Introduction
Coronary artery-related cardiac ischemia is the most
common cause of cardiomyopathy and heart failure, with high rates
of morbidity and mortality worldwide (1). Coronary artery stenosis and/or
occlusion lead to myocardial perfusion reduction, cardiomyocyte
remodeling and fibrosis, dilated ventricles and even heart failure.
Exploring the pathogenesis and regulatory mechanisms of ischemia
may provide significant benefits in its diagnosis, treatment, and
prognosis. Cardiac ischemia is a type of complex cellular and
molecular activity that occurs at multiple levels (2-8).
However, its mechanisms are yet to be fully elucidated. The heart
is an organ rich in mitochondria and thus particularly vulnerable
to ischemia. Due to the role of the 'energy power plant',
mitochondrial homeostasis is crucial for vital cellular
activities.
In the past few decades, omics technology has made a
remarkable contribution to illustrating the pathogenesis and
consequences of cardiac ischemia. Proteomics is a type of omics
approach that focuses on systemic and dynamic molecular changes,
and explores potential crosstalk between multiple molecular levels
and pathways. Expression analysis via liquid chromatography-tandem
mass spectrometry (LC-MS/MS) is used to investigate persistent and
chronic myocardial ischemia (MI) (9-13).
Transient coronary clamps cause severe cardiac ischemia and injury,
accompanied by metabolic disturbances (14). Our recent unpublished experiments
showed that 15 min of ligation of the left anterior descending
(LAD) artery resulted in impaired mitochondrial morphology. Based
on these results, it was hypothesized that mitochondrial proteome
disturbance may occur in the very early stages of ischemia. In the
present study, the mitochondrial proteome profiles of mice hearts
that underwent short-term LAD ligation were evaluated using
LC-MS/MS proteome expression analysis. More than 1,700 proteins
were screened, and 119 differentially expressed proteins were
identified. A number of proteins were involved in the activities
and pathways that regulate oxidative phosphorylation (OXPHOS),
complement activation and mitochondrial autophagy. These results
indicated that mitochondrial proteomic changes may serve as
potential biomarkers for a very early stage of ischemic cardiac
injury and provide novel insights into the mechanism of myocardial
remodeling.
Materials and methods
Animals and grouping
The animals used were 8-week-old male C57BL/6N mice
and their body weights were 20-25 g. The animals were purchased
from the Beijing Vital River Laboratory Animal Technology Co., Ltd.
The mice were housed under a 12 h light/dark cycle at 22±2˚C and at
a relative humidity of 40-60%. They had free access to standard
mouse chow and tap water. After one week of adaptive feeding, the
24 mice were randomly divided into an acute MI group and a sham
group. All experiments and procedures were approved by the
Institutional Animal Care and Use Committee Review Board of the
General Hospital of Ningxia Medical University (approval no.
2020-01) and complied with the guidelines of the National
Institutes of Health Animal Care and Use Committee.
Acute MI model establishment
The MI mouse model was established by ligation of
the LAD, following previously reported methods (15-17).
The mice were anesthetized with a 2% isoflurane (RWD) by a delivery
machine (Pat.4879997, VAPOMATIC™, CWE13-13000) at an airflow rate
of 0.8 l/min. Deep sedation was confirmed by the absence of corneal
and toe pinch reflexes. After disinfecting the surgical area, a 1-2
cm skin cut was made over the left side of the chest. After
dissection and retraction of the pectoral major and minor muscles,
the fourth intercostal space was exposed. A small hole was made at
the fourth intercostal space with a mosquito clamp to open the
pleural membrane and pericardium. Then the heart was smoothly and
gently ‘popped out’ through the hole. The LAD was quickly ligated
using a 6-0 silk suture ~1 mm distal to the left atrial appendage
and 2 mm in width and depth. The ST-segment elevation on the ECG
recording (PowerLab System, AD Instruments) was used to confirm
successful occlusion of the LAD. The heart was placed back into the
intrathoracic space immediately after ligation. The muscles and
skin were closed. The operation in the sham group was the same,
except no LAD ligation was performed. After 15 min of ligation, the
animals were euthanized under anesthesia of 5% isoflurane by a
delivery machine at an airflow rate of 1.0 l/min, and the left
ventricles were collected immediately.
Preparation of samples for
transmission electron microscopy (TEM) examination
A small piece (1 mm3) of the fresh left
ventricle myocardium was rinsed in ice-cold PBS, and the buffer was
then removed using clean filter paper. The tissues were fixed in 2%
glutaraldehyde for 2 h, washed with 0.1 M dimethyl sodium arsenate
three times, fixed in 4% osmic acid for 2 h, rinsed again in 0.1 M
dimethyl sodium arsenate and successively dehydrated with 30-50-70%
alcohol solutions. These processes were performed at 4˚C. The fixed
tissues were infiltrated with propylene oxide and embedded in epoxy
resin at 60˚C for 48 h. The tissues were then cut into ultrathin
sections and observed and imaged by TEM (Hitachi HT-7800; Hitachi,
Ltd.; magnifications, x1,500, x5,000 and x10,000) after staining
with 2% uranyl acetate for 15 min at room temperature and lead
citrate for 5 min at room temperature.
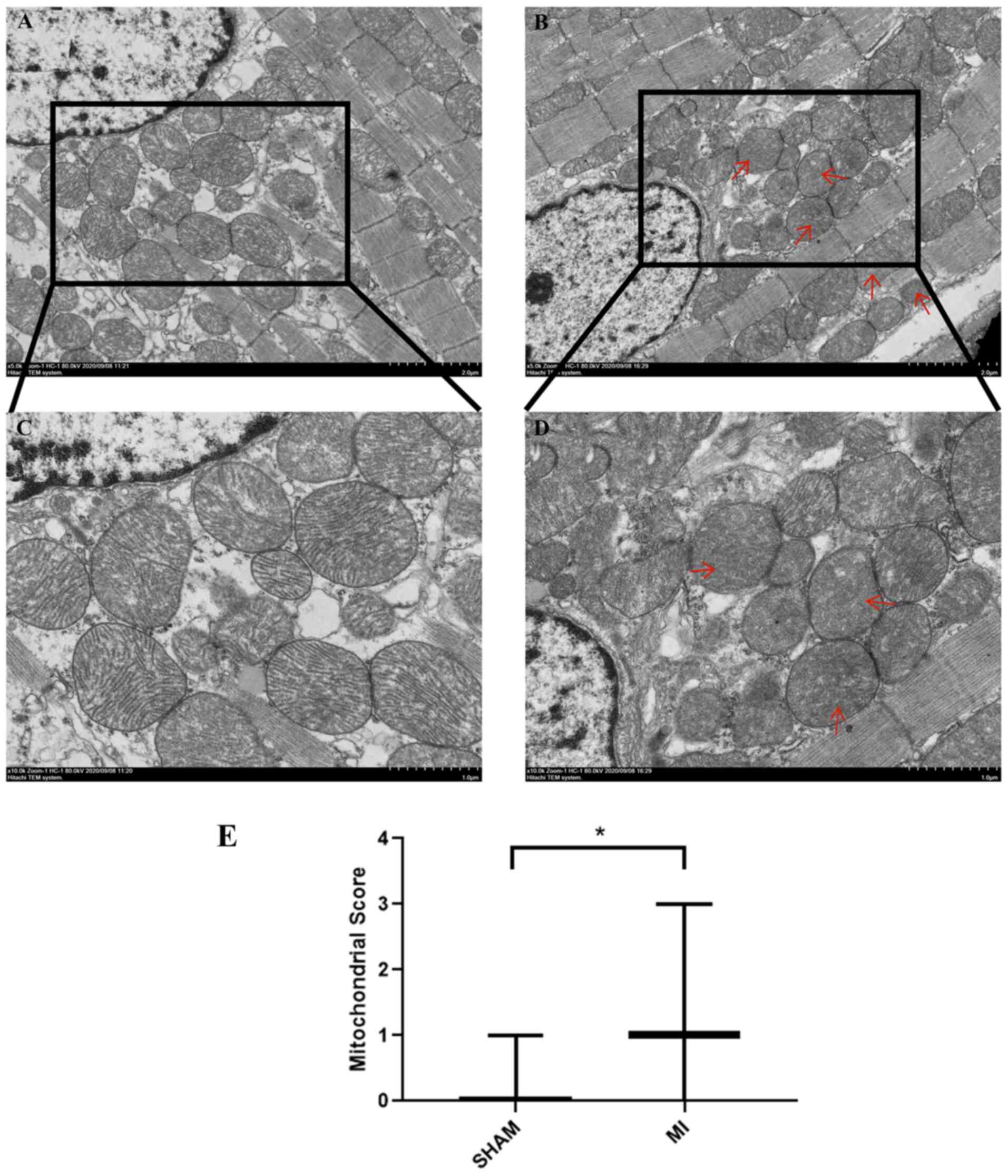
Flameng's classification method was used to quantify
mitochondrial injury under TEM as previously reported (18). At the same magnification (x5,000),
5 fields were randomly selected for imaging, and ~20 mitochondria
were randomly selected from each field for analysis in each mouse.
According to the damage degree, the mitochondria were graded on a
scale of 0 to 4 as follows: 0, normal ultrastructure of the
mitochondrion; 1, normal ultrastructure of the crests and matrix
but absence of granular deposits; 2, loss of matrix granules and
clarification of the matrix without breaking of crests; 3,
disruption of mitochondrial crests with clarification as well as
condensation of the matrix; and 4, disruption of crests, and loss
of integrity of the mitochondrial membranes. The final score is
presented as the median and was analyzed using a Mann-Whitney U
test. The experiment was repeated 3 times.
