active chemotherapy drug,
represents a fourth-line therapy in prolactinomas after DA,
transphenoidal selective hypophysectomy and radiotherapy (49). A specific time frame for its
introduction into the patient regime varies, but prompt
intervention is recommended (50).
As pointed by the 2018 ESE guideline, temozolomide as a single
medication is the first-line medical therapy after specific
standard therapies have failed; 3 cycles are needed in order to
decide if the patient is a responder, in which case therapy is
prolonged for 6 months (4).
However, following this article, controversies emerged at
temozolomide withdrawn since an efficient fifth-line therapy (or
second-line treatment) after temozolomide is still lacking and
re-starting the same medication usually fails to achieve a relevant
clinical and imaging response. Thus, the length of the therapy may
go up to 12 cycles in many studies and even up to 14-59 cycles if
it is well tolerated (a cycle means a daily oral dose of 50-200
mg/sqm, 5 out of 28 days) (51).
Prolonged administration is under consideration in many cases
despite guidelines based on an individual multidisciplinary
decision (52).
Almalki et al described rates of response
following temozolomide therapy as following: 76% of patients with
tumor reduction, 75% with serum prolactin level reduction, 8% with
normalization of prolactin, and a failure rate of 20.6% of subjects
(53). It seems that APRLs had a
higher rate of response to temozolomide than corticotropinomas
(60%) and somatotropinomas (26.7%) (54). The assay of O(6)-methylguanine-DNA methyltransferase
(MGMT) status may serve as a surrogate to anticipate the
temozolomide response rate (50).
5. Future considerations
As mentioned, APRL is part of a syndromic
combination similar to that seen in MEN1 syndrome, and in this
particular circumstance the aggressive profile cannot be predicted
by a specific gene mutation configuration itself but by a
constellation of factors. However, the presence of other tumors
such as associated neuroendocrine neoplasia of the pancreas or
adrenals are direct contributors to a more severe overall prognosis
(55,56).
Once temozolomide is inefficient or it is not
tolerated by a patient with APRLs, the therapeutic options are
extremely limited (57). A few
cases of prolactinomas, respective corticotropinomas with very
aggressive profile have been recently reported as candidates for
new drugs such as checkpoint inhibitors (ipilimumab, nivolumab)
after temozolomide has failed to control the APT (58). The ErbB pathway is newly detected as
correlated with aggressive profile of prolactinoma and DA
resistance in APRL; thus, therapy with lapatinib, an oral tyrosine
kinase inhibitor (TKI) targeting ErbB1-epidermal growth factor
receptor (EGFR) is under evaluation, already being included in
Phase 2a clinical trials (59).
Somatostatin analogue therapy for APRLs has demonstrated promising
results, as well as its use in DA-resistant prolactinomas which are
not APRLs (60-62).
If a pituitary neuroendocrine tumor with prolactin production has
somatostatin receptor (SSTR) expression, especially type 5 more
than type 2, pasireotide long-acting release (LAR) (40 mg per
month) may be beneficial (61,62).
Of course, a mixed secretion of both prolactin and
growth hormone is associated with a better response to somatostatin
analogues (63). In APRLs, cystic
transformation and tumor necrosis have been reported under
pasireotide LAR (64). Pasireotide,
outside the fact that it represents a second-line medical therapy
in acromegaly, is also used in Cushing disease and some pancreatic
neuroendocrine neoplasia (65,66).
In addition, isolated case reports have introduced pasireotide as
an alternative to prolactinomas that are resistant to standard
therapies, not necessarily APRLs (67). The use of SSTR immunostaining after
neurosurgery may predict a potential response to this class of
drugs, as currently being used in acromegaly treatment (68-70).
Moreover, estrogen modulators and metformin have
been suggested for APRLs, but the findings still require
statistical validation since the current level of evidence is low
(60). The mammalian target of
rapamycin (mTOR) inhibitor, everolimus, was applied as an off-label
therapy in one case of APRL in combination with cabergoline and the
results were encouraging (71).
Generally, everolimus has been approved for pancreatic
neuroendocrine neoplasia also in combination with somatostatin
analogues including ocreotide or lanreotide (72,73).
The mTOR pathway is involved in the development of tumors derived
from pituitary cells through intervention of the pituitary tumor
transforming gene (PTTG1) (74).
The observation is consistent for GH-secreting tumors as well
(75). Murine experiments suggest
that inhibition of the mTOR pathway may increase the cytotoxicity
induced by temozolomide (76). The
use of temozolomide for APTs dates since 2006, and it is generally
considered to be well tolerated (the most common side effects are
nausea, fatigue, different types of cytopenia) (77). It is usually recommended for
treating brain cancers such as glioblastoma (78,79).
