Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disease that primarily affects joints and other organ systems,
often disabling the patient (1). RA
pathogenesis lies in the excessive and continuous activation of
effector cells by self-antigens, which causes progressive
inflammation and damages tissues and organs (2). Regulatory T cells (Tregs) are primary
immunosuppressive cells that keep immunity balanced and maintain
tolerance to self-antigens (3). It
is assumed that dysfunction of these cells contributes to RA
pathogenesis (4). The quantitative
data currently available on Treg populations in RA are
contradictory, due to a high level of heterogeneity in disease
duration and therapy approaches in patients with RA (5-7).
There is no consensus on the suppressor activity of Treg cells in
RA either, which may be due to research on the topic being limited
(8-11).
The present study sought to analyze the Treg subpopulation and
functional activity in RA.
Treg cells provide a broad range of regulatory
mechanisms by contact or humoral interactions (3). For that reason, the present study
investigated not only their ability to suppress the proliferation
of responder cells, but also the expression of CTLA-4 and PD-L1
molecules involved in contact suppression. The Treg ability to
produce regulatory cytokines, TGF-β and IL-10, was also studied. In
addition, the researchers studied the Treg migration potential via
CCR4 expression (12) and the
extent of their activation by HLA-DR expression (13,14).
Given that T-cell receptors (TCRs) are involved in the
trans-endocytosis of CD80/86 molecules with CTLA-4 involvement
(15), to the best of our
knowledge, the present study was the first to propose using CD86
molecules as a marker of the Treg/antigen presenting cell (APC)
interaction intensity, which may be representative of the contact
suppressor activity of Treg cells against a specific range of
antigens.
An increasing amount of data is available on the
negative effects of converting Treg cells in RA-associated
inflammation, including decreasing FoxP3 expression and triggering
the expression of the RORyt transcription factor to Th17 cells. For
example, exFoxP3 lymphocytes have stronger osteoclastogenic
potential compared with that in Th17 originating from Th0; they are
less susceptible to Treg suppression (16,17).
In addition, exFoxP3-Th17 lymphocytes play a key role in the
inflammation and articular damage in RA, due to the intense
production of inflammatory cytokines, IL-17, IL-21 and
IL-22(18). To effectively assess
the Treg subpopulation and their ability to differentiate to Th17
cells, the present study counted transitional
FoxP3+RORyt+ lymphocytes and
CD4+RORyt+ lymphocytes in the peripheral
blood of RA patients compared with that in the donor group. As
RORyt is the main transcriptional factor of Th17 cells, then
bi-positive FoxP3+RORyt+ and
CD4+RORyt+ lymphocytes may have potential for
IL-17 expression (19), and might
be involved in RA pathogenesis. Thus, the present study evaluated
Treg cells in terms of their suppressor potential, the extent of
activation and the degree of differentiation into pathogenic
exFoxP3-Th17 lymphocytes.
Materials and methods
Patients
The present study used peripheral blood samples from
22 conditionally healthy donors (controls) and 21 patients affected
by oligo- or polyarticular RA, with low, moderate and high disease
activity (20); the duration of
disease was 7.1±4.1 years on average. Disease activity was
evaluated by the Disease Activity Score (DAS)-28 criterion
(21), which is an indicator that
takes into account erythrocyte sedimentation rate (ESR) and number
of inflamed or painful joints, as well as a subjective assessment
of the patient's state of health through a patient global
assessment based on patient reported outcomes (22). Additionally, ESR, level of
C-reactive Protein (CRP), and level of rheumatoid factor (RF) were
evaluated in the laboratory. Blood was sampled in cases of
manifestation of RA or exacerbation upon hospitalization of each
patient to the immunopathology unit. The general patient group
according to disease duration included: Patients with new-onset RA
[disease-modifying anti-rheumatic drugs (DMARD)-naïve] 7 persons;
and patients on DMARD therapy, 14 persons. DMARDs included basic
commonly accepted treatments, methotrexate and glucocorticoids.
