Introduction
Acute myocardial infarction (AMI) is a
cardiovascular disease with common and potentially fatal
presentation (1). Although there
was a relative decline in AMI of 48.7% over a study period
(2000-2014), AMI still is a leading cause of morbidity and
mortality worldwide (2). The
pathogenesis of myocardial ischemia-reperfusion (I/R) injury is
considered one of the main research topics in the field of vascular
disease (3,4). The mechanism underlying myocardial I/R
injury has not been fully elucidated. Ca2+ overload and
oxidative stress are the primary factors associated with this
disease, and inflammatory immune responses and induction of
apoptosis are also involved (5,6).
During I/R, AMP-activated protein kinase (AMPK)
signaling serves a role in oxidative stress, cell apoptosis and
cardiac dysfunction, and was indicated to mediate beneficial
influence over adiponectin in cardiac damage induced by I/R
(7-10).
Adiponectin serves a vital role in the cardiac protection
mechanism, the decline of which was indicated to enhance I/R injury
and the ineffectiveness of ischemia post-conditioning (11). The activation of AMPK is mediated by
adaptor protein phosphotyrosine interacting with PH domain and
leucine zipper 1 (APPL1), which binds to both adiponectin receptor
(AdipoR)1 and R2(12). Adiponectin
has been reported to protect myocardial cells from I/R injury by
regulating AMPK and cyclooxygenase 2 (COX2) expression (13). In addition, following myocardial I/R
injury induction in rats, adiponectin expression has been indicated
to regulate the activation of the AMPK/sirtuin 1 axis (14). A previous study has demonstrated
that adiponectin protected H9c2 cells from hypoxia-reoxygenation
(H/R)-induced apoptosis, but it could also activate APPL1(15).
Reactive oxygen species (ROS) are the main cause of
oxidative stress and the primary inducer of I/R injury (16). Under normoxic conditions, the
production and clearance of ROS in myocardial cells are balanced.
However, during myocardial ischemia and hypoxia, the increased
production of ROS can directly result in the oxidation of lipids,
proteins and DNA, which further damages the cellular structure
(17). As a result, the products of
lipid peroxidation and the activity levels of the antioxidant
enzymes, such as superoxide dismutase (SOD), are increased, leading
to myocardial injury (18). Nuclear
factor erythroid 2-related factor 2 (NRF2) is involved in the
regulation of redox balance, stress response and inflammation
(19). It has been reported that
NRF2 mediated the modulation of oxidative stress in cerebral
oxygen-glucose deprivation/reoxygenation (20). In addition, an interplay between
NRF2 and NF-κB has been demonstrated, which involved
counterbalancing the activity of one another in cerebrovascular
disorders (21).
Based on this evidence, the present study aimed to
investigate the role and mechanism of action of APPL1 in H9c2 cells
following H/R injury.
Materials and methods
Cell lines and establishment of the
H/R model
H9c2 (rat myocardial cells) were purchased from The
Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences and were routinely cultured in DMEM supplemented with 10%
FBS (each, Gibco; Thermo Fisher Scientific, Inc.) at 37˚C with 5%
CO2. H9c2 cells were subjected to hypoxia for 24 h in an
incubator at 37˚C (82% N2, 18% CO2 and
<0.5% O2) and reoxygenation for 1 h in an incubator
at 37˚C (5% CO2 and 21% O2) to simulate
myocardial cell injury caused by I/R, and a H/R model was
established (H/R group). Cells in control group were routinely
cultured in DMEM supplemented with 10% FBS at 37˚C with 5%
CO2. H9c2 cells were pretreated with a specific AMPK
activator (100 µM; product name, MK-3903; cat. no. GC31361; GlpBio
Technology) and AMPK inhibitor (10 µM; product name, compound C;
cat. no. HY-13418A; MedChemExpress) for 24 h.
Western blot analysis
H9c2 cells were exposed to specific conditions (H/R,
APPL1 overexpression model, AMPK inhibitor or AMPK activator) and
the cell was lysed using RIPA lysis buffer containing 1% PMSF
(Beijing Solarbio Science & Technology Co., Ltd.) on ice for 30
min. The supernatant was collected after centrifugation at 4˚C for
15 min at 12,000 x g, and the protein concentration was determined
via the BCA method. Subsequently, the proteins were transferred to
PVDF membranes following 12% SDS-PAGE (50 µg proteins/lane were
loaded on the gel). The membranes were blocked with 5% skimmed milk
powder solution for 2 h at room temperature. The following primary
antibodies were incubated with the membranes at 4˚C overnight: AMPK
(1:1,000; cat. no. ab214425; Abcam), phosphorylated (p)-AMPK
(1:1,000; cat. no. ab133448; Abcam), SOD2 (1:1,000; cat. no.
ab68155; Abcam), SOD3 (1:1,000; cat. no. ab83108; Abcam), COX2
(1:1,000; cat. no. ab179800; Abcam), APPL1 (1:1,000; cat. no.
ab250150; Abcam), GAPDH (1:5,000; cat. no. ab181602; Abcam), NRF2
(1:1,000; cat. no. ab89443; Abcam), Bcl2 (1:1,000; cat. no.
ab196495; Abcam), heme oxygenase 1 (HO-1; 1:10,000; cat. no.
ab68477; Abcam), p-liver kinase B1 (p-LKB1; cat. no. 3482; 1,1000;
Cell Signaling Technology, Inc.), LKB1 (cat. no. 3047; 1,1000; Cell
Signaling Technology, Inc.), p-acetyl-CoA carboxylase α (p-ACC;
cat. no. 3661; 1,1000; Cell Signaling Technology, Inc.), ACC (cat.
no. 3662; 1,1000; Cell Signaling Technology, Inc.), cleaved
caspase-3 (cat. no. 9664; 1,1000; Cell Signaling Technology, Inc.),
cleaved poly (ADP-ribose) polymerase (PARP; cat. no. 94885; 1,1000;
Cell Signaling Technology, Inc.), NF-κB p65 (cat. no. 8242; all
1:1,000; Cell Signaling Technology, Inc.). The membranes were
washed with TBST and then incubated with a secondary antibody
solution [goat anti-rabbit IgG H&L (HRP); 1:10,000; cat. no.
ab97051; Abcam] for 2 h at room temperature. Following incubation,
a chemiluminescence detection kit kit (Advansta, Inc.) was used to
visualize the proteins. Quantity One software (v4.6.6; Bio-Rad
Laboratories, Inc.) was used to analyze the gray scale of the
protein bands. The ratio of gray value of the target protein was
normalized to GAPDH to represent the relative expression levels of
the target protein.
SOD activity
H9c2 cells were collected and lysed using RIPA lysis
buffer (Beijing Solarbio Science & Technology, Co., Ltd.). The
cell supernatant was then obtained following centrifugation at
3,000 x g for 15 min at 4˚C. SOD activity was detected
according to the manufacturer's protocol of the SOD assay kit (cat.
no. A001-3-2; WST-1 method; Nanjing Jiancheng Bioengineering
Institute). The absorbance at a wavelength of 450 nm was detected
using a microplate reader.