6. Conclusions
APRLs represent a challenging condition that
requires a multimodal approach; in addition to a standard
three-line therapy, temozolomide medication is required.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AV drafted the manuscript and critically revised the
final form. FS researched the literature data, and AP critically
revised the manuscript in light of the literature findings. MCD is
the corresponding author and revised the literature findings. MC
drafted the manuscript and reviewed the references. RCP researched
the literature and AG approved the final form in light of the
literature data. All authors read and approved the final manuscript
for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chatzellis E, Alexandraki KI, Androulakis
II and Kaltsas G: Aggressive pituitary tumors. Neuroendocrinology.
101:87–104. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Iglesias P, Magallón R, Mitjavila M,
Rodríguez Berrocal V, Pian H and Díez JJ: Multimodal therapy in
aggressive pituitary tumors. Endocrinol Diabetes Nutr (Engl Ed).
67:469–485. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ilie MD, Jouanneau E and Raverot G:
Aggressive pituitary adenomas and carcinomas. Endocrinol Metab Clin
North Am. 49:505–515. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Raverot G, Burman P, McCormack A, Heaney
A, Petersenn S, Popovic V, Trouillas J and Dekkers OM: European
Society of Endocrinology. European society of endocrinology
clinical practice guidelines for the management of aggressive
pituitary tumours and carcinomas. Eur J Endocrinol. 178:G1–G24.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Thakkar K, Sarathi V and Shah NS: Current
Status of diagnosis and management for functioning pituitary
tumors: Part I. Neurol India. 68 (Suppl 1):S13–S19. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mehta GU and Lonser RR: Management of
hormone-secreting pituitary adenomas. Neuro Oncol. 19:762–773.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Araujo-Castro M, Berrocal VR and
Pascual-Corrales E: Pituitary tumors: Epidemiology and clinical
presentation spectrum. Hormones (Athens). 19:145–155.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Poiana C, Chirita C, Carsote M, Hortopan D
and Goldstein A: Galactocele and prolactinoma-a pathogenic
association? Maturitas. 6:98–102. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Samperi I, Lithgow K and Karavitaki N:
Hyperprolactinaemia. J Clin Med. 8(2203)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Greenman Y: Prolactinomas and menopause:
Any changes in management? Pituitary. 23:58–64. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tirosh A and Shimon I: Management of
macroprolactinomas. Clin Diabetes Endocrinol. 1(5)2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Iliesiu A, Ungureanu IA, Petca A,
Constantin MM, Petca RC, Sandru F, Constantin T and Dumitrascu MC:
Paraganglioma presenting as a mesenteric cystic mass: A case
report. Exp Ther Med. 20:2489–2492. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nassiri F, Cusimano MD, Scheithauer BW,
Rotondo F, Fazio A, Syro LV, Kovacs K and Lloyd RV: Prolactinomas:
Diagnosis and treatment. Expert Rev Endocrinol Metab. 7:233–241.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Van den Broek MF, van Nesselrooij BP,
Verrijn Stuart AA, van Leeuwaarde RS and Valk GD: Clinical
relevance of genetic analysis in patients with pituitary adenomas:
A systematic review. Front Endocrinol (Lausanne).
10(837)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Barry S and Korbonits M: Update on the
genetics of pituitary tumors. Endocrinol Metab Clin North Am.
49:433–452. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Carty DM, Harte R, Drummond RS, Ward R,
Magid K, Collier D, Owens M and Korbonits M: AIP variant causing
familial prolactinoma. Pituitary. 24:48–52. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Daly AF, Cano DA, Venegas-Moreno E,
Petrossians P, Dios E, Castermans E, Flores-Martínez A, Bours V,
Beckers A and Soto-Moreno A: AIP and MEN1 mutations and AIP
immunohistochemistry in pituitary adenomas in a tertiary referral
center. Endocr Connect. 8:338–348. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Schernthaner-Reiter MH, Trivellin G and
Stratakis CA: Interaction of AIP with protein kinase A
(cAMP-dependent protein kinase). Hum Mol Genet. 27:2604–2613.
2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cuny T, Barlier A, Feelders R, Weryha G,
Hofland LJ, Ferone D and Gatto F: Medical therapies in pituitary
adenomas: Current rationale for the use and future perspectives.
Ann Endocrinol (Paris). 76:43–58. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Honegger J, Nasi-Kordhishti I, Aboutaha N
and Giese S: Surgery for prolactinomas: A better choice? Pituitary.