Donor and patient groups were comparable in sex and age; the donors
were aged 52±11 years; the patients were aged 51±16 years (Table I). RA was diagnosed following the
1987 American College of Rheumatology criteria (23).
 | Table IClinical parameters of patients with
RA and healthy donors. |
Table I
Clinical parameters of patients with
RA and healthy donors.
| | | Patients with
RA |
|---|
| Parameter | Healthy donors | Receiving DMARD
therapy | New-onset |
|---|
| Sex |
|
Male | 11 | 5 | 2 |
|
Female | 11 | 9 | 5 |
| Mean age ± SD,
years | 52±11 | 55±13 | 45±14 |
| Mean CRP ± SD,
mg/dl | - | 17.4±12.7 | 22.8±14.2 |
| Mean ESR ± SD,
mm/h | - | 34.8±13.4 | 32.29±14.2 |
| Mean RF ± SD,
IU/ml | - | 372.7±216.1 | 386.7±219.1 |
| Mean DAS-28 ±
SD | - | 3.6±2.1 | 3.9±2.4 |
| Duration of
treatmenta | - | 6.2±4.5 years | <2 weeks |
Patients were recruited between November 2018 and
December 2019 at the Clinic of Immunopathology of the Research
Institute of Fundamental and Clinical Immunology (RIFCI;
Novosibirsk, Russia).
The exclusion criteria were as follows: i) Pregnancy
or lactation; ii) an acute infection; iii) any vaccination within 3
months prior to the study; iv) any severe infection or somatic
pathology; an v) the use of any of genetically engineered
biological drugs. In all cases, blood was sampled upon written
voluntary informed consent and the local Ethics Committee's
Approval (protocol no. 110: October 11, 2018; RIFCI Ethics
Committee).
Flow cytometry
Peripheral blood mononuclear cells (PBMCs) were
obtained via Ficoll-Urografin (ρ=1,077 g/l) density gradient
centrifugation (Ficoll® was supplied by Biolot; cat.
1.2.8.1., Urografin was supplied by Bayer AG), at 1,153 RCF for 25
min at room temperature (24,25).
Phenotypic traits of Treg-cells
(CD3+CD4+CD25+FoxP3+)
from the PBMCs were tested using monoclonal antibodies: CD3 using
FITC (cat. no. 300406), CD4 using APC/Cy7 (cat. no. 300518), CD25
using APC (cat. no. 302610), FoxP3 using PE (cat. no. 320108),
CTLA-4 using PE/Cy7 (cat. no. 349914), PD-L1 using PE/Cy7 (cat. no.
329718), HLA-DR using PerCP (cat. no. 307628), CD86 using PE/Cy7
(cat. no. 305422), and CCR4 using PE/Cy7 (cat. no. 335405), which
were all supplied by BioLegend, Inc. These antibodies were used at
a volume of 5 µl per 106 cells. Cells were stained for
RORyt using PerCP (R&D Systems, Inc.; cat. no. IC6006C; 10 µl
per 106 cells). For intracellular staining, the present
study used a True-Nuclear Transcription Factor Buffer Set according
to the manufacturer's instructions (BioLegend, Inc.; cat. no.
424401). CTLA-4 expression analysis estimated the surface and total
(membrane + intracellular) expression of this molecule. When
studying the phenotypical traits of Treg-cells while experimentally
suppressing the proliferation of responder T cells, intracellular
markers were not used as the cultures were not rich in these cells.
The phenotype
(CD3+CD4+CD25+CD127lo)
was evaluated using the following monoclonal antibodies: CD3 using
PE/Cy7 (cat. no. 300420), CD4 using APC/Cy7 (cat. no. 300518), CD25
using PE (cat. no. 302606), CD127 using PerCP/Cy5.5 (cat. no.
351322), CTLA-4 using APC (cat. no. 349908), and PD-L1 using APC
(cat. no. 329708), which were all supplied by BioLegend, Inc., at a
volume of 5 µl per 106 cells. To assess the positive
population when testing the expression of the studied molecules,
the team applied fluorescence minus one controls, as well as
isotype controls to assess the expression of RORyt. Additionally,
the percentage of subpopulations of Treg cells with low and high
CD25 expression was evaluated (CD25loFoxP3+
and CD25hiFoxP3+ respectively). Mean
fluorescence intensity (MFI) was used for evaluation of expression
density of CTLA-4 and PD-L1 on the Treg surface. Flow cytometry was
performed on the BD FACS Canto II cytometer (BD Biosciences).