Mitochondrial isolation
Cardiac mitochondria were extracted using a
mitochondrial isolation kit (Nanjing KeyGen Biotech Co., Ltd.; cat.
no. KGA827) according to the manufacturer's instructions. The fresh
left ventricle was immediately placed on ice, rinsed with cold
saline and drained with filter paper. Then, 40-60 mg of tissue was
cut into small pieces using ophthalmic scissors and transferred
into a small volume (2 ml) glass homogenizer. Next, ice-cold lysis
buffer (1 M Tris-HCl, 25 mM MgCl2, 0.5 M KCl) was added
to the homogenizer until the accumulated volume was 6-fold that of
the tissue pieces. The tissue was ground 20 times at 4˚C. After
adding medium buffer (2 M sucrose), the homogenate was centrifuged
at 1,200 x g for 5 min at 4˚C twice. The supernatant was collected
and centrifuged at 7,000 x g for 10 min at 4˚C. The pellet
contained mitochondria. Finally, the purified mitochondria were
obtained by centrifugation at 9,500 x g for 5 min at 4˚C after
adding suspension buffer (1 M Tris-HCl, 25 mM MgCl2, 2 M
sucrose). Part of the freshly purified mitochondria was used for
the respiration function study, and the rest was stored at -80˚C
for proteomic analysis.
Western blotting
The mitochondrial proteins were extracted using a
mitochondrial protein isolation kit (Nanjing KeyGen Biotech Co.,
Ltd.; cat. no. KGP850) according to the manufacturer's
instructions. All steps were performed in an ice bath. Protein
concentration was determined using a bicinchoninic acid (BCA)
protein detection kit (Nanjing KeyGen Biotech Co., Ltd.; cat. no.
KGPBCA). Proteins (40 µg/lane) were loaded and resolved on 10%
SDS-PAGE and transferred to a nitrocellulose membrane.
Mitochondrial transmembrane protein voltage-dependent
anion-selective channel protein 1 (VDAC1) antibody (Abcam; cat. no.
ab154856; 1:5,000) was used to confirm the efficiency of
mitochondrial isolation and rabbit anti-GAPDH (BIOSS; cat. no.
bs-2188R; 1:5,000) was used as the loading control. The
nitrocellulose membrane was blocked in 10% skim milk for 1 h at
22±2˚C and then incubated with anti-VDAC1 overnight at 4˚C. The
VDAC1 membrane was stripped and reblotted with GAPDH overnight at
4˚C. Horseradish peroxidase-labeled anti-rabbit secondary antibody
(Abbkine, Inc.; cat. no. A21020; 1:5,000) was used to detect the
primary antibodies.
Measurement of mitochondrial membrane
potential (MMP)
MMP was analyzed using a fluorescent probe JC-1
assay kit (Beyotime Institute of Biotechnology; cat. no. C2006).
JC-1 working solution was added to the purified mitochondria and
mixed. The mixture was then added to a 96-well plate. Readings were
measured using a fluorescence microplate reader (VICTOR Nivo;
PerkinElmer, Inc.) at excitation and emission wavelengths of 485
and 590 nm, respectively.
Mitochondria proteome LC-MS/MS
analysis
Protein digestion. The purified mitochondria
were powdered in liquid nitrogen and transferred to a 5 ml
centrifuge tube. Four volumes of lysis buffer (8 M urea, 1%
protease inhibitor cocktail) were added to the powder and sonicated
three times on ice using a high-intensity ultrasonic processor
(Scientz). The remaining debris were removed by centrifugation at
12,000 x g for 10 min at 4˚C. Finally, the supernatant was
collected, and the protein concentration was determined using a BCA
assay. For digestion, the protein solution was reduced with 5 mM
dithiothreitol for 30 min at 56˚C and alkylated with 11 mM
iodoacetamide for 15 min at room temperature in the dark. Then, the
urea concentration in the protein sample was diluted to <2 M
with 100 mM NH4HCO3. Finally, trypsin was
added at a ratio of 1:50 trypsin: protein mass for the first
digestion overnight and a ratio of 1:100 trypsin: protein mass for
the second 4 h of digestion.
LC-MS/MS analysis. Tryptic peptides
were dissolved in 0.1% formic acid and then loaded. The high
performance liquid chromatography gradient increased from 6 to 23%
(0.1% formic acid in 98% acetonitrile) over 26 min, 23 to 35% over
8 min, and increased to 80% over 3 min, then held at 80% for 3 min.
On the EASY-nLC 1,000 ultra-performance LC (UPLC) system, the
constant flow rate was 400 nl/min. The peptides were subjected to a
nanojet ionization source followed by MS/MS in the Q Exactive™ Plus
(Thermo Fisher Scientific, Inc.) coupled online to the UPLC. The
electrospray voltage applied was 2.0 kV. The resolution of the
intact peptide detected in Orbitrap was 70,000 Da. Then, the
peptides were selected with normalized collision energy setting a
value of 28 for MS/MS, and the fragments were detected in the
Orbitrap at a resolution of 17,500. Automatic gain control was set
at 5E4. For MS scans, the m/z scan range was 350-1,800. Label-free
quantification was performed as described in a previous study
(19-21).
Database search and bioinformatics analysis.
The MS/MS data were processed using the MaxQuant search engine
(v.1.5.2.8). The original data were obtained via database
retrieval. Unsupervised principal component analysis (PCA) was used
to observe the overall distribution of proteins among the samples
and the stability of the analytical process. Proteins were
classified by Gene Ontology (GO) annotation (22,23)
into the three following categories: Cellular component, molecular
function, and biological process. The Kyoto Encyclopedia of Genes
and Genomes (KEGG) was used to analyze pathway enrichment (24). For each category, a two-tailed
Fisher's exact test was used to assess the enrichment of
differentially expressed proteins vs. all identified proteins. The
GO and KEGG terms with a corrected P<0.05 were considered
statistically significant. The -log10(P-value) represents
enrichment degree. The cluster membership was visualized using a
heatmap.
Statistical analysis
All data were analyzed using SPSS v23.0 (IBM Corp)
and GraphPad Prism v5.0 (GraphPad Software, Inc.). The results are
presented as the mean ± standard deviation. An unpaired t-test was
used to compare the means between the groups. The test significance
level was α=0.05, and P<0.05 was considered to indicate a
statistically significant difference. The differentially expressed
proteins were screened by fold change (FC) and an unpaired t-test.
The differential screening conditions were FC=1.2x and P<0.05.
Both FC=0 and FC=inf belong to the difference of ‘with or without’.
A two-tailed Fisher's exact test was applied to test the protein
enrichment analysis, and a corrected P-value <0.05 was
considered to be significant. These enrichment analyses were
performed according to the database of identified proteins and a
two-tailed Fisher's exact test was employed. All terms with
corrected a P-value <0.05 were considered significantly enriched
differentially expressed proteins. The number of experimental
replications was 3.
Results
Acute MI induced mitochondrial
morphological changes
First, TEM was used to examine the myocardium and
mitochondrial morphology following MI. As shown in Fig. 1A and C, the sarcomeres were intact, the muscle
filaments were neatly arranged, the nuclear membrane was intact
(Fig. S1), and the chromatin was
uniformly arranged. Mitochondria were normal and ridges were
visible and not dissolved or ruptured in the sham group. The outer
membrane dissolution and break down of the inner membrane (cristae)
in some mitochondria was observable in the MI group (Fig. 1B and D; red arrows). The mitochondrial score in
the MI group increased significantly (P<0.05) compared with the
sham group (Fig. 1E). These
results indicated that acute MI induced mitochondrial damage.
Acute MI decreases MMP
Fig. 2A shows that
VDAC1 protein expression was strong (the original uncropped western
blots shown in Fig. S2) and the
mitochondria were efficiently isolated in both groups (Fig. 2B). The MMP was evaluated using a
JC-1 assay. The results showed that the MMP of the MI group was
lower than that of the sham group (Fig. 2C, P<0.05).