23:45–51. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kuo M, Maya MM, Bonert V and Melmed S:
Prospective evaluation of incidental pituitary imaging findings in
the Sella Turcica. J Endocr Soc. 5(bvaa186)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chanson P and Maiter D: The epidemiology,
diagnosis and treatment of Prolactinomas: The old and the new. Best
Pract Res Clin Endocrinol Metab. 33(101290)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lasolle H, Ilie MD and Raverot G:
Aggressive prolactinomas: How to manage? Pituitary. 23:70–77.
2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Donoho DA and Laws ER Jr: The role of
surgery in the management of prolactinomas. Neurosurg Clin N Am.
30:509–514. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Dai C, Liu X, Ma W and Wang R: The
treatment of refractory pituitary adenomas. Front Endocrinol
(Lausanne). 10(334)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hauser BM, Lau A, Gupta S, Bi WL and Dunn
IF: The Epigenomics of pituitary adenoma. Front Endocrinol
(Lausanne). 10(290)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Trouillas J, Delgrange E, Wierinckx A,
Vasiljevic A, Jouanneau E, Burman P and Raverot G: Clinical,
pathological, and molecular factors of aggressiveness in lactotroph
tumours. Neuroendocrinology. 109:70–76. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vroonen L, Daly AF and Beckers A:
Epidemiology and management challenges in prolactinomas.
Neuroendocrinology. 109:20–27. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yoo F, Chan C, Kuan EC, Bergsneider M and
Wang MB: Comparison of male and female prolactinoma patients
requiring surgical intervention. J Neurol Surg B Skull Base.
79:394–400. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Olarescu NC, Perez-Rivas LG, Gatto F, Cuny
T, Tichomirowa MA, Tamagno G and Gahete MD: EYRC (ENEA Young
Researcher Committee). Aggressive and malignant prolactinomas.
Neuroendocrinology. 109:57–69. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Glezer A and Bronstein MD: Prolactinomas.
Endocrinol Metab Clin North Am. 44:71–78. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Langlois F, McCartney S and Fleseriu M:
Recent progress in the medical therapy of pituitary tumors.
Endocrinol Metab (Seoul). 32:162–170. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Bettencourt-Silva R, Queirós J, Pereira J
and Carvalho D: Giant prolactinoma, germline BRCA1 mutation, and
depression: A case report. J Med Case Rep. 12(360)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huynh PP, Ishii LE and Ishii M:
Prolactinomas. JAMA. 325(195)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Giraldi EA and Ioachimescu AG: The role of
dopamine agonists in pituitary adenomas. Endocrinol Metab Clin
North Am. 49:453–474. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Shimon I: Giant prolactinomas.
Neuroendocrinology. 109:51–56. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sahakian N, Castinetti F, Dufour H,
Graillon T, Romanet P, Barlier A, Brue T and Cuny T: Clinical
management of difficult to treat macroprolactinomas. Expert Rev
Endocrinol Metab. 14:179–192. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Vroonen L, Daly AF and Beckers A:
Challenges and controversies in the treatment of prolactinomas.
Expert Rev Endocrinol Metab. 9:593–604. 2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Salenave S, Ancelle D, Bahougne T, Raverot
G, Kamenický P, Bouligand J, Guiochon-Mantel A, Linglart A, Souchon
PF, Nicolino M, et al: Macroprolactinomas in children and
adolescents: Factors associated with the response to treatment in
77 patients. J Clin Endocrinol Metab. 100:1177–1186.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Vermeulen E, D'Haens J, Stadnik T, Unuane
D, Barbe K, Van Velthoven V and Gläsker S: Predictors of dopamine
agonist resistance in prolactinoma patients. BMC Endocr Disord.
20(68)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Maiter D: Management of dopamine
agonist-resistant prolactinoma. Neuroendocrinology. 109:42–50.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Espinosa-Cárdenas E, Sánchez-García M,
Ramírez-Rentería C, Mendoza-Zubieta V, Sosa-Eroza E and Mercado M:
High biochemical recurrence rate after withdrawal of cabergoline in
prolactinomas: Is it necessary to restart treatment? Endocrine.
70:143–149. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Vermeulen E, D'Haens J, Stadnik T, Unuane
D, Barbe K, Van Velthoven V and Gläsker S: Predictors of dopamine
agonist resistance in prolactinoma patients. BMC Endocr Disord.