Treg suppressor activity
Treg functional activity was investigated by
evaluating the suppression of CD4+ and CD8+
proliferation in vitro upon stimulation. A pure Treg
population was extracted using immunomagnetic separation from PBMCs
by the Miltenyi Biotec Ltd. MACS Treg Isolation kit; the population
was isolated to have a
CD3+CD4+CD25+CD127low
phenotype. The purity of magnetic sorting was 93.2±4%, on average.
The Treg-depleted PBMC fraction was stained with a
carboxyfluorescein succinimidyl ester (CFSE) vital stain
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. Then, Treg cells were cultured with
PBMCs, at a 1:1 ratio (30,000 Treg per 30,000 PBMC) over 4 days, in
round-bottomed plates with RPMI-1640 medium (Sigma-Aldrich; Merck
KGaA), supplemented with 10% FCS (HyClone; Cytiva) and antibiotics
at 37˚C and 5% CO2. IL-2 (25 U/ml (BIOTECH, Ltd.)
coupled with anti-CD3 antibodies (0.25 µg/ml) (Sorbent), were used
as proliferation stimulants. The cells were also cultured without
stimulants as controls. CFSE-labeled PBMCs were cultured under the
same conditions without Treg cells. Supernatants were sampled on
day 3 to determine ELISA for IL-10 (Vector-Best) and TGFβ
(BioLegend, Inc.) concentrations as these are the main Treg
suppressor cytokines. CTLA-4 and PD-L1 expression on Treg cells was
calculated on day 4; the Treg suppression index was calculated for
CD4+ and CD8+ cells using the following
formula (26):
Statistical analysis
The data was analyzed using Statistica v6.0 (TIBCO
Software, Inc.) and two non-parametric methods, the Wilcoxon test
for dependent samples and the Mann-Whitney test for independent
samples, and one parametric method, a two-tailed Student's t-test.
For multiple independent groups (>2), ANOVA was used (the
Kruskal-Wallis test in case of non-parametric distribution),
followed by post hoc analysis (Tukey's and Dunn's multiple
comparison tests for parametric and non-parametric distribution,
respectively). The Shapiro-Wilk test was used for investigating
normality distribution. For correlation analysis, the Spearman's
rank correlation test was used. P<0.05 was considered to
indicate a statistically significant difference. Non-parametric
methods were used for descriptive statistics in cases of
non-parametric distribution: The median and interquartile ranges
were calculated at 25 and 75% percentiles. Parametric methods were
used for descriptive statistics in cases of Gaussian distribution:
Mean ± SD. Graphs were generated using GraphPad Prism (v7.03;
GraphPad Software, Inc.).