Plasmid transfection
H9c2 cells (10x104 cells/ml) were
transfected with APPL1 overexpression plasmids (5 nM; ov-APPL1;
Genomeditech) or empty plasmids (5 nM; pcDNA3.1; Genomeditech)
using Lipofectamine® 3000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
At 24 h after transfection, the cells were incubated at
37˚C for 12 h and then exposed to H/R conditions, in
which H9c2 cells were subjected to hypoxia for 24 h in an incubator
at 37˚C (82% N2, 18% CO2 and <0.5%
O2) followed by reoxygenation for 1 h in an incubator at
37˚C (5% CO2, 21% O2) to simulate myocardial
cell injury).
Reverse transcription-quantitative PCR
(RT-qPCR)
After H/R induction, H9c2 cells were collected for
RT-qPCR analysis. Total RNA was extracted with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and its
concentration was measured at a wavelength of 260 nm by
spectrophotometry. In accordance with their respective
manufacturer's protocols, a high-Capacity cDNA Reverse
Transcription kit (Thermo Fisher Scientific, Inc.) and
SYBR™ Green quantitative kit (Thermo Fisher Scientific,
Inc.) were used to reverse transcribe RNA into cDNA and for qPCR
analysis, respectively, and GAPDH was used as the internal
reference. The primers used in the present study were as follows:
APPL1 forward, 5'-GCCCGCAGACAAGGTCTTTA-3' and reverse,
5'-TGAGGTCAGGTGTGTTGCTG-3'; TNF-α forward,
5'-TGAGCACAGAAAGCATGATC-3' and reverse,
5'-CATCTGCTGGTACCACCAGTT-3'; monocyte chemoattractant protein 1
(MCP-1) forward, 5'-TCCACCACTATGCAGGTCTC-3' and reverse,
5'-TGGACCCATTCCTTATTGGG-3'; IL-1β, forward,
5'-GACCTGTTCTTTGAGGCTGAC-3', and reverse,
5'-TCCATCTTCTTCTTTGGGTATTGTT-3'; and GAPDH forward,
5'-AGGGGCCATCCACAGTCTTC-3' and reverse, 5'-CAGTGCCAGCCTCGTCTCAT-3'.
The qPCR thermocycling conditions were as follows: Initial
denaturation at 94˚C for 3 min; followed by 34 cycles of annealing
at 55˚C for 45 sec and extension at 72˚C for 2 min; and a final
extension at 72˚C for 7 min. The results of the experiments were
analyzed using the 2-ΔΔCq method (22).
Cell counting kit-8 (CCK-8) assay
H9c2 cells were seeded into 96-well plates
(1x107 cells/ml). A total of 100 µl cell suspension was
added to each well. Following incubation, 10 µl CCK-8 solution
(GlpBio Technology) was added to each well and incubated for 2 h at
37˚C. The absorbance was measured at a wavelength of 450 nm with a
microplate reader (Thermo Fisher Scientific, Inc.).
Lactate dehydrogenase (LDH) assay
When cells undergo apoptosis or necrosis, the cell
membrane ruptures and intracellular LDH is released into the medium
(23). The cell supernatant was
collected after centrifugation at 400 x g at 4˚C for 5
min and LDH activity was detected using a LDH kit (cat. no.
ab65393; Abcam) according to the manufacturer's instructions. The
absorbance value of each well was measured at a wavelength of 450
nm using a microplate reader, and the activity levels of LDH were
quantitatively detected based on pyruvate levels. During the
experiment, three wells were used per experimental condition.
ROS staining
H9c2 cells (1x106 cells/well) were seeded
into 6-well plates and treated with H/R (three wells for each
experimental condition). A total of 1 µl ROS probe (cat. no.
HY-D0940; MedChemExpress) was added for a 30 min incubation at
37˚C. The culture medium was discarded, and the cells were rinsed
twice with PBS. Four random fields of view were selected and imaged
using a fluorescence microscope (Olympus Corporation;
magnification, x200). The fluorescence intensity was detected using
ImageJ 1.52r software (National Institutes of Health).
TUNEL assay
The apoptotic cells were stained using the TUNEL kit
(cat. no. C1086; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. H9c2 cells were collected, washed
with PBS and subsequently fixed using 4% paraformaldehyde at room
temperature for 30 min. Following incubation with 0.3% Triton X-100
for 5 min at room temperature, the cells were further incubated
with TUNEL solution at 37˚C for 60 min. DAPI (0.1 µg/ml) was added
to counterstain nuclei at room temperature in the dark for 5 min.
Four random fields were selected for analysis and the apoptotic
cells was observed using a fluorescence microscope (Olympus
Corporation; magnification, x200).
Statistical analysis
SPSS 22.1 software (IBM Corp.) was used for analysis
of all data. GraphPad Prism 7.0 (GraphPad Software, Inc.) was used
for production of the relevant graphs. The data are presented as
the mean ± SD. Each experiment was at least repeated three times.
One-way ANOVA was used for comparisons among multiple groups,
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
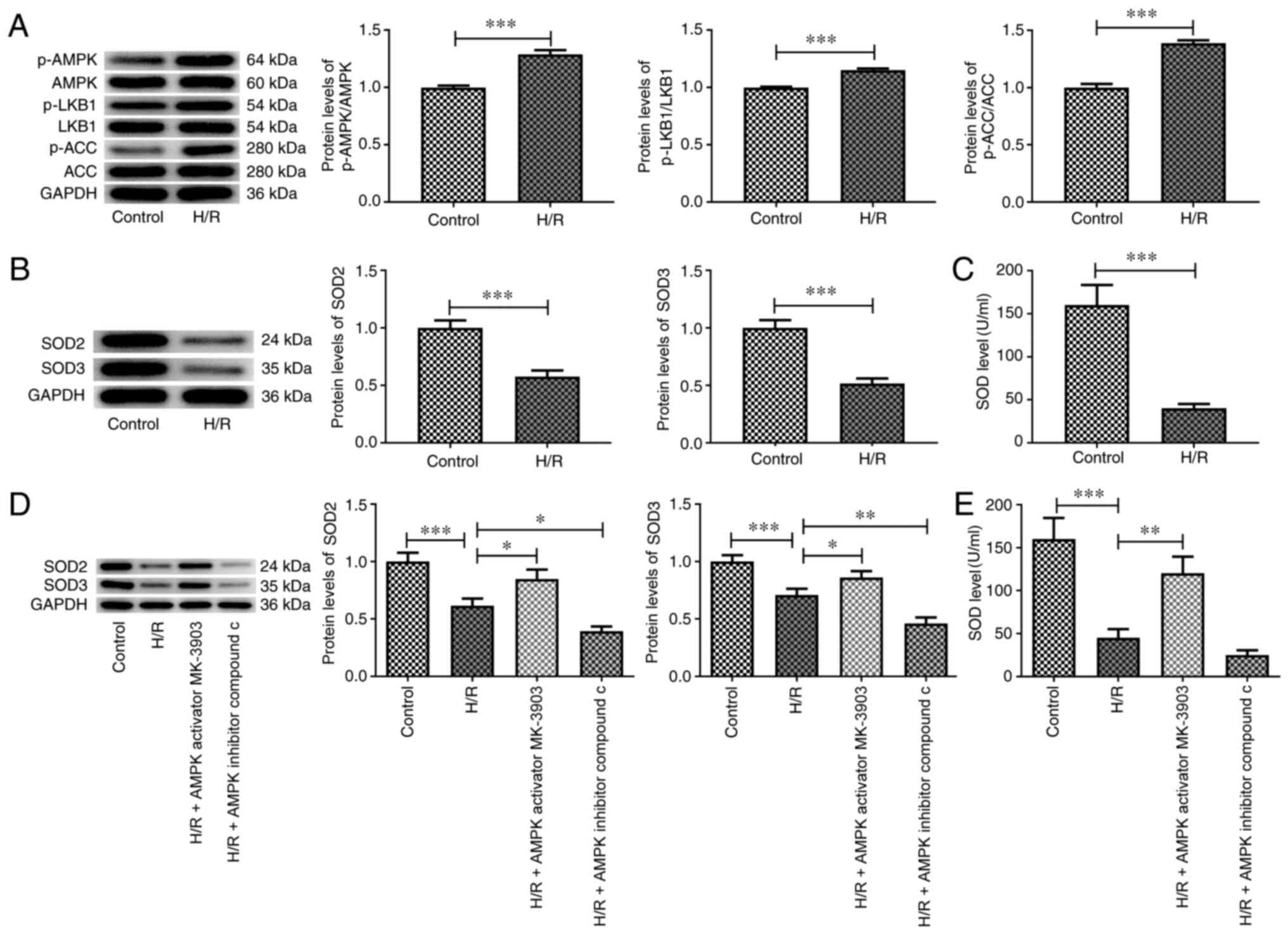
H/R affects the AMPK signaling pathway
and the expression levels of antioxidant enzymes
AMPK activation has been reported to serve a
protective role in decreasing the death of myocardial cells induced
by I/R (4,24). The expression levels of the total
and phosphorylated forms of I/R-associated proteins, including
AMPK, LKB1 and ACC, were assessed using western blot analysis. The
phosphorylation levels of these proteins were significantly
increased in H/R-treated H9c2 cells compared with those in the
control group (Fig. 1A).