Mitochondrial proteomics
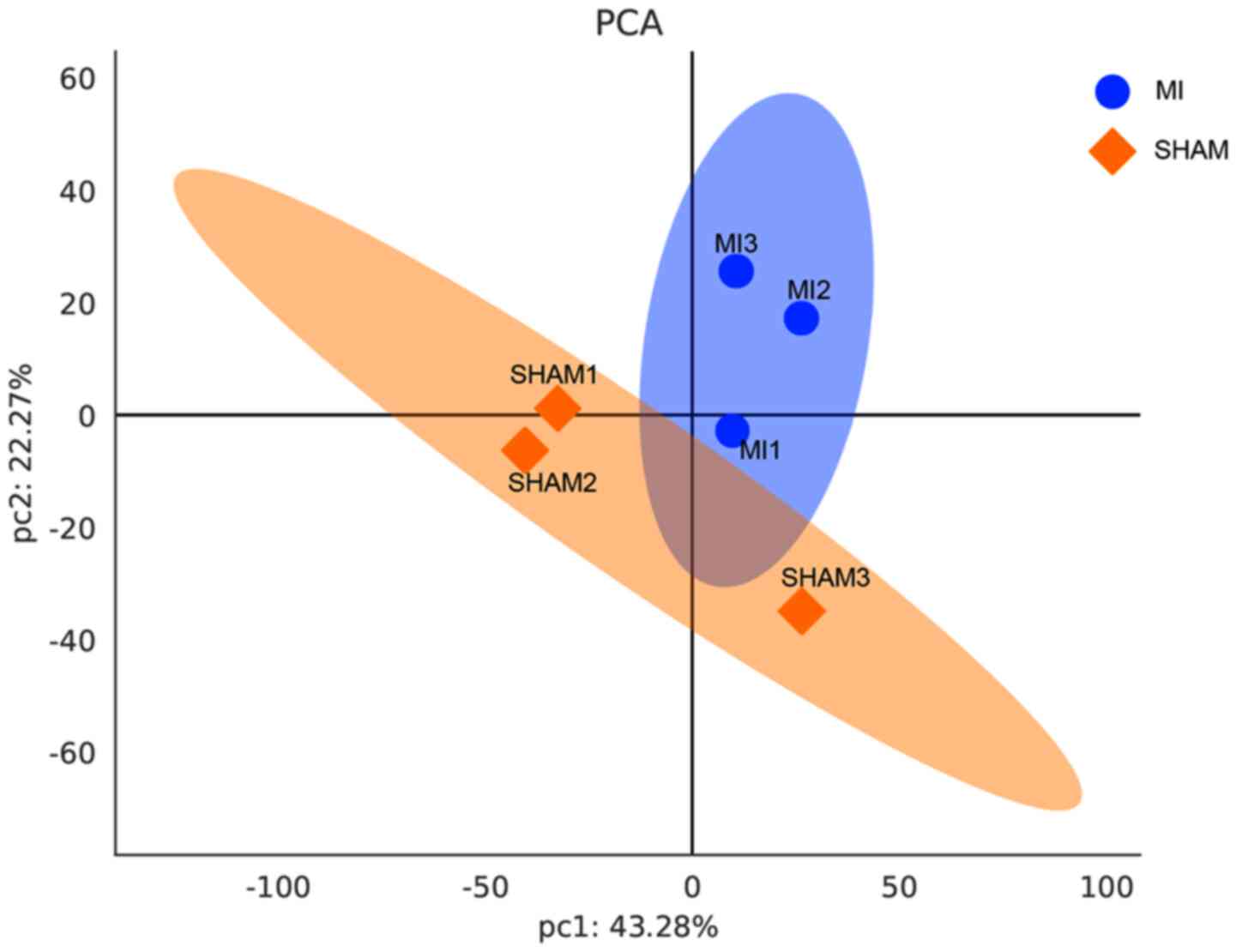
First, the expression of trusted proteins was
analyzed using PCA (Fig. 3). The
results showed substantial differences between the two groups. More
than 1,700 trusted proteins were identified in this study. Through
the analysis and synchronous comparison of myocardial mitochondria
in three separated samples of the sham and MI groups, 119
differential proteins were confirmed. Overall, 27 of the 119
proteins were upregulated and 16 were downregulated compared with
the sham group (Table I), and the
remaining 76 proteins showed ‘yes’ or ‘no’ differences between the
two groups (Tables II and
III). These results identified
LAD ligation/ischemia as the principal source of variance. The
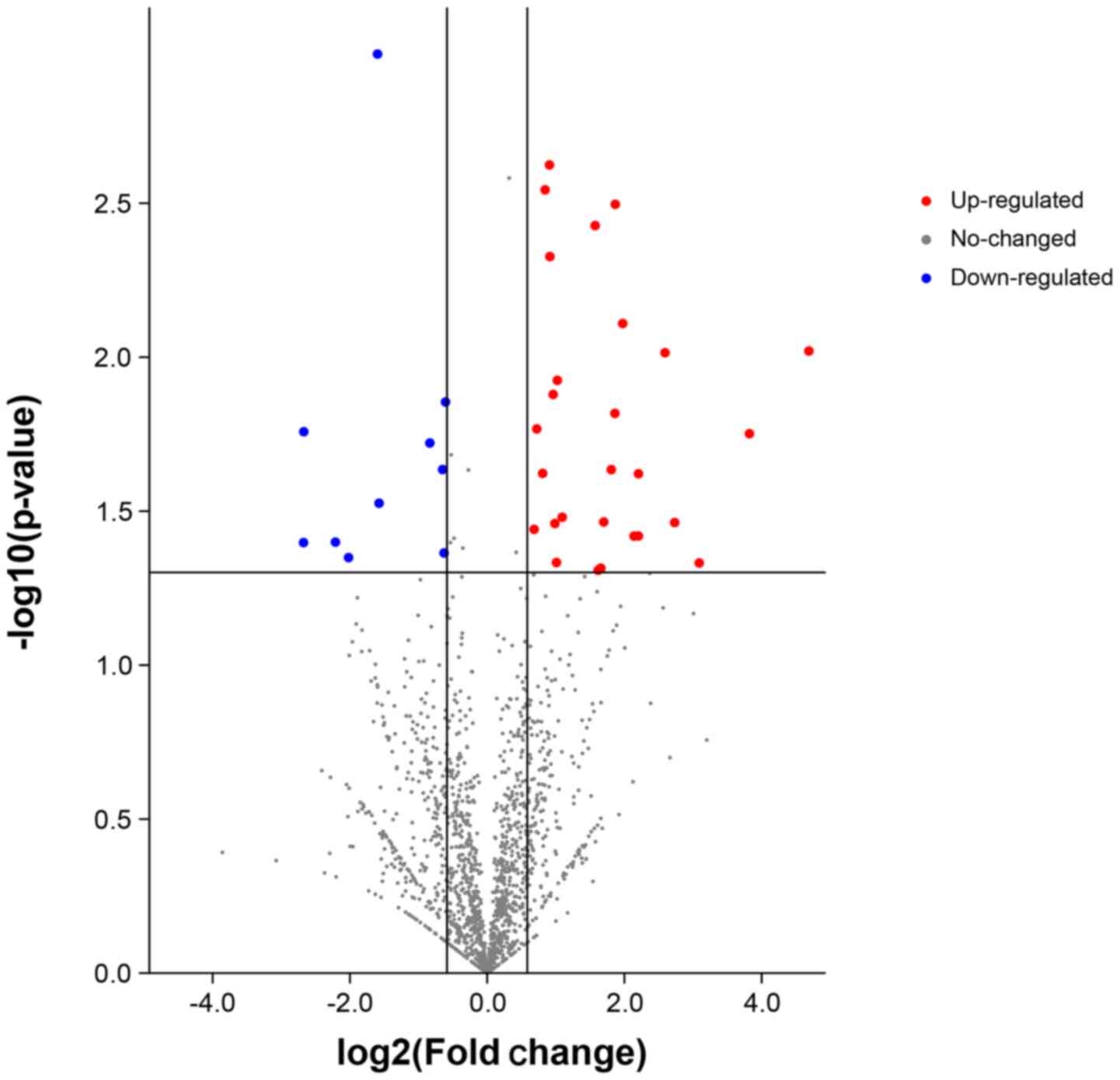
volcano map shows the overall distribution of the proteins
(Fig. 4). The differentially
expressed proteins were screened using FC=1.2 and P<0.05 as the
screening conditions. The blue and red dots represent the
significantly downregulated and upregulated proteins, respectively.
The gray dots represent non-significant proteins. Unsupervised
hierarchical clustering was used to group differentially expressed
proteins according to their expression profiles. The results are
shown in a heatmap (Fig. S3).
Each line in the heatmap represents a protein with mean
upregulation (red) or downregulation (purple) in expression within
the two groups. The proteins that did not exhibit significant
differences in expression are indicated in white. NADH
dehydrogenase (ubiquinone) iron-sulfur protein 6 (NDUFS6),
cytochrome b-c1 complex subunit 1 (UQCRC1), thrombospondin-1
(THBS1) and pyruvate dehydrogenase phosphatase regulatory subunit
(PDPR) were upregulated, and dynactin subunit 2 (DCTN2) and
a-kinase anchor protein 1 (AKAP1) were downregulated in the
ischemic group. Cytochrome C oxidase (COX) assembly protein COX11,
mitochondrion adenosine triphosphate (ATP) synthase F(0) complex
subunit C2 (ATP5G2), mitochondria ubiquitin ligase activator of
NF-κB1 (MUL1) were identified in the MI group and E3
ubiquitin-protein ligase UBR3 was identified in the sham group.
 | Table IDownregulated and upregulated
proteins in the MI group. |
Table I
Downregulated and upregulated
proteins in the MI group.
| Accession no. | Gene | Protein
description | FC | P-value | Type |
|---|
| Q99KJ8 | DCTN2 | Dynactin subunit
2 | 0.315 | 0.001c | Down |
| P31725 | S100A9 | Protein
S100-A9 | 0.323 | 0.015a | Down |
| Q3UH59 | MYH10 | Myosin-10 | 0.370 | 0.020a | Down |
| Q62448 | EIF4G2 | Eukaryotic
translation initiation factor 4γ2 | 0.423 | 0.026a | Down |
| Q8VCQ8 | CALD1 | Caldesmon 1 | 0.492 | 0.028a | Down |
| P19157 | GSTP1 | Glutathione
S-transferase P1 | 0.542 | 0.010b | Down |
| O08715 | AKAP1 | A-kinase anchor
protein 1, mitochondrial | 0.593 | 0.048a | Down |
| A0A0A6YX73 | PRKAR2α | cAMP-dependent
protein kinase type II-α regulatory subunit | 0.619 | 0.036a | Down |
| Q9CQS4 | SLC25A46 | Solute carrier
family 25 member 46 | 0.620 | 0.035a | Down |
| Q8R2P8 | KARS | Lysine-tRNA
ligase | 0.635 | 0.013a | Down |
| P05132 | PRKACα | cAMP-dependent
protein kinase catalytic subunit α | 0.652 | 0.047a | Down |
| P49312 | HNRNPA1 | Heterogeneous
nuclear ribonucleoprotein A1 | 0.665 | 0.050a | Down |
| P55096 | ABCD3 | ATP-binding
cassette sub-family D member 3 | 0.666 |
<0.001c | Down |
| P14733 | LMNB1 | Lamin-B1 | 0.684 | 0.029a | Down |
| Q9ET78 | JPH2 | Junctophilin-2 | 0.745 | 0.035a | Down |
| Q6ZWN5 | RPS9 | 40S ribosomal
protein S9 | 0.823 | 0.044a | Down |
| Q9D1G1 | RAB1B | Ras-related protein
Rab-1B | 1.201 | 0.031a | Up |
| Q9CZ13 | UQCRC1 | Cytochrome b-c1
complex subunit 1, mitochondrial | 1.479 | 0.048a | Up |
| Q99MR8 | MCCC1 | Methylcrotonoyl-CoA
carboxylase subunit α, mitochondrial | 1.496 | 0.043a | Up |
| Q8R5L1 | C1QBP | Complement
component 1 Q subcomponent-binding protein, mitochondrial | 1.591 | 0.027a | Up |
| Q3ULD5 | MCCC2 | Methylcrotonoyl-CoA
carboxylase β chain, mitochondrial | 1.612 | 0.013a | Up |
| Q9ER35 | FN3K |
Fructosamine-3-kinase | 1.654 | 0.013a | Up |
| P84096 | RHOG | Rho-related
GTP-binding protein RhoG | 1.676 | 0.031a | Up |
| Q7TSQ8 | PDPR | Pyruvate
dehydrogenase phosphatase regulatory subunit, mitochondrial | 1.724 | 0.046a | Up |
| Q921I1 | TF |
Serotransferrin | 1.731 | 0.025a | Up |
| Q5FWK3 | ARHGAP1 | Rho
GTPase-activating protein 1 | 1.800 | 0.017a | Up |
| A0A0R4J0I1 | SERPINA3K | Serine protease
inhibitor A3K | 1.810 | 0.005b | Up |
| Q00897 | SERPINA1D | α-1-antitrypsin
1-4 | 1.820 | 0.026a | Up |
| Q8BJ03 | COX15 | Cytochrome c
oxidase assembly protein COX15 homolog | 1.878 | 0.017a | Up |
| O08528 | HK2 | Hexokinase-2 | 1.912 | 0.015a | Up |
| P48036 | ANXA5 | Annexin A5 | 1.951 | 0.049a | Up |
| Q60994 | ADIPOQ | Adiponectin | 1.951 | 0.031a | Up |
| P07724 | ALB | Serum albumin | 1.969 | 0.031a | Up |
| P52503 | NDUFS6 | NADH dehydrogenase
[ubiquinone] iron-sulfur protein 6, mitochondrial | 2.175 | 0.046a | Up |
| A0A0R4J0X5 | SERPINA1C | Alpha-1-antitrypsin
1-3 | 2.536 | 0.045a | Up |
| Q61838 | A2M |
Alpha-2-macroglobulin | 2.764 | 0.019a | Up |
| O35639 | ANXA3 | Annexin A3 | 2.867 | 0.002b | Up |
| Q91VB8 | HBA-A1 | Alpha globin 1 | 3.001 | 0.037a | Up |
| P05064 | ALDOA |
Fructose-bisphosphate aldolase A | 3.409 | 0.026a | Up |
| A8DUK4 | HBB-BS | β-globin | 3.730 | 0.004b | Up |
| E9QNT8 | ANK1 | Ankyrin-1 | 3.885 | 0.040a | Up |
| P01027 | C3 | Complement C3 | 4.137 | 0.044a | Up |
| Q80YQ1 | THBS1 |
Thrombospondin-1 | 5.096 | 0.036a | Up |
| Table IIProteins specific to the SHAM
group. |
Table II
Proteins specific to the SHAM
group.