Results
Peripheral blood Treg subpopulations
in RA
To analyze the population of Treg cells in
peripheral blood, the total CD25+FoxP3+ pool
was calculated as a percentage, as were the subpopulations with low
and high CD25 expression: CD25lo and CD25hi,
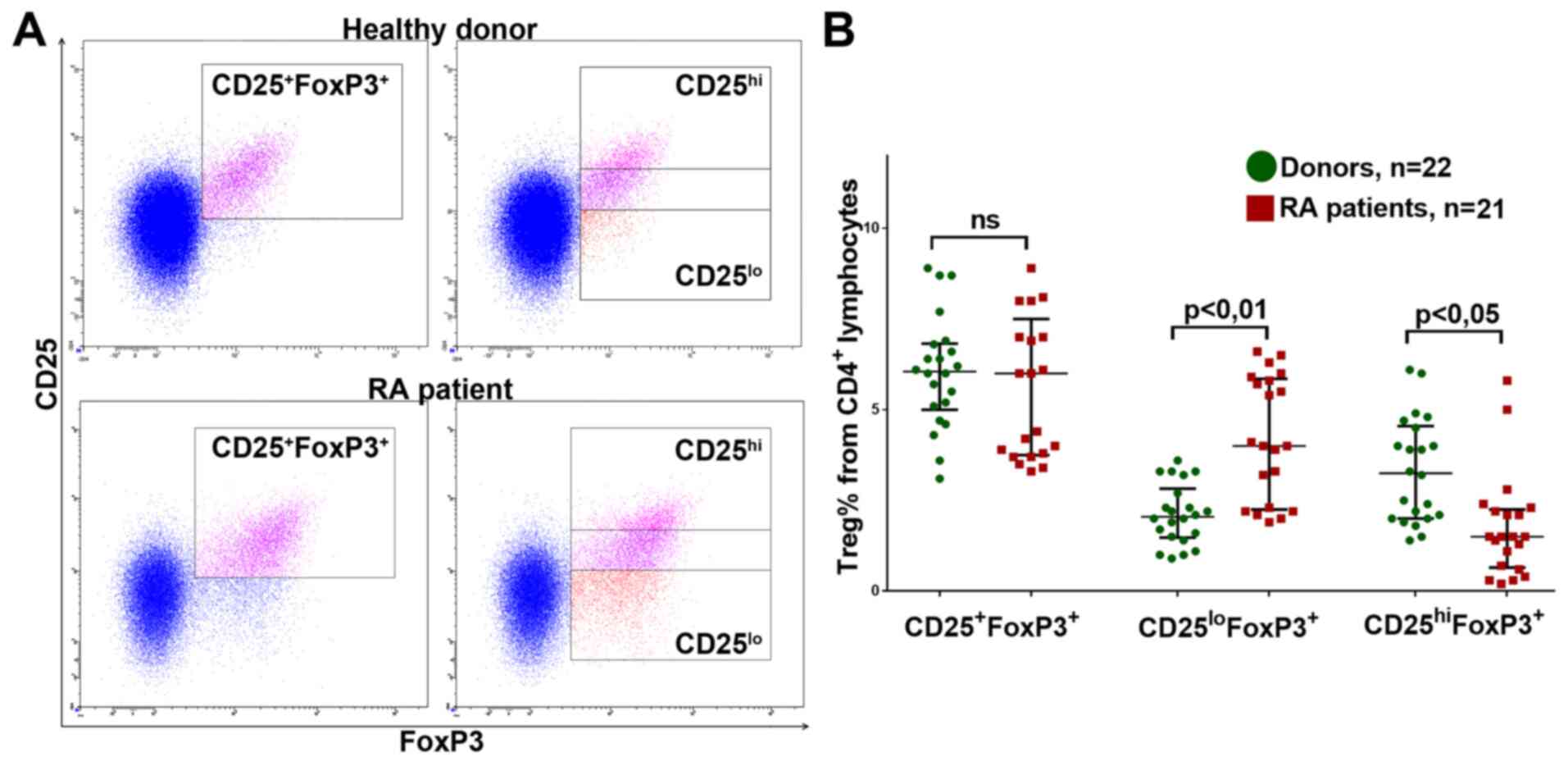
respectively (Fig. 1A).
The percentage of CD25+FoxP3+
Treg lymphocytes did not significantly differ from that in RA
patients in either the new-onset RA or DMRAD therapy groups (data
not shown). However, patients in both groups had significantly more
CD25loFoxP3+Tregs and significantly fewer
CD25hiFoxP3+Tregs (Fig. 1B; data shown for donors and the
general group of RA patients).