Furthermore, the expression levels of SOD2 and SOD3, and SOD
activity were significantly decreased by H/R treatment compared
with those in the control group (Fig.
1B and C). It has been reported
that AMPK/GSK3β-Nrf2 is implicated in affecting oxidative stress
(25,26). To verify the effects of the AMPK
signaling pathway on the expression of the antioxidant enzymes,
MK-3903 or compound C were added to the cells. The results
indicated that the expression levels of SOD2 and SOD3, and SOD
activity were tightly regulated by the AMPK signaling pathway
(Fig. 1D and E).
APPL1 overexpression upregulates the
expression levels of LKB1/AMPK/ACC signaling pathway-related
proteins
The protein and mRNA expression levels of APPL1 were
detected by western blot analysis and RT-qPCR, respectively. The
results revealed that the mRNA and protein expression levels of
APPL1 were significantly downregulated following H/R exposure in
H9c2 cells compared with those in the control group (Fig. 2A and B). To explore the mechanism of action
underlying APPL1, this protein was overexpressed in H9c2 cells with
or without H/R induction (Fig.
2C-F). The phosphorylation levels of LKB1, AMPK and ACC were
increased after APPL1 overexpression compared with the H/R and
Ov-NC cotreatment group (Fig. 2G).
In addition, the expression levels of SOD2 and SOD3, and levels of
SOD activity were also significantly increased by APPL1
overexpression compared with the H/R and Ov-NC cotreatment group
(Fig. 2H and I).
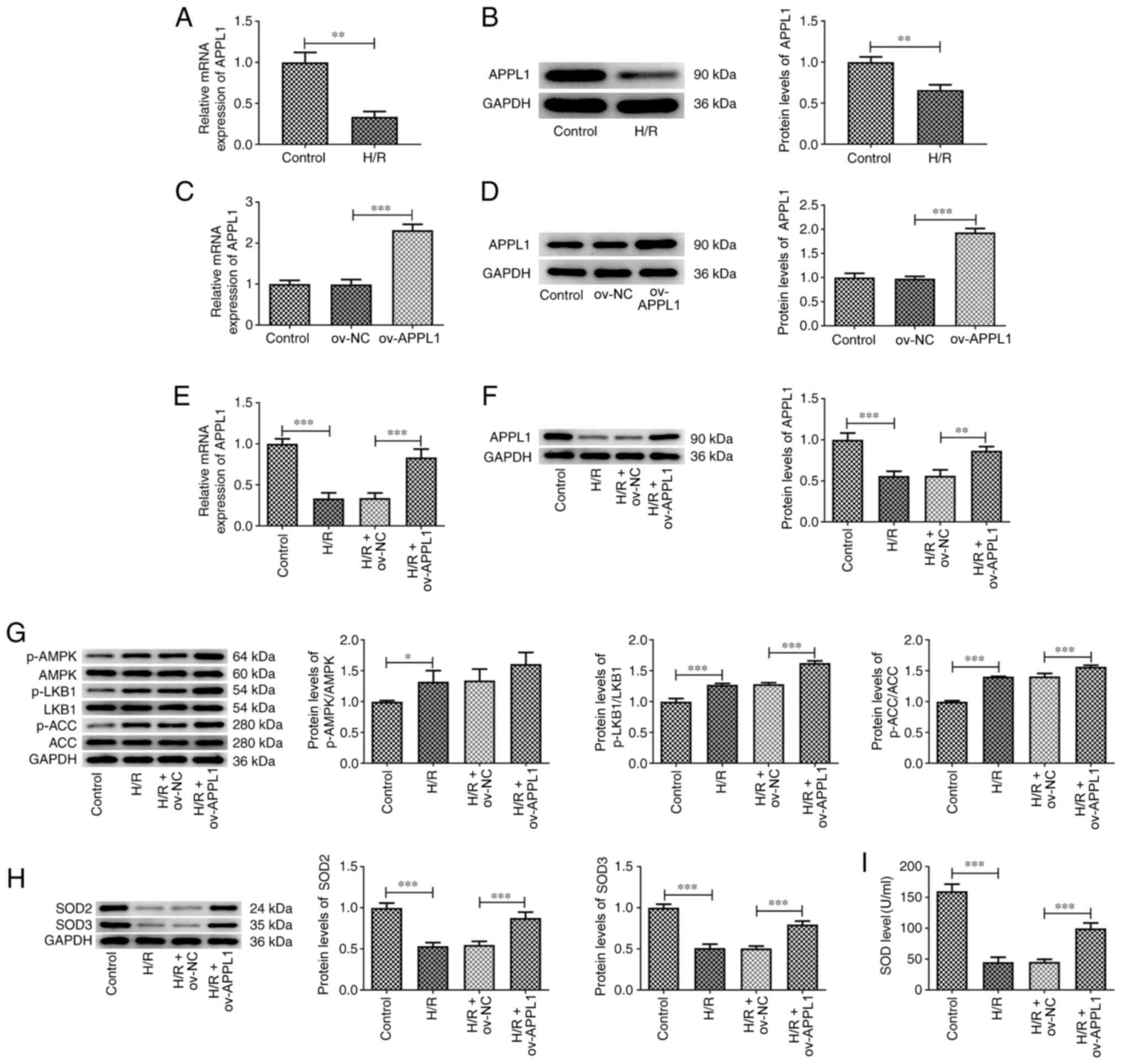 | Figure 2APPL1 overexpression increases the
phosphorylation levels of LKB1, AMPK and ACC, as well as SOD
levels. (A) mRNA and (B) protein expression levels of APPL1 in
H/R-induced H9c2 cells. (C) mRNA and (D) protein expression levels
of APPL1 in H9c2 cells with or without ov-APPL1 transfection. (E)
mRNA and (F) protein expression levels of APPL1 in H/R-induced H9c2
cells overexpressing APPL1. Determination of the expression levels
of (G) p-LKB1, LKB1, p-AMPK, AMPK, p-ACC, ACC, (H) SOD2 and SOD3.