| Accession no. | Gene | Protein
descriptions | Type |
|---|
| Q8K0M3 | SORBS3 | Vinexin | Up |
| A2A848 | ACOX1 | Peroxisomal
acyl-coenzyme A oxidase 1 | Up |
| A8Y5P4 | MAP7D1 | MAP7
domain-containing protein 1 | Up |
| A2AS45 | PKP4 | Plakophilin-4 | Up |
| A4QPC5 | CMA1 | Chymase | Up |
| D3YYT1 | GLYR1 | Putative
oxidoreductase GLYR1 | Up |
| D3Z598 | LTBP4 | Latent-transforming
growth factor beta-binding protein 4 | Up |
| E9Q6Q8 | TBC1D4 | TBC1 domain family
member 4 | Up |
| Q5U430 | UBR3 | E3
ubiquitin-protein ligase UBR3 | Up |
| F7A1B4 | ENG | Endoglin | Up |
| O54774 | AP3D1 | AP-3 complex
subunit delta-1 | Up |
| O55022 | PGRMC1 | Membrane-associated
progesterone receptor component 1 | Up |
| O88587 | COMT | Catechol
O-methyltransferase | Up |
| P13541 | MYH3 | Myosin-3 | Up |
| P63163 | SNRPN | Small nuclear
ribonucleoprotein-associated protein N | Up |
| P37804 | TAGLN | Transgelin | Up |
| Q8BVQ9 | PSMC2 | 26S protease
regulatory subunit 7 | Up |
| Q3U962 | COL5A2 | Collagen alpha-2(V)
chain | Up |
| Q921L6 | CTTN | Src substrate
cortactin | Up |
| Q60972 | RBBP4 | Histone-binding
protein RBBP4 | Up |
| Q6P549 | INPPL1 |
Phosphatidylinositol 3,4,5-trisphosphate
5-phosphatase 2 | Up |
| Q6ZWQ9 | MYL12A | Myosin, light chain
12A, regulatory, non-sarcomeric | Up |
| Q8BGU5 | CCNY | Cyclin-Y | Up |
| Q8VCI5 | PEX19 | Peroxisomal
biogenesis factor 19 | Up |
| Q99N92 | MRPL27 | 39S ribosomal
protein L27, mitochondrial | Up |
| Q9D666 | SUN1 | SUN
domain-containing protein 1 | Up |
| Q9DAW9 | CNN3 | Calponin-3 | Up |
| Q9JIK5 | DDX21 | Nucleolar RNA
helicase 2 | Up |
| Q9JKY7 | CYP2D22 | Cytochrome P450
CYP2D22 | Up |
| Q9JLB0 | MPP6 | MAGUK p55 subfamily
member 6 | Up |
| Q9JLH8 | TMOD4 | Tropomodulin-4 | Up |
| Q9JMG7 | HDGFRP3 | Hepatoma-derived
growth factor-related protein 3 | Up |
| Q9Z0P5 | TWF2 | Twinfilin-2 | Up |
| Q9Z2X1 | HNRNPF | Heterogeneous
nuclear ribonucleoprotein F | Up |
| Table IIIProteins specific to the MI
group. |
Table III
Proteins specific to the MI
group.
| Accession no. | Gene | Protein
descriptions | Type |
|---|
| A0A075B6A0 | IGHM | Ig mu chain C
region | Up |
| A0A087WQS5 | ATP5G2 | ATP synthase F(0)
complex subunit C2, mitochondrial | Up |
| E9QP56 | APOC3 | Apolipoprotein
C-III | Up |
| F8VPK5 | ROCK2 | Rho-associated
protein kinase; Rho-associated protein kinase 2 | Up |
| A0A2I3BPW0 | NDRG2 | N-myc
downstream-regulated gene 2 protein | Up |
| A0A3B2WBH9 | TJP2 | Tight junction
protein ZO-2 | Up |
| A0A494BB95 | EIF1AX | Eukaryotic
translation initiation factor 1A | Up |
| A2AEX8 | FHL1 | Four and a half LIM
domains protein 1 | Up |
| B7ZCL8 | MPP1 | 55 kDa erythrocyte
membrane protein | Up |
| B8JJM5 | CFB | Complement factor
B | Up |
| D6RGQ0 | CFH | Complement factor
H | Up |
| O88986 | GCAT |
2-amino-3-ketobutyrate coenzyme A ligase,
mitochondrial | Up |
| F7AAP4 | ATP2B4 |
Calcium-transporting ATPase | Up |
| Q91YX5 | LPGAT1 | Acyl-CoA:
lysophosphatidylglycerol acyltransferase 1 | Up |
| Q8CIZ8 | VWF | von Willebrand
factor; von Willebrand antigen 2 | Up |
| O55042 | SNCA |
Alpha-synuclein | Up |
| O70194 | EIF3D | Eukaryotic
translation initiation factor 3 subunit D | Up |
| P01837 | IGKC | Igκ chain C
region | Up |
| P06684 | C5 | Complement C5 | Up |
| P07759 | SERPINA3K | Serine protease
inhibitor A3K | Up |
| P19221 | F2 | Prothrombin | Up |
| P29699 | AHSG |
Alpha-2-HS-glycoprotein | Up |
| P29788 | VTN | Vitronectin | Up |
| P35550 | FBL | rRNA
2-O-methyltransferase fibrillarin | Up |
| P40142 | TKT | Transketolase | Up |
| P43883 | PLIN2 | Perilipin-2 | Up |
| P49722 | PSMA2 | Proteasome subunit
alpha type-2 | Up |
| Q8CE80 | CAST | Calpastatin | Up |