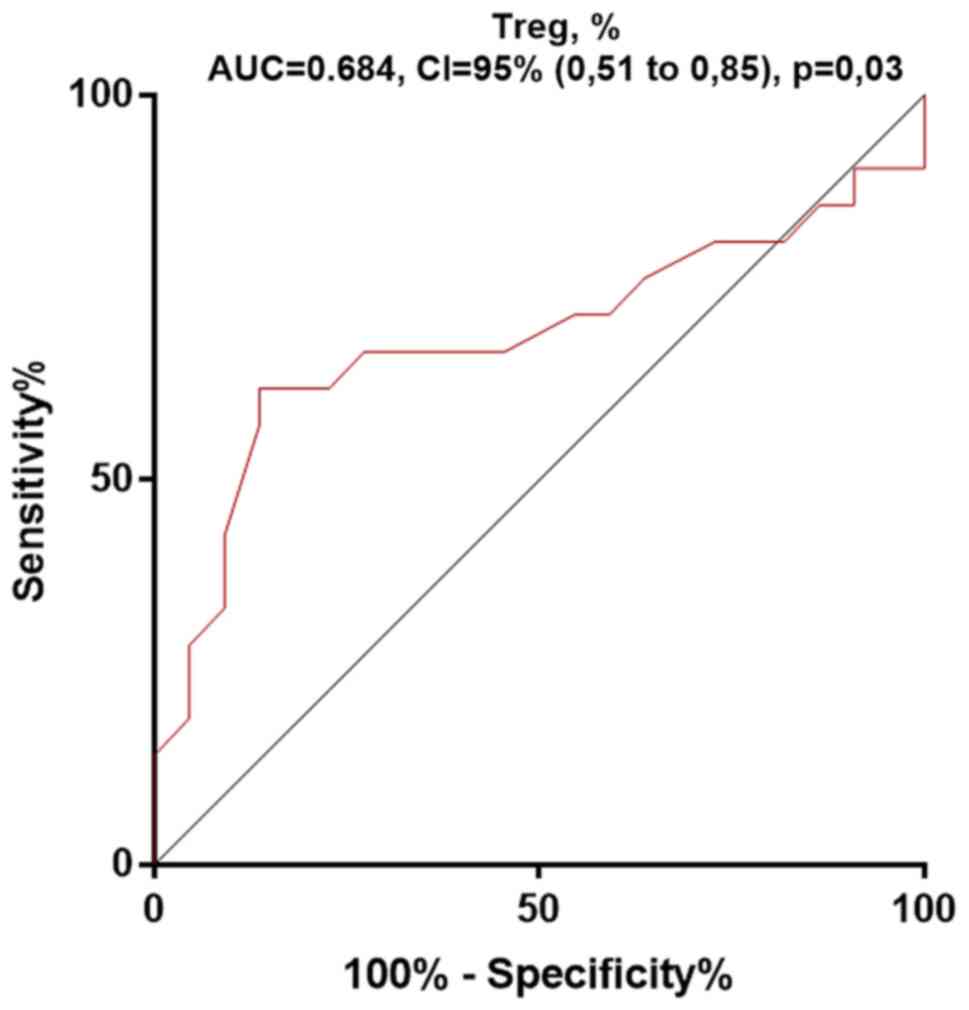
In addition, receiver operating characteristics
(ROC) curves were plotted for the Treg percentage (Fig. 2). The area under the ROC curve, of
the Treg percentage for the RA prediction was 0.68, showing its low
predictive ability (P=0.03). This confirmed the absence of a
significant difference between donors and RA patients in the
percentage of Tregs.
The IL-2R/Jak/STAT5 pathway is important to the
stable expression of FoxP3(27),
which might be disrupted by RA-related inflammation, due to the
effects of IL-6 and TGF-β (17), in
which case the RORyt transcription factor is expressed and Treg
cells differentiate into exFoxP3-Th17 lymphocytes (28). Thus, a lower expression of CD25
(IL-2R) might be associated with the conversion of Treg cells to
Th17. As a result, the expression of RORyt using CD4+
lymphocytes in the blood samples from donors and patients with RA,
as a marker of early differentiation to Th17 lymphocytes.