(I) Determination of the activity levels of SOD. The experimental
data are presented as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. APPL1, adaptor
protein phosphotyrosine interacting with PH domain and leucine
zipper 1; ACC, acetyl-CoA carboxylase; H/R, hypoxia-reoxygenation;
SOD, superoxide dismutase; LKB1, liver kinase B1; AMPK,
AMP-activated protein kinase; p, phosphorylated; ov,
overexpression; NC, negative control. |
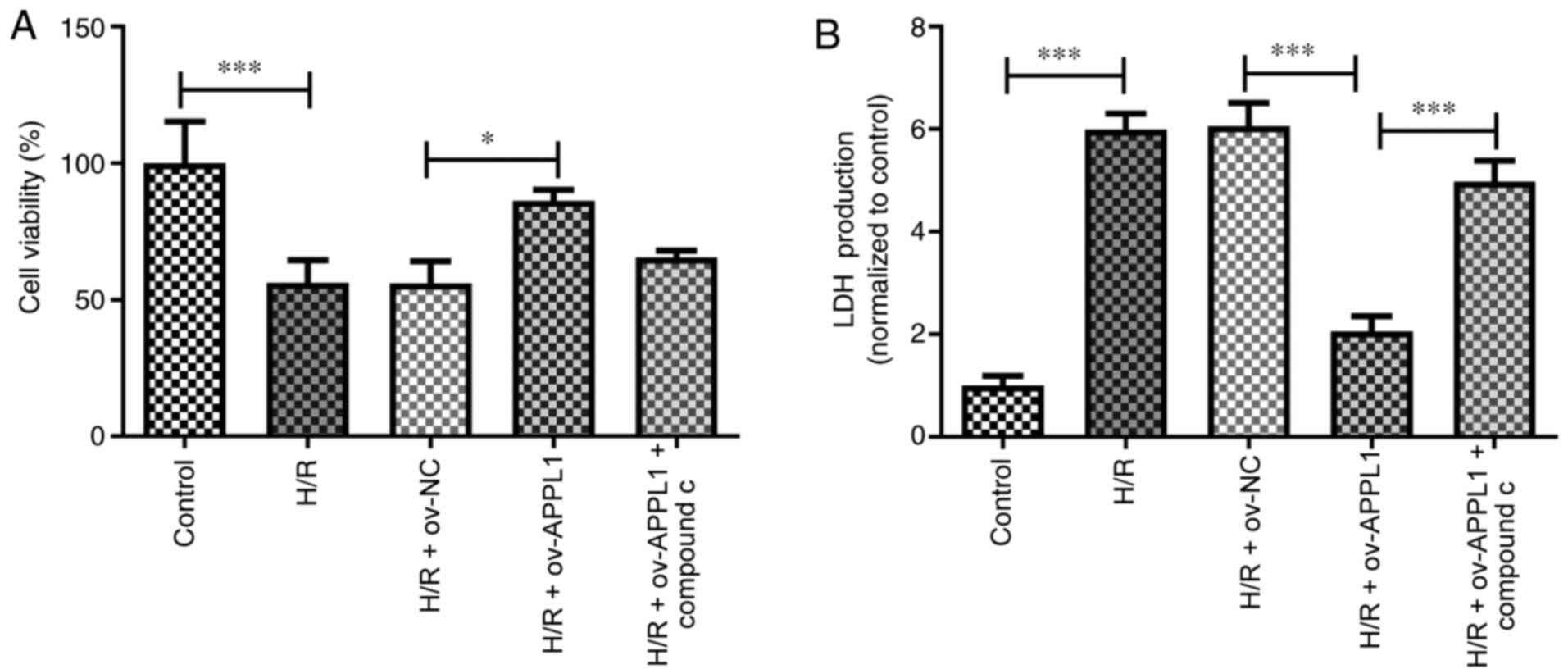
APPL1 increases cell viability and
reduces LDH release via the AMPK signaling pathway
To assess whether AMPK activation was associated
with APPL1 overexpression in H/R-treated H9c2 cells, compound C was
used to inhibit the activation of AMPK. Cell viability was detected
using a CCK-8 assay. Following H/R treatment, the viability of H9c2
cells was significantly decreased compared with that in the control
group. When the cells were transfected with ov-APPL1 plasmids for
12 h, the results of cell viability indicated that APPL1
overexpression significantly reduced H/R-induced cell injury
compared with the H/R + ov-NC group, displaying no apparent
cytotoxic effect (Fig. 3A). When
cardiomyocytes are damaged, they release several different proteins
into the bloodstream, such as LDH. The release of LDH from H9c2
myocardial cells was significantly increased in the H/R group
compared with that in the control group (Fig. 3B). However, the effect of APPL1
overexpression on cell viability and LDH release was significantly
counteracted by inhibiting AMPK via co-treatment of the cells with
compound C.