| P61514 | RPL37A | 60S ribosomal
protein L37a | Up |
| P62835 | RAP1A | Ras-related protein
Rap-1A | Up |
| Q00519 | XDH | Xanthine
dehydrogenase/oxidase | Up |
| Q5SRC5 | COX11 | Cytochrome c
oxidase assembly protein COX11, mitochondrial | Up |
| Q61578 | FDXR | NADPH: adrenodoxin
oxidoreductase, mitochondrial | Up |
| Q6PA06 | ATL2 | Atlastin-2 | Up |
| Q6PDI5 | ECM29;AI314180 |
Proteasome-associated protein ECM29
homolog | Up |
| Q8VE37 | RCC1 | Regulator of
chromosome condensation | Up |
| Q7TNL9 | CHCHD10 |
Coiled-coil-helix-coiled-coil-helix
domain-containing 10 | Up |
| Q8BXZ1 | TMX3 | Protein
disulfide-isomerase TMX3 | Up |
| Q8VCM5 | MUL1 | Mitochondrial
ubiquitin ligase activator of NF-κB1 | Up |
| Q91X72 | HPX | Hemopexin | Up |
| Q99LB4 | CAPG | Macrophage-capping
protein | Up |
| Q9EST5 | ANP32B | Acidic leucine-rich
nuclear phosphoprotein 32 family member B | Up |
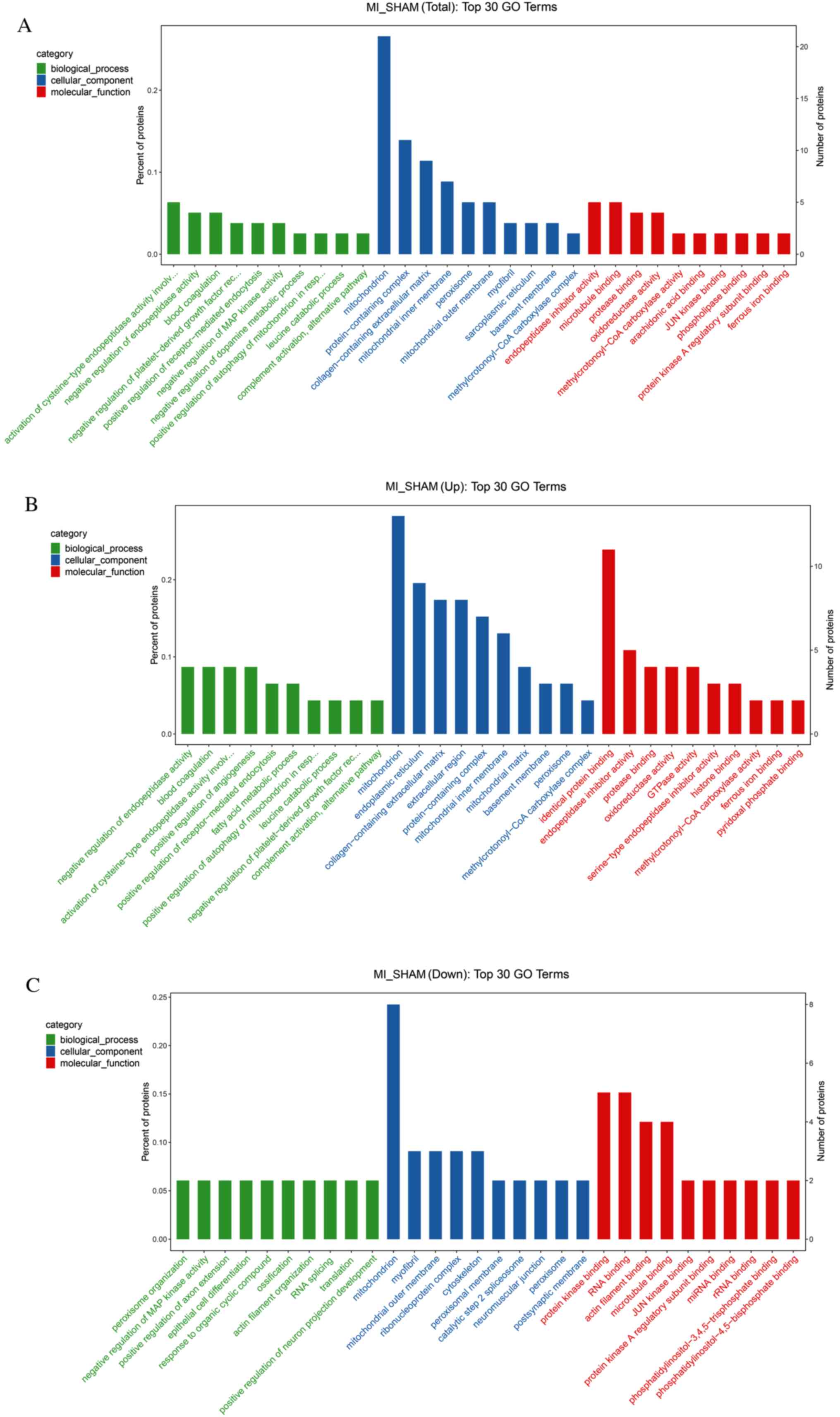
According to the GO database, the enrichment of the
total, upregulated, and downregulated differentially expressed
proteins were analyzed (Fig.
5A-C). For each category, a two-tailed Fisher's exact test was
used to test the enrichment of differentially expressed proteins
vs. all identified proteins. The GO term with a corrected P-value
<0.05 was considered statistically significant. Proteins were
classified by GO annotation into the following three categories:
Biological processes, cellular components and molecular functions.
Pathway enrichment of the top 30 GO terms of the total
differentially expressed proteins is shown in Fig. 5A. The most perturbed biological
processes were endopeptidase activity regulation and blood
coagulation. The mitochondria, protein-containing complex,
collagen-containing extracellular matrix and mitochondria inner
membrane were the most relevant cellular components. Endopeptidase
inhibitor activity and microtubule binding were the most relevant
molecular functions.
For the upregulated proteins, the most relevant
biological processes were the negative regulation of endopeptidase
activity, blood coagulation, the activation of cysteine-type
endopeptidase activity and the positive regulation of angiogenesis.
The most relevant cellular components included the endoplasmic
reticulum, collagen-containing extracellular matrix and
extracellular region. The top four molecular functions were
identical protein binding, endopeptidase inhibitor activity,
protease binding and oxidoreductase activity (Fig. 5B).
For the downregulated proteins, the closely related
biological processes included peroxisome organization, negative
regulation of MAP kinase activity and positive regulation of axon
extension. Except for mitochondria, the most highly relevant
cellular components were myofibrils, the mitochondrial outer
membrane, the ribonucleoprotein complex and the cytoskeleton.
Closely related molecular functions included protein kinase
binding, RNA binding, actin filament binding and
microtubule-binding (Fig. 5C).
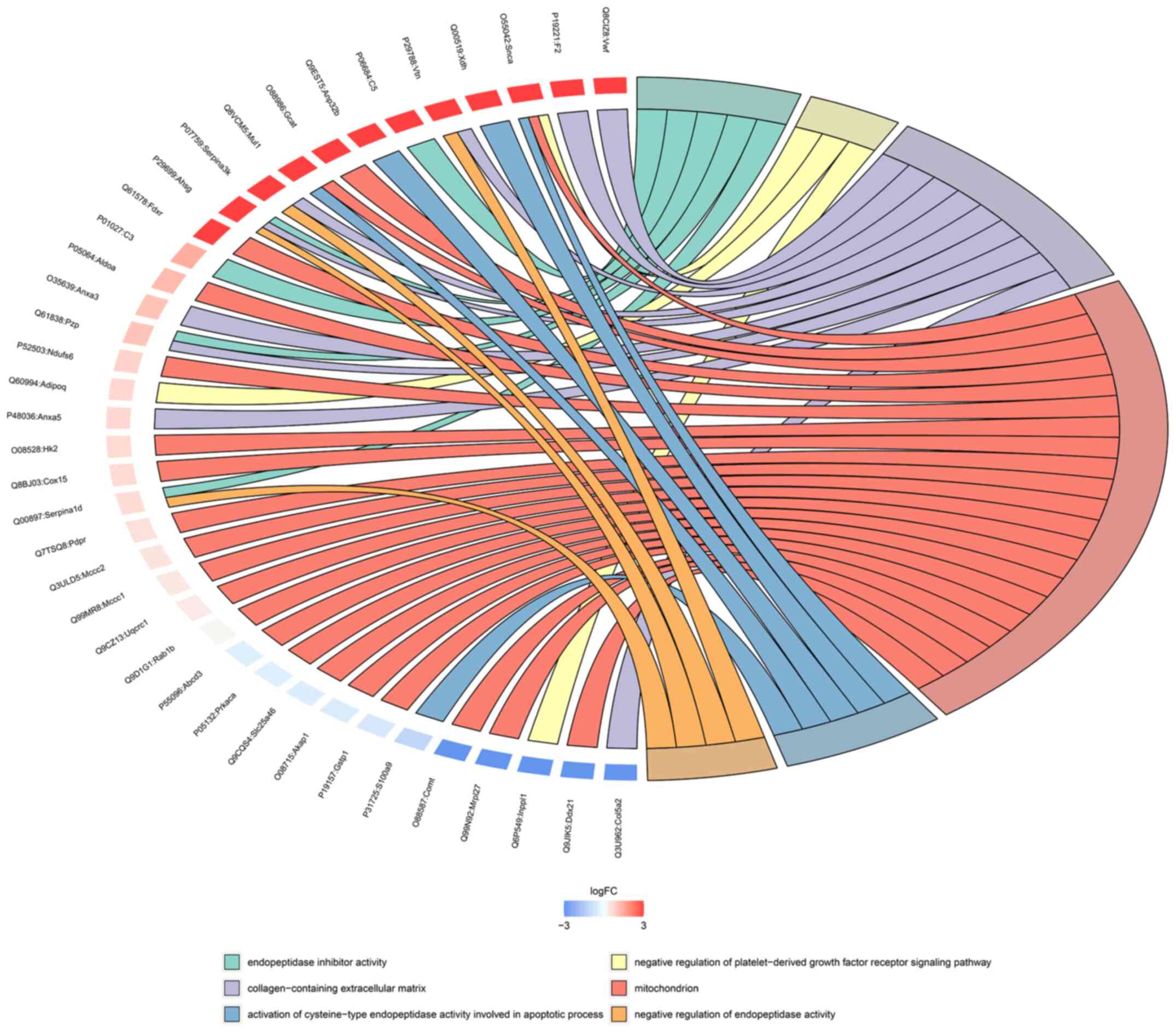
The relationship between the enriched GO terms and
the differentially expressed proteins is displayed as a chord
diagram in Fig. 6. A small arc on
the left side of the chord diagram represents one protein. The
color represents the difference: Blue indicates downregulation and
red indicates upregulation. The GO terms are shown on the
right-hand side of the diagram. Different colors represent
different GO terms; that is, different functions. GO terms
correspond to multiple differentially expressed proteins, and a
protein can participate in multiple GO terms; the mitochondria and
collagen-containing extracellular matrices possessed the most
enriched proteins.