20(68)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lu C, Liu Y, Lu Z and Huan C: Ki-67 and
clinical correlations in patients with resistant prolactinomas. Ann
Clin Lab Sci. 50:199–204. 2020.PubMed/NCBI
|
|
45
|
Stiles CE, Steeds RP and Drake WM:
Monitoring patients receiving dopamine agonist therapy for
hyperprolactinaemia. Ann Endocrinol (Paris). 82:182–186.
2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Souteiro P, Belo S and Carvalho D:
Dopamine agonists in prolactinomas: When to withdraw? Pituitary.
23:38–44. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Castinetti F: Radiation techniques in
aggressive pituitary tumours and carcinomas. Rev Endocr Metab
Disord. 21:287–292. 2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Pérez Pinzón J, González-Devia D, Kattah
Calderón W, López Panqueva RDP and Jiménez Hakim E: Unusual course
of an aggressive pituitary prolactinoma: Case report and review of
the literature. Case Rep Neurol. 11:148–156. 2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Barkhoudarian G, Palejwala SK, Ogunbameru
R, Wei H, Eisenberg A and Kelly DF: Early recognition and
initiation of temozolomide chemotherapy for refractory, invasive
pituitary macroprolactinoma with long-term sustained remission.
World Neurosurg. 118:118–124. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chen C, Yin S, Zhang S, Wang M, Hu Y, Zhou
P and Jiang S: Treatment of aggressive prolactinoma with
temozolomide: A case report and review of literature up to date.
Medicine (Baltimore). 96(e8733)2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Lizzul L, Lombardi G, Barbot M, Ceccato F,
Gardiman MP, Regazzo D, Bellu L, Mazza E, Losa M and Scaroni C:
Long-course temozolomide in aggressive pituitary adenoma: Real-life
experience in two tertiary care centers and review of the
literature. Pituitary. 23:359–366. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Elbelt U, Schlaffer SM, Buchfelder M,
Knappe UJ, Vila G, Micko A, Deutschbein T, Unger N, Lammert A,
Topuzoglu-Müller T, et al: Efficacy of temozolomide therapy in
patients with aggressive pituitary adenomas and carcinomas-a German
survey. J Clin Endocrinol Metab. 105(dgz211)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Almalki MH, Aljoaib NN, Alotaibi MJ,
Aldabas BS, Wahedi TS, Ahmad MM and Alshahrani F: Temozolomide
therapy for resistant prolactin-secreting pituitary adenomas and
carcinomas: A systematic review. Hormones (Athens). 16:139–149.
2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Aydoğan Bİ, Ünlütürk U, Emral R and Güllü
S: Course of aggressive somatotroph, corticotroph and mammotroph
tumors under temozolomide; Report of three cases and review of the
literature. Turk Neurosurg: May 7, 2017 (Epub ahead of print).
|
|
55
|
Mele C, Mencarelli M, Caputo M, Mai S,
Pagano L, Aimaretti G, Scacchi M, Falchetti A and Marzullo P:
Phenotypes associated with MEN1 syndrome: A focus on
genotype-phenotype correlations. Front Endocrinol (Lausanne).
11(591501)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Miulescu R, Balaban DV, Sandru F and Jinga
M: Cutaneous manifestations in pancreatic diseases-A review. J Clin
Med. 9(2611)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ortiz LD, Syro LV, Scheithauer BW, Rotondo
F, Uribe H, Fadul CE, Horvath E and Kovacs K: Temozolomide in
aggressive pituitary adenomas and carcinomas. Clinics (Sao Paulo).
67 (Suppl 1):S119–S123. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Duhamel C, Ilie MD, Salle H, Nassouri AS,
Gaillard S, Deluche E, Assaker R, Mortier L, Cortet C and Raverot
G: Immunotherapy in corticotroph and lactotroph aggressive tumors
and carcinomas: Two case reports and a review of the literature. J
Pers Med. 10(88)2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Cooper O, Bonert VS, Rudnick J, Pressman
BD, Lo J, Salvatori R, Yuen KCJ, Fleseriu M and Melmed S:
EGFR/ErbB2-targeting lapatinib therapy for aggressive
prolactinomas. J Clin Endocrinol Metab. 106:e917–e925.
2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Souteiro P and Karavitaki N: Dopamine
agonist resistant prolactinomas: Any alternative medical treatment?