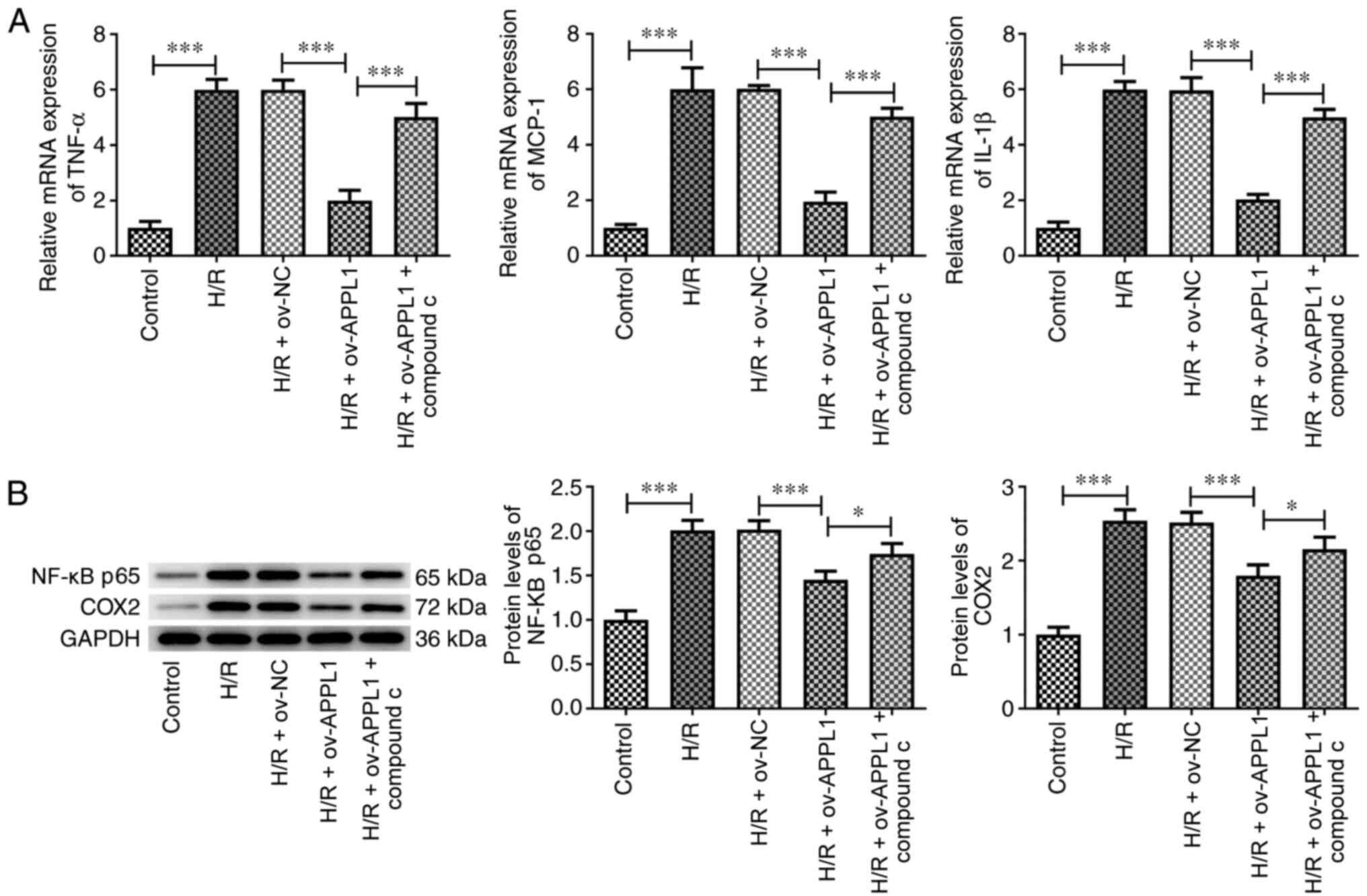
APPL1 overexpression reduces the
expression levels of proinflammatory markers via AMPK
signaling
APPL1 overexpression significantly decreased the
expression levels of the proinflammatory markers TNF-α, MCP-1 and
IL-1β in H/R-induced H9c2 cells, which was significantly reversed
following compound C treatment (Fig.
4A). Similar effects were also observed after H/R treatment and
APPL1 overexpression in the expression levels of p65 and COX2
(Fig. 4B), indicating that APPL1
could regulate the NF-κB signaling pathway via AMPK.
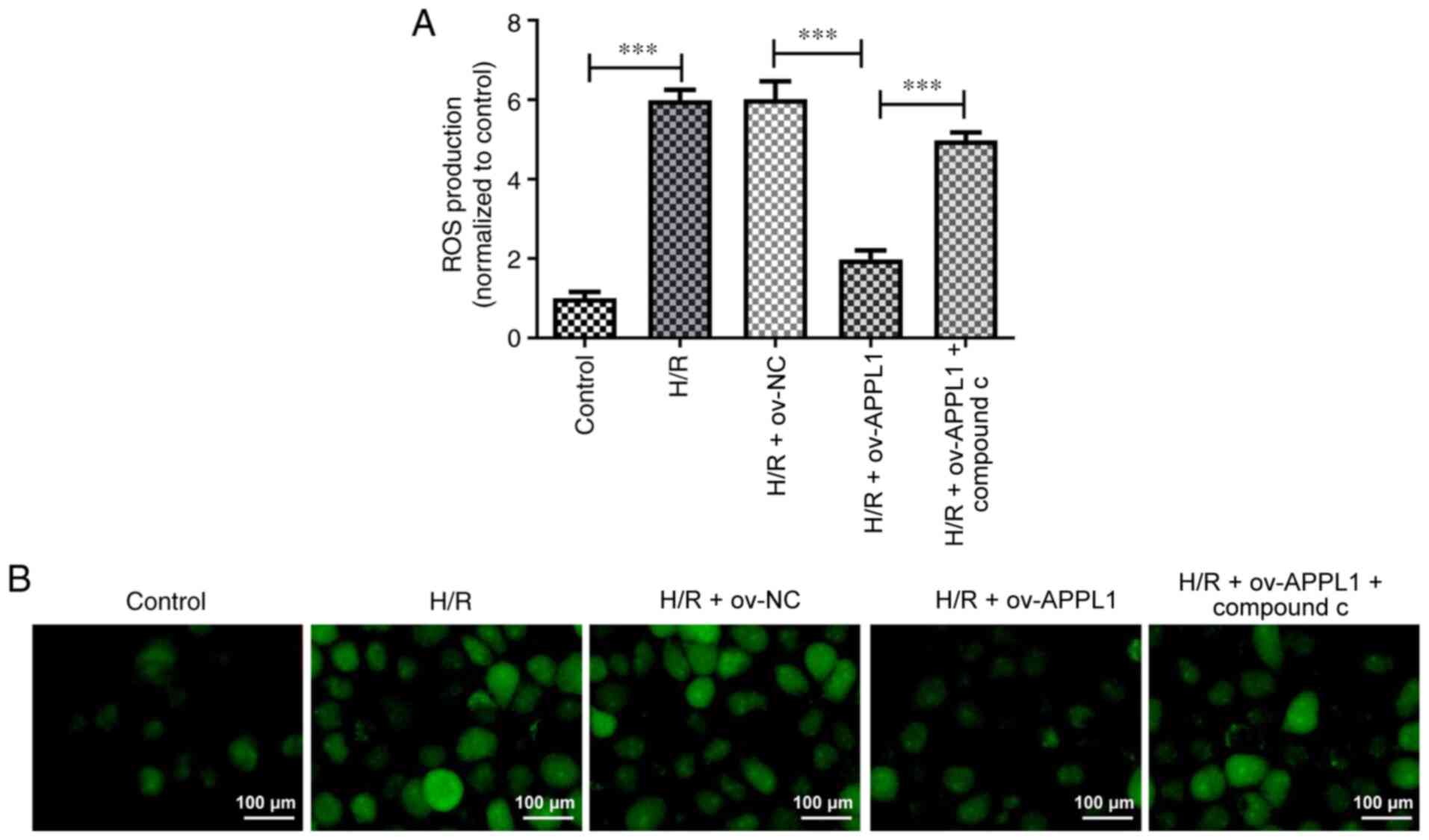
APPL1 overexpression decreases ROS
regeneration via the AMPK signaling pathway
Previous studies have indicated that H/R can induce
ROS production and oxidative damage, leading to cardiomyocyte death
and apoptosis (27,28). In the present study, it was further
revealed that ROS production was significantly decreased by APPL1
overexpression in H/R-induced H9c2 cells, as determined by ROS
staining (Fig. 5A and B). However, this effect was significantly
inhibited by compound C treatment.