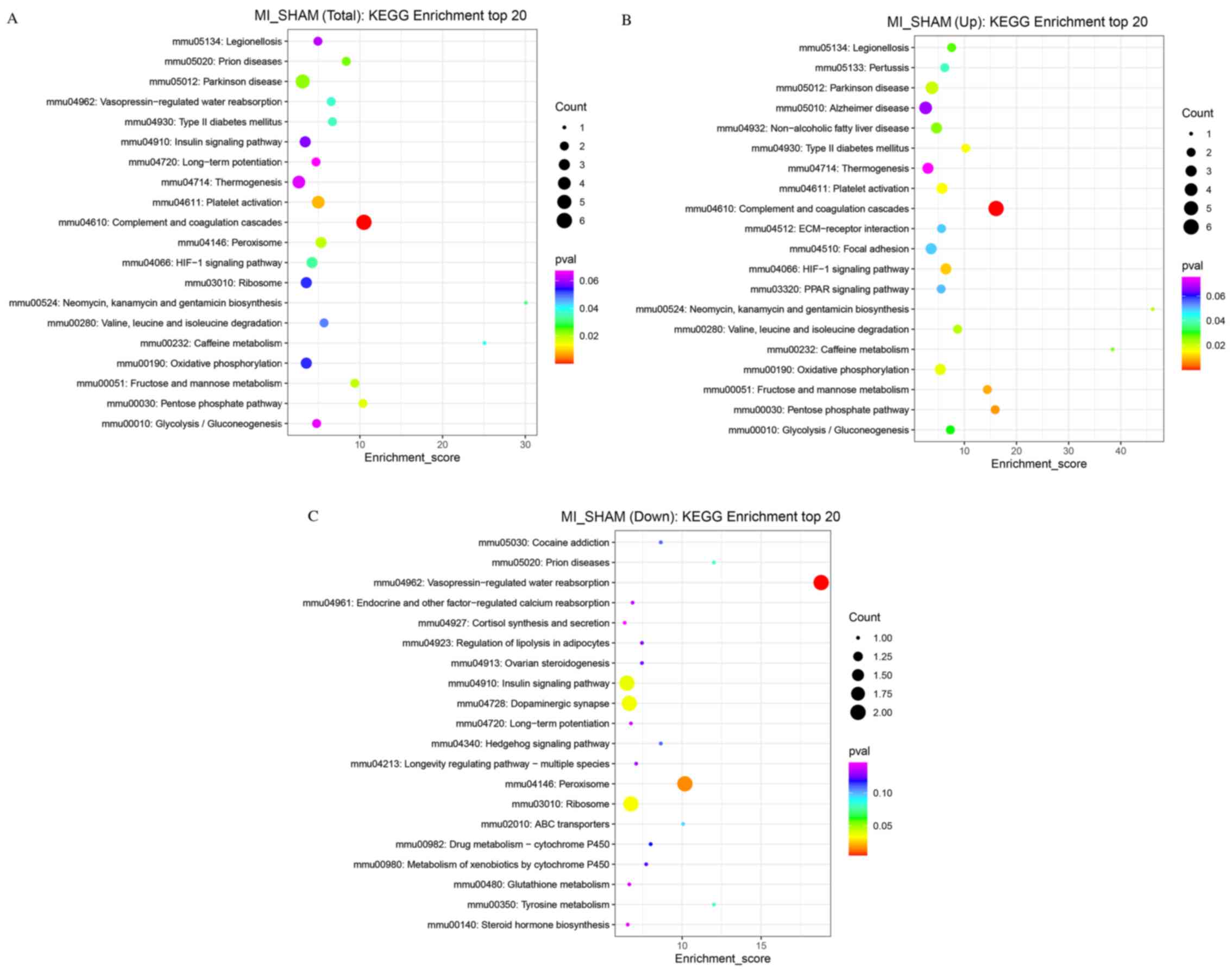
The KEGG database was used to analyze the metabolic
pathways of the proteins. Biological functions related to these
proteins were obtained from KEGG enrichment analysis (Fig. 7). The pathways with the most
relevant differentially expressed proteins included Parkinson's
disease, prion diseases, platelet activation, complement and
coagulation cascades, peroxisome, hypoxia-inducible factor-1
(HIF-1) signaling, type 2 diabetes mellitus, fructose and mannose
metabolism (Fig. 7A). For
upregulated proteins, the most relevant biological functions
included Parkinson's disease, non-alcoholic fatty liver disease,
focal adhesion, complement, coagulation cascades, the peroxisome
proliferator-activated receptor (PPAR) signaling pathway, OXPHOS,
the pentose phosphate pathway and the HIF-1 signaling pathway
(Fig. 7B). The closely related
biological functions of downregulated proteins included
vasopressin-regulated water reabsorption, the insulin signaling
pathway, dopaminergic synapses, ribosomes, and peroxisomes
(Fig. 7C).
Discussion
Ischemic heart disease is the leading cause of heart
failure worldwide (1).
Disturbances to ATP synthesis resulting from MI leads to energy
starvation in the myocardium, resulting in the remodeling of
myocardial energy metabolism (25). The heart is one of the most
metabolically active organs in the body (26). In the resting state, the human
heart uses 6 kg of ATP per day (27). Cardiac function declines when
mitochondrial OXPHOS and ATP production fail to keep up with the
demand for energy consumption. Mitochondrial disorders are a sign
of cardiovascular disease and heart failure (27). Early ultrastructural changes
include the depletion of cytoplasmic glycogen particles, and the
swelling of cardiomyocyte mitochondria can be observed after a few
minutes of ischemia, although these changes are reversible if
ischemia lasts no longer than 15-20 min (28). Any change in mitochondrial
structure is associated with impaired mitochondrial function, such
as the alteration of metabolic substrate utilization, impaired
activity of the mitochondrial electron transport chain, increased
formation of reactive oxygen species (ROS), and altered ion
homeostasis (26). Consequently,
cardiac function is affected by these adverse effects (29). Proteins are the ultimate
‘executors’ of cellular activity and function. To the best of our
knowledge, the proteomic characteristics of cardiac mitochondria
that suffer from short-term acute ischemia have not previously been
reported.
In the present study, cardiac mitochondrial injury,
such as outer membrane dissolution, inner membrane (cristae) loss
and rupture were observed in the MI group 15 min after LAD
ligation. However, the cardiomyocyte membrane, sarcomeres and
nuclei remained intact. Decreased MMP measurements also showed
damaged mitochondria in the MI group. These results indicated that
mitochondria are extremely sensitive to ischemia and may be a
potential marker of ischemia onset. Moreover, the results suggested
that proteome disturbances are inevitable during this period. To
elucidate the proteome changes, the proteome expression profile of
the left ventricle mitochondria was evaluated using LC-MS/MS
analysis. More than 1,700 proteins were screened, and 119
differentially expressed proteins were confirmed, with 27
upregulated and 16 downregulated proteins in the MI group. In
addition, 34 proteins were present in the sham group and 42 in the
MI group. Once the differential proteins were identified, GO
annotation and the KEGG pathway enrichment analyses were performed
to elucidate the proteins' function. According to the GO
annotation, the pathway categories include cellular component,
molecular function and biological process. Several differentially
expressed proteins were involved in the activities and pathways of
OXPHOS, complement activation, positive regulation of mitochondrial
autophagy, and oxidative dismutase activity. These proteins
included numerous upregulated proteins with FC ≥1.47, such as
UQCRC1, pyruvate dehydrogenase phosphatase regulatory subunit, rho
GTPase-activating protein 1, NDUFS6, Annexin A3 and A5, complement
1Q subcomponent-binding and protein complement C3,
fructose-bisphosphate aldolase A and ankyrin-1, and the
downregulated dynactin subunit 2, myosin 10, A-kinase anchor
protein 1, cAMP-dependent protein kinase type II-α regulatory
subunit and cAMP-dependent protein kinase catalytic subunit α.
Interestingly, a number of proteins were present only in the sham
or MI group. For example, UBR3, cAMP-dependent protein kinase type
II-α regulatory subunit (PRKAR2α), A-kinase anchor protein 1
(AKAP1, mitochondrial), and Junctophilin-2 (JPH2) were expressed
only in the sham group. These genes were primarily involved in
inflammatory response, microtubule binding and protease binding.
Dozens of the proteins were related to cellular components. ATP5G2,
calcium-transporting ATPase, COX11, adrenodoxin oxidoreductase and
MUL1 were expressed in the MI group only. Some of the proteins that
were present in the MI group are subunits of the mitochondrial
respiratory chain complex. The ‘presence’ difference between the
sham and MI groups indicated that mitochondrial proteome
disturbance was extensive and distinct as early as minutes after MI
occurrence.
To the best of our knowledge, there are no studies
that have elucidated the relationship between vasopressin-regulated
water reabsorption, dopaminergic synapses, peroxisomes and MI.
Studies regarding the relevance of Parkinson's disease,
non-alcoholic fatty liver disease and prion diseases to MI are
lacking. Platelet activation, coagulation dysfunction and abnormal
glucose metabolism may be involved in MI. THBS1, involved in
coagulation dysfunction, also underwent the most substantial
changes. The link between these pathways and MI remains to be fully
elucidated and requires further investigation. Interestingly,
OXPHOS, the HIF-1 signaling pathway, pentose phosphate pathway and
PPAR signaling pathway are closely related to electron transport
and energy metabolism, suggesting that the disorder of energy
metabolism in cardiomyocytes is the initial pathophysiological
feature of acute MI (30-32).
Mitochondrial adaptation plays a vital role in
protecting key cellular components from ischemic damage. In
addition, this is the main entry point for electrons to enter the
process of OXPHOS, and is helpful in establishing the proton
gradient needed for a large number of cells to synthesize ATP
(33-34). Complex I (ubiquinone
oxidoreductase) is the largest complex of the mitochondrial
respiratory chain, which is composed of >40 subunits and is
located in the mitochondrial inner membrane. Mutations that lead to
complex I defects are the most common cause of neurodegeneration,
myopathies and heart failure (35-37).
Complex I is the most vulnerable enzyme, and the first to be
damaged (38). The most common
diagnosis in patients with energy production disorders is complex I
activity deficiency (39). NDUFS6
is a highly conserved subunit of complex I (40,41).