Pituitary. 23:27–37. 2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Raverot G, Vasiljevic A, Jouanneau E and
Lasolle H: Confirmation of a new therapeutic option for aggressive
or dopamine agonist-resistant prolactin pituitary neuroendocrine
tumors. Eur J Endocrinol. 181:C1–C3. 2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Gomes-Porras M, Cárdenas-Salas J and
Álvarez-Escolá C: Somatostatin analogs in clinical practice: A
review. Int J Mol Sci. 21(1682)2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lv L, Jiang Y, Yin S, Hu Y, Chen C, Ma W,
Jiang S and Zhou P: Mammosomatotroph and mixed
somatotroph-lactotroph adenoma in acromegaly: A retrospective study
with long-term follow-up. Endocrine. 66:310–318. 2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Coopmans EC, van Meyel SWF, Pieterman KJ,
van Ipenburg JA, Hofland LJ, Donga E, Daly AF, Beckers A, van der
Lely AJ and Neggers SJCMM: Excellent response to pasireotide
therapy in an aggressive and dopamine-resistant prolactinoma. Eur J
Endocrinol. 181:K21–K27. 2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Siddiqui M, Vora A, Ali S, Abramowitz J
and Mirfakhraee S: Pasireotide: A novel treatment for tumor-induced
hypoglycemia due to insulinoma and non-islet cell tumor
hypoglycemia. J Endocr Soc. 5(bvaa171)2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Zhao N, Yang X, Li C, Ma J and Yin X:
Efficacy and safety of pasireotide for Cushing's disease: A
protocol for systematic review and meta-analysis. Medicine
(Baltimore). 99(e23824)2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Lasolle H, Vasiljevic A, Borson-Chazot F
and Raverot G: Pasireotide: A potential therapeutic alternative for
resistant prolactinoma. Ann Endocrinol (Paris). 80:84–88.
2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Oki Y: Medical management of functioning
pituitary adenoma: An update. Neurol Med Chir (Tokyo). 54:958–965.
2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Valea A, Ghervan C, Carsote M, Morar A,
Iacob I, Tomesc F, Pop DD and Georgescu C: Effects of combination
therapy: Somatostatin analogues and dopamine agonists on GH and
IGF1 levels in acromegaly. Clujul Med. 88:310–313. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Valea A, Carsote M, Ghervan C and
Georgescu C: Glycemic profile in patients with acromegaly treated
with somatostatin analogue. J Med Life. 8:82–86. 2015.PubMed/NCBI
|
|
71
|
Zhang D, Way JS, Zhang X, Sergey M,
Bergsneider M, Wang MB, Yong WH and Heaney AP: Effect of everolimus
in treatment of aggressive prolactin-secreting pituitary adenomas.
J Clin Endocrinol Metab. 104:1929–1936. 2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zhang J, Francois R, Iyer R, Seshadri M,
Zajac-Kaye M and Hochwald SN: Current understanding of the
molecular biology of pancreatic neuroendocrine tumors. J Natl
Cancer Inst. 105:1005–1017. 2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wolin EM: The expanding role of
somatostatin analogs in the management of neuroendocrine tumors.
Gastrointest Cancer Res. 5:161–168. 2012.PubMed/NCBI
|
|
74
|
Chen R, Duan J, Li L, Ma Q, Sun Q, Ma J,
Li C, Zhou X, Chen H, Jing Y, et al: mTOR promotes pituitary tumor
development through activation of PTTG1. Oncogene. 36:979–988.
2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Gorshtein A, Rubinfeld H, Kendler E,
Theodoropoulou M, Cerovac V, Stalla GK, Cohen ZR, Hadani M and
Shimon I: Mammalian target of rapamycin inhibitors rapamycin and
RAD001 (everolimus) induce anti-proliferative effects in
GH-secreting pituitary tumor cells in vitro. Endocr Relat Cancer.
16:1017–1027. 2009.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Dai C, Zhang B, Liu X, Ma S, Yang Y, Yao
Y, Feng M, Bao X, Li G, Wang J, et al: Inhibition of PI3K/AKT/mTOR
pathway enhances temozolomide-induced cytotoxicity in pituitary
adenoma cell lines in vitro and xenografted pituitary adenoma in
female nude mice. Endocrinology. 154:1247–1259. 2013.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Whitelaw BC: How and when to use
temozolomide to treat aggressive pituitary tumours. Endocr Relat
Cancer. 26:R545–R552. 2019.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Schreck KC and Grossman SA: Role of
temozolomide in the treatment of cancers involving the central
nervous system. Oncology (Williston Park). 32:555–560.
2018.PubMed/NCBI
|
|
79
|
Arora A and Somasundaram K: Glioblastoma
vs temozolomide: Can the red queen race be won? Cancer Biol Ther.
20:1083–1090. 2019.PubMed/NCBI View Article : Google Scholar
|