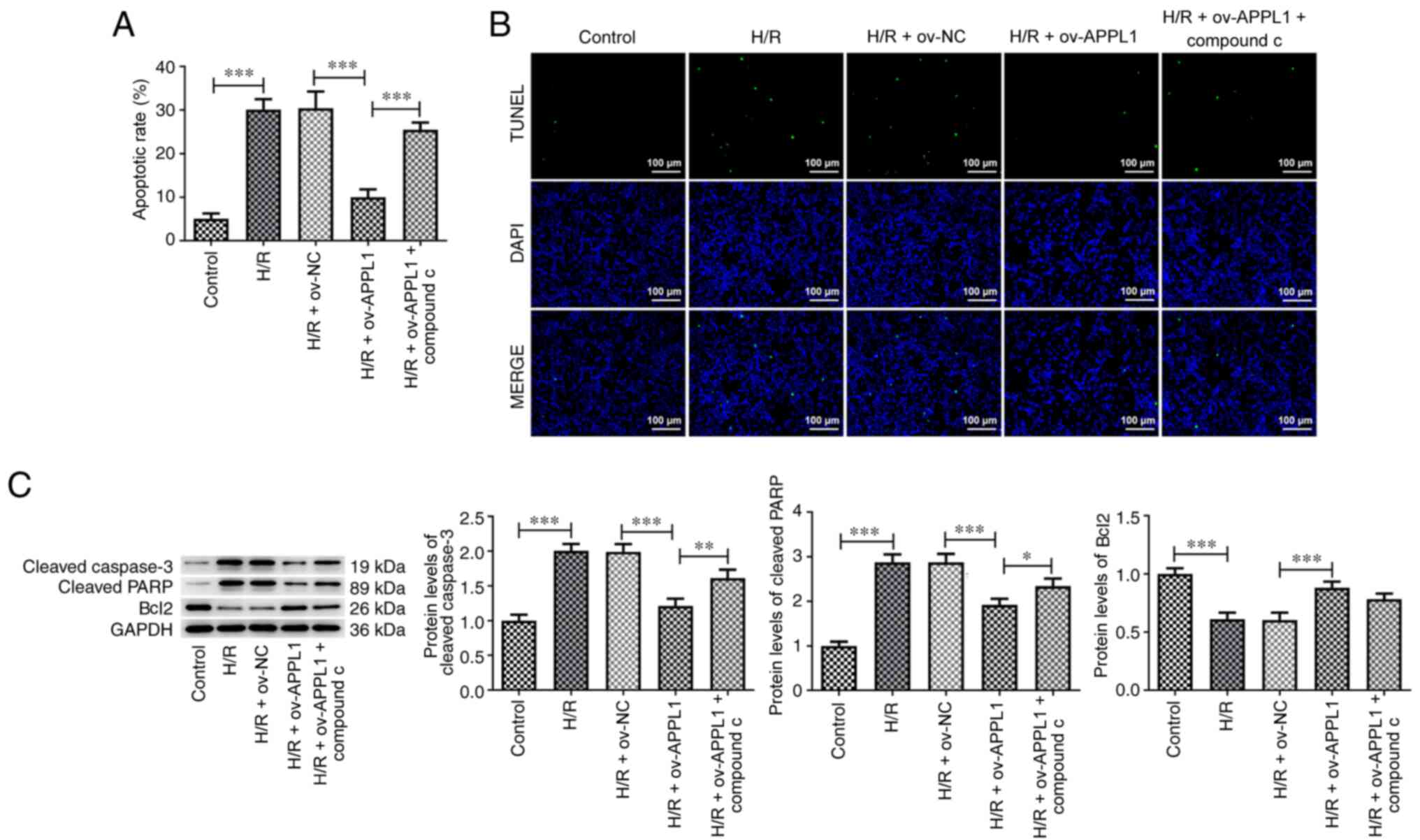
APPL1 overexpression suppresses cell
apoptosis induced by H/R
TUNEL staining was used to detect the apoptosis of
H9c2 cells. The apoptotic rate of the ov-APPL1 group was
significantly reduced compared with that of the H/R + ov-NC group.
Western blot analysis was used to detect the expression levels of
apoptosis-associated proteins. The data demonstrated that the
expression levels of cleaved caspase-3 and cleaved PARP were
significantly reduced, whereas the expression levels of Bcl2 were
significantly increased following APPL1 overexpression in
H/R-induced H9c2 cells. This effect was reversed by compound C
treatment (Fig. 6C); however, no
effect was observed on Bcl-2 expression.
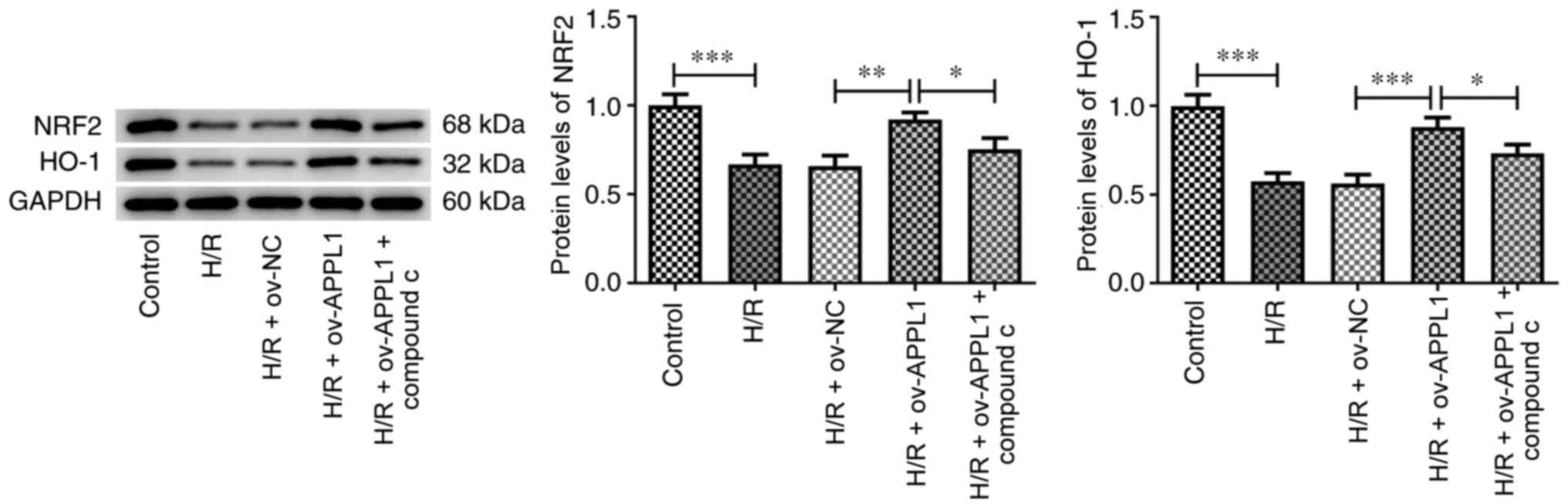
APPL1 overexpression upregulates the
expression of NRF2
Previous studies have reported that the activation
of AMPK causes a further activation of the NRF2 signaling pathway
and protects cells from oxidative damage via NRF2 (26,29).