NDUFS4 and NDUFS6 are crucial to the final step of complex I
assembly and may provide a connection point for the integration of
other subunits (42). Knockout of
the NDUFS6 gene can lead to complex I instability and functional
defects (43). The deletion of
NDUFS6 increases ROS production and damages mitochondria through
complex I deficiency (44). A
previous study established NDUFS6gt/gt mice with complex
I deficiency, which revealed decreased NDUFS6 protein expression
levels (43). The mice developed
cardiomyopathy and heart failure. Isolated heart studies have shown
that resting contractile function is severely impaired even in
NDUFS6gt/gt mice without heart failure. UQCRC1 is a
subunit of complex III in the respiratory chain, is encoded by
nuclear DNA and consists of 480 amino acids (45-48).
Disrupting one UQCRC1 allele resulted in decreased UQCRC1 mRNA and
protein expression in mice. In addition, complex III formation,
activity, and ATP content were decreased in the brain at baseline.
Under ischemic conditions, the brain tissue showed decreased MMP
and ATP content as well as increased free radical levels compared
with wild-type mice. These results indicated that UQCRC1 plays a
key role in complex III (49-51).
Overexpression of UQCRC1 can enhance the activity of complex III in
mice (52). Specific loss of
UQCRC1 in cells of epithelial origin can lead to mitochondrial
dysfunction. The overexpression of UQCRC1 helps to maintain the
mitochondrial function of H9C2 cells and regulates
apoptosis-related proteins by mediating the PI3K/Akt/GSK-3b pathway
to protect H9C2 cells from simulated ischemia/reperfusion injury
(53). These studies suggest that
UQCRC1 plays an important role in mitochondrial function, and may
have a protective effect on cardiomyocytes. In the present study,
short-term cardiac ischemia caused significant upregulation of
NDUFS6 and UQCRC1 compared with the sham group. This might be
compensatory regulation for protecting the heart from metabolic
dysfunction due to mitochondrial injury. COX consists of 13
subunits; the subunits COX I, COX II and COX III are encoded by
mitochondrial DNA (54-56).
COX11 is an endogenous mitochondrial membrane protein that is
essential for the assembly of active COX (57,58).
The ATP synthase F (0) complex consists of subunits C1 (ATP5G1), C2
(ATP5G2) and C3 (ATP5G3) (59). A
previous study identified a substantial increase in ATP5G1 in a rat
model of ischemic heart failure (60). Only ATP5G2 was identified in the
hearts of the ischemic mice. However, its effect on mitochondrial
hemostasis needs to be further elucidated.
In conclusion, comprehensive and significant
mitochondrial proteomic changes occurred after a short period of
acute MI. However, the screened proteins should be confirmed by
parallel reaction monitoring. Due to the limited output of
mitochondrial protein in one mouse heart, the differential proteins
will be validated by ELISA and/or PCR in blood samples in the
future. There are still other limitations in the present study; for
example, detection of serum CKMB level is necessary for confirming
establishment of the MI model and will enrich the data. Among the
differentially expressed proteins, the mitochondrial respiratory
chain protein is the major focus of our lab. These differential
proteins are involved in cellular component, oxidative
phosphorylation and ubiquitination, which may regulate the
post-translational modification of mitochondrial proteins.
Illustrating their downstream effects and possible molecular
mechanisms by overexpression/knockdown, western blotting,
co-immunoprecipitation and confocal microscopy analysis in a
hypoxic cardiomyocyte model will be performed in our future
studies.
In conclusion, short-term acute ischemia can result
in apparent cardiac mitochondrial damage and extensive disturbances
to the proteome. A series of altered proteins are closely
correlated with the pathways involved in OXPHOS, complement
activation and mitochondrial autophagy. The results of the present
study showed that mitochondrial changes may be regarded as
biomarkers of the very early stages of ischemic cardiac injury. In
addition, this study provides novel insights into the mechanism of
myocardial remodeling targeting the mitochondrial proteome.
Supplementary Material
Cardiomyocytes under transmission
electron microscopy. Cardiomyocytes in the (A) Sham and (B) MI
groups. Scale bar, 10 μm. MI, myocardial ischemia.
Original uncropped western blots shown
in for Fig. 2A. VDAC1,
voltage.dependent anion.selective channel protein 1.
Differentially expressed protein
analysis. Clustering heatmap. MI, myocardial ischemia. FC, fold
change.
Acknowledgements
We would like to thank Miss Yipin Han (Bloomberg
School of Public Health, Department of Epidemiology-Cardiovascular
and Clinical Epidemiology, Johns Hopkins University, Baltimore USA)
and Mr. Mewand Khan (School of Clinical Medicine, Ningxia Medical
University, Yinchuan, Ningxia, China) for their suggestions and
discussions.
Funding
Funding: This study was supported by the National Natural
Science Foundation (grant no. 81660045) and Ningxia Natural Science
Foundation (grant no. 2020AAC02037).
Availability of data and materials
Additional details regarding the LC-MS/MS analysis
are available in the public database proteomecentral.proteomexchange.org/cgi/GetDataset?
(accession no. PXD026207; lingfeng_zeng@163.com, Password:
sUdIhJj4). The datasets used and/or analyzed during the current
study are also available from the corresponding author on
reasonable request.
Authors' contributions
JW and JH confirm the authenticity of all the raw
data. JW and YF collected the tissues, prepared the specimens, and
performed the western blotting and statistical analysis. QL and FX
extracted the mitochondria and performed the western blotting. JW,
RH and RY established the mouse model. JH and JW designed the
experiments. JW and JH wrote the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All experiments and procedures were approved by the
Institutional Animal Care and Use Committee Review Board of the
General Hospital of Ningxia Medical University (approval no.
2020-01) and complied with the guidelines of the National
Institutes of Health Animal Care and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Farthing DE, Farthing CA and Xi L: Inosine
and hypoxanthine as novel biomarkers for cardiac ischemia: From
bench to point-of-care. Exp Biol Med (Maywood). 240:821–831.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shvedova M, Anfinogenova Y,
Atochina-Vasserman EN, Schepetkin IA and Atochin DN: c-Jun
N-terminal kinases (JNKs) in myocardial and cerebral
ischemia/reperfusion injury. Front Pharmacol. 9(715)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Oyama Y, Bartman CM, Gile J, Sehrt D and
Eckle T: The circadian PER2 enhancer nobiletin reverses the
deleterious effects of midazolam in myocardial ischemia and
reperfusion injury. Curr Pharm Des. 24:3376–3383. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Škrlec I, Milić J and Steiner R: The
impact of the circadian genes CLOCK and ARNTL on myocardial
infarction. J Clin Med. 9(484)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pavo N, Lukovic D, Zlabinger K, Zimba A,
Lorant D, Goliasch G, Winkler J, Pils D, Auer K, Jan Ankersmit H,
et al: Sequential activation of different pathway networks in
ischemia-affected and non-affected myocardium, inducing intrinsic
remote conditioning to prevent left ventricular remodeling. Sci
Rep. 7(43958)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ruiz-Meana M, Abellán A, Miró-Casas E and
Garcia-Dorado D: Opening of mitochondrial permeability transition
pore induces hypercontracture in Ca2+ overloaded cardiac
myocytes. Basic Res Cardiol. 102:542–552. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Santos-Gallego CG, Vahl TP, Goliasch G,
Picatoste B, Arias T, Ishikawa K, Njerve IU, Sanz J, Narula J,
Sengupta PP, et al: Sphingosine-1-phosphate receptor agonist
fingolimod increases myocardial salvage and decreases adverse
postinfarction left ventricular remodeling in a porcine model of
ischemia/reperfusion. Circulation. 133:954–966. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Saito T, Nah J, Oka SI, Mukai R, Monden Y,
Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, et al: An
alternative mitophagy pathway mediated by Rab9 protects the heart
against ischemia. J Clin Invest. 129:802–819. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jacquet S, Yin X, Sicard P, Clark J,
Kanaganayagam GS, Mayr M and Marber MS: Identification of cardiac
myosin-binding protein C as a candidate biomarker of myocardial
infarction by proteomics analysis. Mol Cell Proteomics.
8:2687–2699. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kim N, Lee Y, Kim H, Joo H, Youm JB, Park
WS, Warda M, Cuong DV and Han J: Potential biomarkers for ischemic
heart damage identified in mitochondrial proteins by comparative
proteomics. Proteomics. 6:1237–1249. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wu J, Zhang Y, Wu Q, Xie D, Dai W, Zhang
X, Yang Z and Wang D: Integrative analyses of myocardial lipidome
and proteome implicate mitochondrial dysfunction in lethal
ventricular tachyarrhythmia (LVTA) induced by acute myocardial
ischemia (AMI). J Proteomics. 197:14–22. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cao TH, Jones DJL, Voors AA, Quinn PA,
Sandhu JK, Chan DC, Parry HM, Mohan M, Mordi IR, Sama IE, et al:
Plasma proteomic approach in patients with heart failure: Insights
into pathogenesis of disease progression and potential novel
treatment targets. Eur J Heart Fail. 22:70–80. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang Y, Li C, Chuo W, Liu Z, Ouyang Y, Li
D, Han J, Wu Y, Guo S and Wang W: Integrated proteomic and
metabolomic analysis reveals the NADH-mediated TCA cycle and energy
metabolism disorders based on a new model of chronic progressive
heart failure. Mol Biosyst. 9:3135–3145. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang G, Zhou B, Zheng Y, Feng K, Rao L,
Zhang J, Xin J, Zhang B and Zhang L: Time course proteomic profile
of rat acute myocardial infarction by SELDI-TOF MS analysis. Int J
Cardiol. 131:225–233. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gao E and Koch WJ: A novel and efficient
model of coronary artery ligation in the mouse. Wound Regeneration
and Repair. Springer, Clifton, NJ, pp299-311, 2013.
|
|
16
|
Lu L, Ma J, Sun M, Wang X, Gao E, Lu L,
Ren J, Yang L and Yang J: Melatonin ameliorates MI-induced cardiac
remodeling and apoptosis through a JNK/p53-dependent mechanism in
diabetes mellitus. Oxid Med Cell Longev.
2020(1535201)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen J, Ceholski DK, Liang L, Fish K and
Hajjar RJ: Variability in coronary artery anatomy affects
consistency of cardiac damage after myocardial infarction in mice.
Am J Physiol Heart Circ Physiol. 313:H275–H282. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Flameng W, Borgers M, Daenen W and
Stalpaert G: Ultrastructural and cytochemical correlates of
myocardial protection by cardiac hypothermia in man. J Thorac
Cardiovasc Surg. 79:413–424. 1980.PubMed/NCBI
|
|
19
|
Wang W, Zhou X, Cui F, Shi C, Wang Y, Men
Y, Zhao W and Zhao J: Proteomic analysis on exosomes derived from
Patients' sera infected with Echinococcus granulosus. Korean
J Parasitol. 57:489–497. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang L, Huang Y, Wang X and Chen Y:
Label-free LC-MS/MS proteomics analyses reveal proteomic changes
accompanying MSTN KO in C2C12 cells. BioMed Res Int.
2019(7052456)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cox J, Neuhauser N, Michalski A, Scheltema
RA, Olsen JV and Mann M: Andromeda: A peptide search engine
integrated into the MaxQuant environment. J Proteome Res.
10:1794–1805. 2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
The Gene Ontology Consortium. The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kanehisa M: Post-Genome Informatics.
Oxford University Press, Oxford, NY, 2000.
|
|
25
|
van Bilsen M, Smeets PJ, Gilde AJ and van
der Vusse GJ: Metabolic remodelling of the failing heart: The
cardiac burn-out syndrome? Cardiovasc Res. 61:218–226.
2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA,
et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Davidson MT, Grimsrud PA, Lai L, Draper
JA, Fisher-Wellman KH, Narowski TM, Abraham DM, Koves TR, Kelly DP
and Muoio DM: Extreme acetylation of the cardiac mitochondrial
proteome does not promote heart failure. Circ Res. 127:1094–1108.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Frangogiannis NG: Pathophysiology of
myocardial infarction. Compr Physiol. 5:1841–1875. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kolb AL, Corridon PR, Zhang S, Xu W,
Witzmann FA, Collett JA, Rhodes GJ, Winfree S, Bready D,
Pfeffenberger ZJ, et al: Exogenous gene transmission of isocitrate
dehydrogenase 2 mimics ischemic preconditioning protection. J Am
Soc Nephrol. 29:1154–1164. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gentillon C, Li D, Duan M, Yu WM,
Preininger MK, Jha R, Rampoldi A, Saraf A, Gibson GC, Qu CK, et al:
Targeting HIF-1α in combination with PPARα activation and postnatal
factors promotes the metabolic maturation of human induced
pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol.
132:120–135. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wu J, Chen P, Li Y, Ardell C, Der T,
Shohet R, Chen M and Wright GL: HIF-1α in heart: Protective
mechanisms. Am J Physiol Heart Circ Physiol. 305:H821–H828.
2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X,
Bhatnagar A, Jones SP and Hill BG: Integration of flux measurements
to resolve changes in anabolic and catabolic metabolism in cardiac
myocytes. Biochem J. 474:2785–2801. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Smeitink JA, Zeviani M, Turnbull DM and
Jacobs HT: Mitochondrial medicine: A metabolic perspective on the
pathology of oxidative phosphorylation disorders. Cell Metab.
3:9–13. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tucker EJ, Compton AG, Calvo SE and
Thorburn DR: The molecular basis of human complex I deficiency.
IUBMB Life. 63:669–677. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsutsui H, Ide T and Kinugawa S:
Mitochondrial oxidative stress, DNA damage, and heart failure.
Antioxid Redox Signal. 8:1737–1744. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Elurbe DM and Huynen MA: The origin of the
supernumerary subunits and assembly factors of complex I: A
treasure trove of pathway evolution. Biochim Biophys Acta.
1857:971–979. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Triepels RH, Van Den Heuvel LP, Trijbels
JM and Smeitink JA: Respiratory chain complex I deficiency. Am J
Med Genet. 106:37–45. 2001.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Forbes JM, Ke BX, Nguyen TV, Henstridge
DC, Penfold SA, Laskowski A, Sourris KC, Groschner LN, Cooper ME,
Thorburn DR, et al: Deficiency in mitochondrial complex I activity
due to Ndufs6 gene trap insertion induces renal disease. Antioxid
Redox Signal. 19:331–343. 2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kmita K, Wirth C, Warnau J,
Guerrero-Castillo S, Hunte C, Hummer G, Kaila VR, Zwicker K, Brandt
U and Zickermann V: Accessory NUMM (NDUFS6) subunit harbors a
Zn-binding site and is essential for biogenesis of mitochondrial
complex I. Proc Natl Acad Sci USA. 112:5685–5690. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lazarou M, McKenzie M, Ohtake A, Thorburn
DR and Ryan MT: Analysis of the assembly profiles for
mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol
Cell Biol. 27:4228–4237. 2007.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zhang Y, Guo L, Han S, Chen L, Li C, Zhang
Z, Hong Y, Zhang X, Zhou X, Jiang D, et al: Adult mesenchymal stem
cell ageing interplays with depressed mitochondrial Ndufs6. Cell
Death Dis. 11(1075)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ke BX, Pepe S, Grubb DR, Komen JC,
Laskowski A, Rodda FA, Hardman BM, Pitt JJ, Ryan MT, Lazarou M, et
al: Tissue-specific splicing of an Ndufs6 gene-trap insertion
generates a mitochondrial complex I deficiency-specific
cardiomyopathy. Proc Natl Acad Sci USA. 109:6165–6170.
2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Schulte U, Arretz M, Schneider H,
Tropschug M, Wachter E, Neupert W and Weiss H: A family of
mitochondrial proteins involved in bioenergetICS and biogenesis.
Nature. 339:147–149. 1989.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shan W, Li J, Xu W, Li H and Zuo Z:
Critical role of UQCRC1 in embryo survival, brain ischemic
tolerance and normal cognition in mice. Cell Mol Life Sci.
76:1381–1396. 2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Unni S, Thiyagarajan S, Srinivas Bharath
MM and Padmanabhan B: Tryptophan oxidation in the UQCRC1 subunit of
mitochondrial complex III (Ubiquinol-Cytochrome C Reductase) in a
mouse model of myodegeneration causes large structural changes in
the complex: A molecular dynamics simulation study. Sci Rep.
9(10694)2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Davis M, Whitely T, Turnbull DM and
Mendelow AD: Selective impairments of mitochondrial respiratory
chain activity during aging and ischemic brain damage. Brain Edema
X. Springer, Berlin, pp56-58, 1997.
|
|
48
|
Moro MA, Almeida A, Bolaños JP and
Lizasoain I: Mitochondrial respiratory chain and free radical
generation in stroke. Free Radic Biol Med. 39:1291–1304.
2005.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Fan H, He Z, Huang H, Zhuang H, Liu H, Liu
X, Yang S, He P, Yang H and Feng D: Mitochondrial quality control
in cardiomyocytes: A critical role in the progression of
cardiovascular diseases. Front Physiol. 11(252)2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Kriaucionis S, Paterson A, Curtis J, Guy
J, Macleod N and Bird A: Gene expression analysis exposes
mitochondrial abnormalities in a mouse model of Rett syndrome. Mol
Cell Biol. 26:5033–5042. 2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Shibanuma M, Inoue A, Ushida K, Uchida T,
Ishikawa F, Mori K and Nose K: Importance of mitochondrial
dysfunction in oxidative stress response: A comparative study of
gene expression profiles. Free Radic Res. 45:672–680.
2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ide T, Tsutsui H, Hayashidani S, Kang D,
Suematsu N, Nakamura K, Utsumi H, Hamasaki N and Takeshita A:
Mitochondrial DNA damage and dysfunction associated with oxidative
stress in failing hearts after myocardial infarction. Circ Res.
88:529–535. 2001.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yi T, Wu X, Long Z, Duan G, Wu Z, Li H,
Chen H and Zhou X: Overexpression of ubiquinol-cytochrome c
reductase core protein 1 may protect h9c2 cardiac cells by binding
with Zinc. BioMed Res Int. 2017(1314297)2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Carr HS, Maxfield AB, Horng YC and Winge
DR: Functional analysis of the domains in Cox11. J Biol Chem.
280:22664–22669. 2005.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sun M, Zuo X, Li R, Wang T and Kang YJ:
Vascular endothelial growth factor recovers suppressed cytochrome c
oxidase activity by restoring copper availability in hypertrophic
cardiomyocytes. Exp Biol Med (Maywood). 239:1671–1677.
2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Palmer DN, Fearnley IM, Medd SM, Walker
JE, Martinus RD, Bayliss SL, Hall NA, Lake BD, Wolfe LS and Jolly
RD: Lysosomal storage of the DCCD reactive proteolipid subunit of
mitochondrial ATP synthase in human and ovine ceroid
lipofuscinoses. Adv Exp Med Biol. 266:211–223. 1989.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Li HS, Zhang JY, Thompson BS, Deng XY,
Ford ME, Wood PG, Stolz DB, Eagon PK and Whitcomb DC: Rat
mitochondrial ATP synthase ATP5G3: Cloning and upregulation in
pancreas after chronic ethanol feeding. Physiol Genomics. 6:91–98.
2001.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Liu T, Chen L, Kim E, Tran D, Phinney BS
and Knowlton AA: Mitochondrial proteome remodeling in ischemic
heart failure. Life Sci. 101:27–36. 2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wadosky KM and Willis MS: The story so
far: Post-translational regulation of peroxisome
proliferator-activated receptors by ubiquitination and SUMOylation.
Am J Physiol Heart Circ Physiol. 302:H515–H526. 2012.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Wang Z, Wang SP, Shao Q, Li PF, Sun Y, Luo
LZ, Yan XQ, Fan ZY, Hu J, Zhao J, et al: Brain-derived neurotrophic
factor mimetic, 7,8-dihydroxyflavone, protects against myocardial
ischemia by rebalancing optic atrophy 1 processing. Free Radic Biol
Med. 145:187–197. 2019.PubMed/NCBI View Article : Google Scholar
|