Therefore, the current study assessed whether APPL1 could activate
the NRF2 signaling pathway in H/R-induced myocardial cells via AMPK
signaling. Nrf2 could directly regulate the promoter activities of
HO-1, a vital antioxidase that is involved in myocardial ischemia
reperfusion injury. Compared with those in the control group, the
expression levels of NRF2 and HO-1(30) were significantly reduced by H/R
treatment and these effects were significantly recovered by APPL1
overexpression (Fig. 7). However,
compound C significantly inhibited the effects of APPL1
overexpression in H/R-induced H9c2 cells. These results indicated
that APPL1 activated the AMPK signaling pathway, thereby enhancing
NRF2 signaling.
Discussion
The activation of ACC catalyzes the synthesis of
malonyl CoA, which induces the transport of long chain fatty
acyl-CoA into the mitochondria for β-oxidation. This process can be
modulated by AMPK (31). The
AMPK/ACC signaling pathway is markedly altered in myocardial I/R as
reported by previous studies (32,33).
The present study provided evidence of this in H9c2 cells exposed
to H/R conditions, which was in agreement with the aforementioned
studies. The phosphorylation levels of AMPK, LKB1 and ACC were
markedly increased following overexpression of APPL1. The
activation of the AMPK signaling pathway has been reported to
mediate the cardioprotective effects of carvedilol (33). Therefore, APPL1 overexpression was
indicated to serve a protective role in HR-induced H9c2 cells.
Further experimental results from the present study confirmed that
SOD2 and SOD3 protein expression levels were increased along with
increased cell viability and decreased LDH production following
overexpression of APPL1 in H/R-induced H9c2 cells. It has also been
previously demonstrated that SOD reduces ROS generation induced by
myocardial I/R and decreases oxidative stress-induced injury
(34,35).
During myocardial I/R, the number of
TUNEL+ cells and the levels of TNF-α, MCP-1 and IL-1β
are notably increased (34). In the
present study, this effect was also demonstrated in H9c2 cells
induced by H/R, which was significantly inhibited by APPL1
overexpression. In addition, compound C reversed these effects of
APPL1 overexpression in H/R-induced H9c2 cells, suggesting that the
suppression of the levels of the proinflammatory markers following
APPL1 overexpression was mediated by AMPK. Similar results were
also observed for NF-κB p65 and COX2. It has been reported that the
inflammatory response caused by the induction of H/R is improved
via AMPK activation, which modulates the JNK-mediated NF-κB
signaling pathway (36). A decrease
in ROS levels has been indicated to be associated with suppressing
the induction of cell apoptosis and reducing the levels of
apoptosis-associated markers in I/R (34). The present study demonstrated that
APPL1 overexpression inhibited ROS production in H/R-induced H9c2
cells, which could lead to the suppression of H/R-induced
apoptosis. This effect was significantly relieved by compound C
treatment. The activation of the Nrf2 signaling pathway serves a
protective role in improving cardiac function and decreasing
myocardial infarct size, which in turn coordinates the upregulation
of antioxidant and anti-inflammatory cellular mechanisms (37). Moreover, in the present study, the
levels of Nrf2 and HO-1 were significantly increased by APPL1
overexpression following H/R, and these effects were suppressed by
compound C treatment, demonstrating the regulation of the Nrf2
signaling pathway by AMPK. The AMPK/Nrf2 signaling pathway is
widely involved in I/R-induced injury, such as cerebral I/R and
myocardial I/R (38-41).
Taken together, the data indicated that APPL1 inhibited oxidative
stress and H/R-induced myocardial injury via the AMPK signaling
pathway. However, whether the regulatory mechanism underlying APPL1
in the AMPK signaling pathway is also present in vivo or in
other cell lines after I/R or H/R still requires further
investigation, which was a limitation of the present study. The
present study indicated that APPL1 may be considered as a potential
therapeutic target for myocardial H/R injury.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Jiangsu
Provincial Key Research and Development Program (grant no.
BE2018611).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, WL, TW and DZ made substantial contributions to
the conception and design of the current study, performed the
experiments, interpreted the data and drafted and revised the
manuscript for important intellectual content. YC and DZ confirmed
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chi GC, Kanter MH, Li BH, Qian L, Reading
SR, Harrison TN, Jacobsen SJ, Scott RD, Cavendish JJ, Lawrence JM,
et al: Trends in acute myocardial infarction by race and ethnicity.
J Am Heart Assoc. 9(e013542)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rout A, Tantry US, Novakovic M, Sukhi A
and Gurbel PA: Targeted pharmacotherapy for ischemia reperfusion
injury in acute myocardial infarction. Expert Opin Pharmacother.
21:1851–1865. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu Y, Liu H and Wang X: Cardioprotection
of pharmacological postconditioning on myocardial
ischemia/reperfusion injury. Life Sci. 264(118628)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chang JC, Lien CF, Lee WS, Chang HR, Hsu
YC, Luo YP, Jeng JR, Hsieh JC and Yang KT: Intermittent hypoxia
prevents myocardial mitochondrial Ca2+ overload and cell
death during ischemia/reperfusion: The role of reactive oxygen
species. Cells. 8(564)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Mitochondrial bioenergetics and cardiolipin
alterations in myocardial ischemia-reperfusion injury: Implications
for pharmacological cardioprotection. Am J Physiol Heart Circ
Physiol. 315:H1341–H1352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ouchi N, Shibata R and Walsh K:
Cardioprotection by adiponectin. Trends Cardiovasc Med. 16:141–146.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Feng Y, Lu Y, Liu D, Zhang W, Liu J, Tang
H and Zhu Y: Apigenin-7-O-β-d-(-6"-p-coumaroyl)-glucopyranoside
pretreatment attenuates myocardial ischemia/reperfusion injury via
activating AMPK signaling. Life Sci. 203:246–254. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu Z, Chen JM, Huang H, Kuznicki M, Zheng
S, Sun W, Quan N, Wang L, Yang H, Guo HM, et al: The protective
effect of trimetazidine on myocardial ischemia/reperfusion injury
through activating AMPK and ERK signaling pathway. Metabolism.
65:122–130. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen
Y and Fu Z: Melatonin attenuates myocardial ischemia-reperfusion
injury via improving mitochondrial fusion/mitophagy and activating
the AMPK-OPA1 signaling pathways. J Pineal Res.
66(e12542)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cao C, Liu HM, Li W, Wu Y, Leng Y, Xue R,
Chen R, Tang LH, Sun Q, Xia Z, et al: Role of adiponectin in
diabetes myocardial ischemia-reperfusion injury and ischemic
postconditioning. Acta Cir Bras. 35(e202000107)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fang H and Judd RL: Adiponectin regulation
and function. Compr Physiol. 8:1031–1063. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shibata R, Sato K, Pimentel DR, Takemura
Y, Kihara S, Ohashi K, Funahashi T, Ouchi N and Walsh K:
Adiponectin protects against myocardial ischemia-reperfusion injury
through AMPK- and COX-2-dependent mechanisms. Nat Med.
11:1096–1103. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Potenza MA, Sgarra L, Nacci C, Leo V, De
Salvia MA and Montagnani M: Activation of AMPK/SIRT1 axis is
required for adiponectin-mediated preconditioning on myocardial
ischemia-reperfusion (I/R) injury in rats. PLoS One.
14(e0210654)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Park M, Youn B, Zheng XL, Wu D, Xu A and
Sweeney G: Globular adiponectin, acting via AdipoR1/APPL1, protects
H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One.
6(e19143)2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng
Q, Wang Y, Yuan Y, Wang X, Tao L, et al: Reduced cardioprotective
action of adiponectin in high-fat diet-induced type II diabetic
mice and its underlying mechanisms. Antioxid Redox Signal.
15:1779–1788. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Farías JG, Molina VM, Carrasco RA, Zepeda
AB, Figueroa E, Letelier P and Castillo RL: Antioxidant therapeutic
strategies for cardiovascular conditions associated with oxidative
stress. Nutrients. 9(966)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016(1656450)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gonçalves AF, Polegato BF, Fernandes AA,
Ishikawa LL, Okoshi K, Bazan SGZ, Minicucci MF, Azevedo PS, Ikoma
MR, Penitenti M, et al: Zinc supplementation attenuates cardiac
remodeling after experimental myocardial infarction. Cell Physiol
Biochem. 50:353–362. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yin H, Wu M and Jia Y: Knockdown of IL-32
protects PC12 cells against oxygen-glucose
deprivation/reoxygenation-induced injury via activation of
Nrf2/NF-κB pathway. Metab Brain Dis. 35:363–371. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sivandzade F, Prasad S, Bhalerao A and
Cucullo L: NRF2 and NF-қB interplay in cerebrovascular and
neurodegenerative disorders: Molecular mechanisms and possible
therapeutic approaches. Redox Biol. 21(101059)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhao D, Sun Y, Tan Y, Zhang Z, Hou Z, Gao
C, Feng P, Zhang X, Yi W and Gao F: Short-duration swimming
exercise after myocardial infarction attenuates cardiac dysfunction
and regulates mitochondrial quality control in aged mice. Oxid Med
Cell Longev. 2018(4079041)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang X, Yang L, Kang L, Li J, Yang L,
Zhang J, Liu J, Zhu M, Zhang Q, Shen Y and Qi Z: Metformin
attenuates myocardial ischemia-reperfusion injury via up-regulation
of antioxidant enzymes. PLoS One. 12(e0182777)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lv H, Liu Q, Wen Z, Feng H, Deng X and Ci
X: Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute
lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox
Biol. 12:311–324. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lv H, Hong L, Tian Y, Yin C, Zhu C and
Feng H: Corilagin alleviates acetaminophen-induced hepatotoxicity
via enhancing the AMPK/GSK3β-Nrf2 signaling pathway. Cell Commun
Signal. 17(2)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guo W, Liu X, Li J, Shen Y, Zhou Z, Wang
M, Xie Y, Feng X, Wang L and Wu X: Prdx1 alleviates cardiomyocyte
apoptosis through ROS-activated MAPK pathway during myocardial
ischemia/reperfusion injury. Int J Biol Macromol. 112:608–615.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu C, Tang M, Zhang X, Li J and Cao G:
Knockdown of miR-665 protects against cardiomyocyte
ischemia/reperfusion injury-induced ros accumulation and apoptosis
through the activation of Pak1/Akt signaling in myocardial
infarction. Int Heart J. 61:347–354. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yu J, Wang WN, Matei N, Li X, Pang JW, Mo
J, Chen SP, Tang JP, Yan M and Zhang JH: Ezetimibe attenuates
oxidative stress and neuroinflammation via the AMPK/Nrf2/TXNIP
pathway after MCAO in rats. Oxid Med Cell Longev.
2020(4717258)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhao Z, Tang Z, Zhang W, Liu J, Li B and
Ding S: Inactivated pseudomonas aeruginosa protects against
myocardial ischemia reperfusion injury via Nrf2 and HO-1. Exp Ther
Med. 19:3362–3368. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Folmes CD and Lopaschuk GD: Role of
malonyl-CoA in heart disease and the hypothalamic control of
obesity. Cardiovasc Res. 73:278–287. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mu F, Duan J, Bian H, Zhai X, Shang P, Lin
R, Zhao M, Hu D, Yin Y, Wen A and Xi M: Metabonomic strategy for
the evaluation of chinese medicine salvia miltiorrhiza and
dalbergia odorifera interfering with myocardial
ischemia/reperfusion injury in rats. Rejuvenation Res. 20:263–277.
2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hu H, Li X, Ren D, Tan Y, Chen J, Yang L,
Chen R, Li J and Zhu P: The cardioprotective effects of carvedilol
on ischemia and reperfusion injury by AMPK signaling pathway.
Biomed Pharmacother. 117(109106)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bae S, Park M, Kang C, Dilmen S, Kang TH,
Kang DG, Ke Q, Lee SU, Lee D and Kang PM: Hydrogen
peroxide-responsive nanoparticle reduces myocardial
ischemia/reperfusion injury. J Am Heart Assoc.
5(e003697)2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hadi NR, Al-Amran F, Yousif M and Zamil
ST: Antiapoptotic effect of simvastatin ameliorates myocardial
ischemia/reperfusion injury. ISRN Pharmacol.
2013(815094)2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen X, Li X, Zhang W, He J, Xu B, Lei B,
Wang Z, Cates C, Rousselle T and Li J: Activation of AMPK inhibits
inflammatory response during hypoxia and reoxygenation through
modulating JNK-mediated NF-κB pathway. Metabolism. 83:256–270.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shen Y, Liu X, Shi J and Wu X: Involvement
of Nrf2 in myocardial ischemia and reperfusion injury. Int J Biol
Macromol. 125:496–502. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hou X, Fu M, Cheng B, Kang Y and Xie D:
Galanthamine improves myocardial ischemia-reperfusion-induced
cardiac dysfunction, endoplasmic reticulum stress-related
apoptosis, and myocardial fibrosis by suppressing AMPK/Nrf2 pathway
in rats. Ann Transl Med. 7(634)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Mo Y, Zhu JL, Jiang A, Zhao J, Ye L and
Han B: Compound 13 activates AMPK-Nrf2 signaling to protect
neuronal cells from oxygen glucose deprivation-reoxygenation. Aging
(Albany NY). 11:12032–12042. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wu WY, Li YD, Cui YK, Wu C, Hong YX, Li G,
Wu Y, Jie LJ, Wang Y and Li GR: The natural flavone acacetin
confers cardiomyocyte protection against hypoxia/reoxygenation
injury via AMPK-mediated activation of Nrf2 signaling pathway.
Front Pharmacol. 9(497)2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhou F, Wang M, Ju J, Wang Y, Liu Z, Zhao
X, Yan Y, Yan S, Luo X and Fang Y: Schizandrin A protects against
cerebral ischemia-reperfusion injury by suppressing inflammation
and oxidative stress and regulating the AMPK/Nrf2 pathway
regulation. Am J Transl Res. 11:199–209. 2019.PubMed/NCBI
|