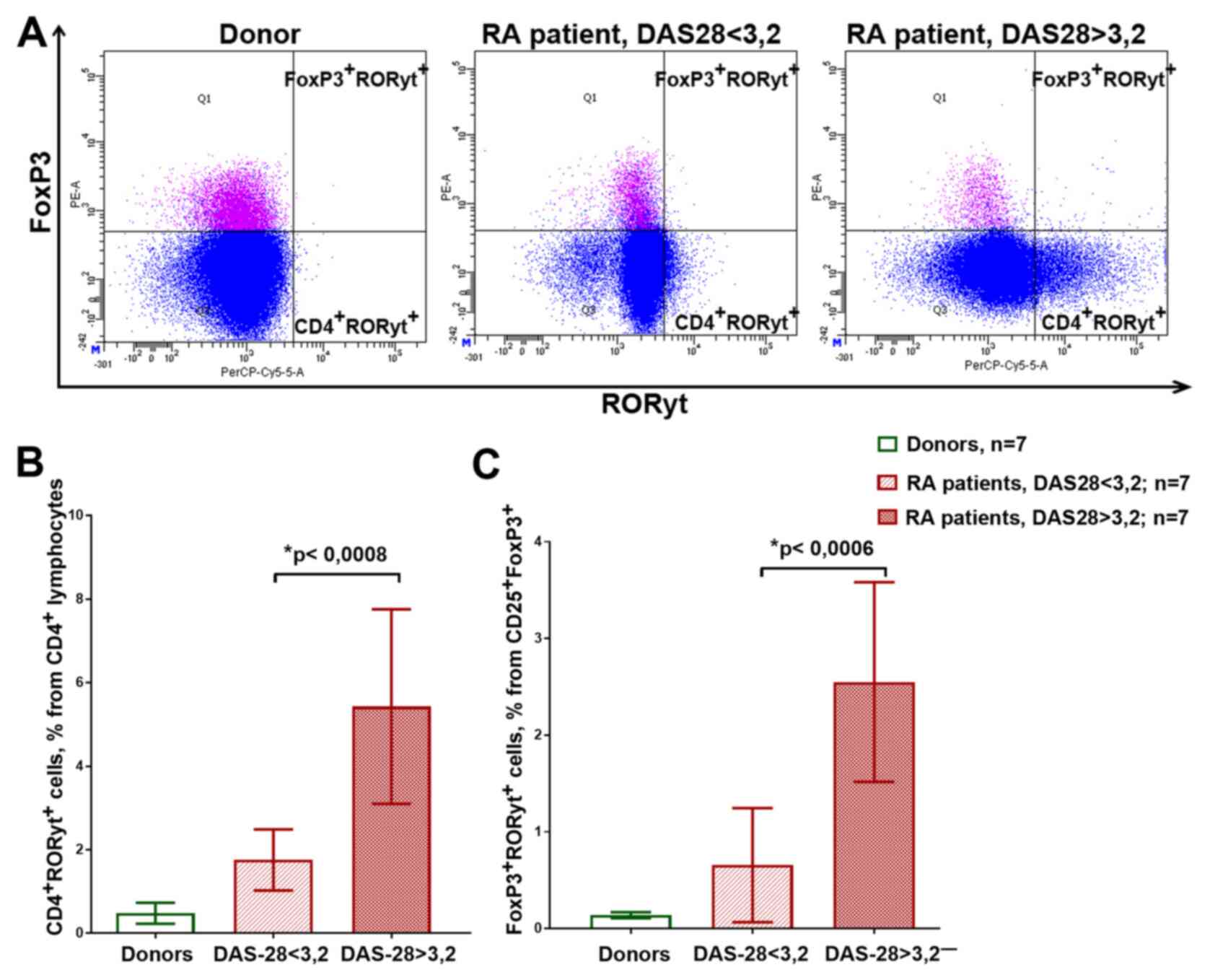
Fig. 3A shows the
examples of the flow cytometry plots reflecting the gating of the
CD4+RORyt+ and
FoxP3+RORyt+ cells. Patients with medium and
high disease activity had significantly more RORyt-expressing cells
compared with that in the healthy donors and patients with low
activity, both in CD4+ cells and in
CD25+FoxP3+Treg cells (Fig. 3B and C). At the same time,
FoxP3+RORyt+ had relatively low CD25
expression (data not shown).
RORyt is the main transcription factor for Th17
lymphocytes; therefore, FoxP3+RORyt+ cells
are the Treg-to-Th17 transition stage (29). Thus, Treg cells in the patients were
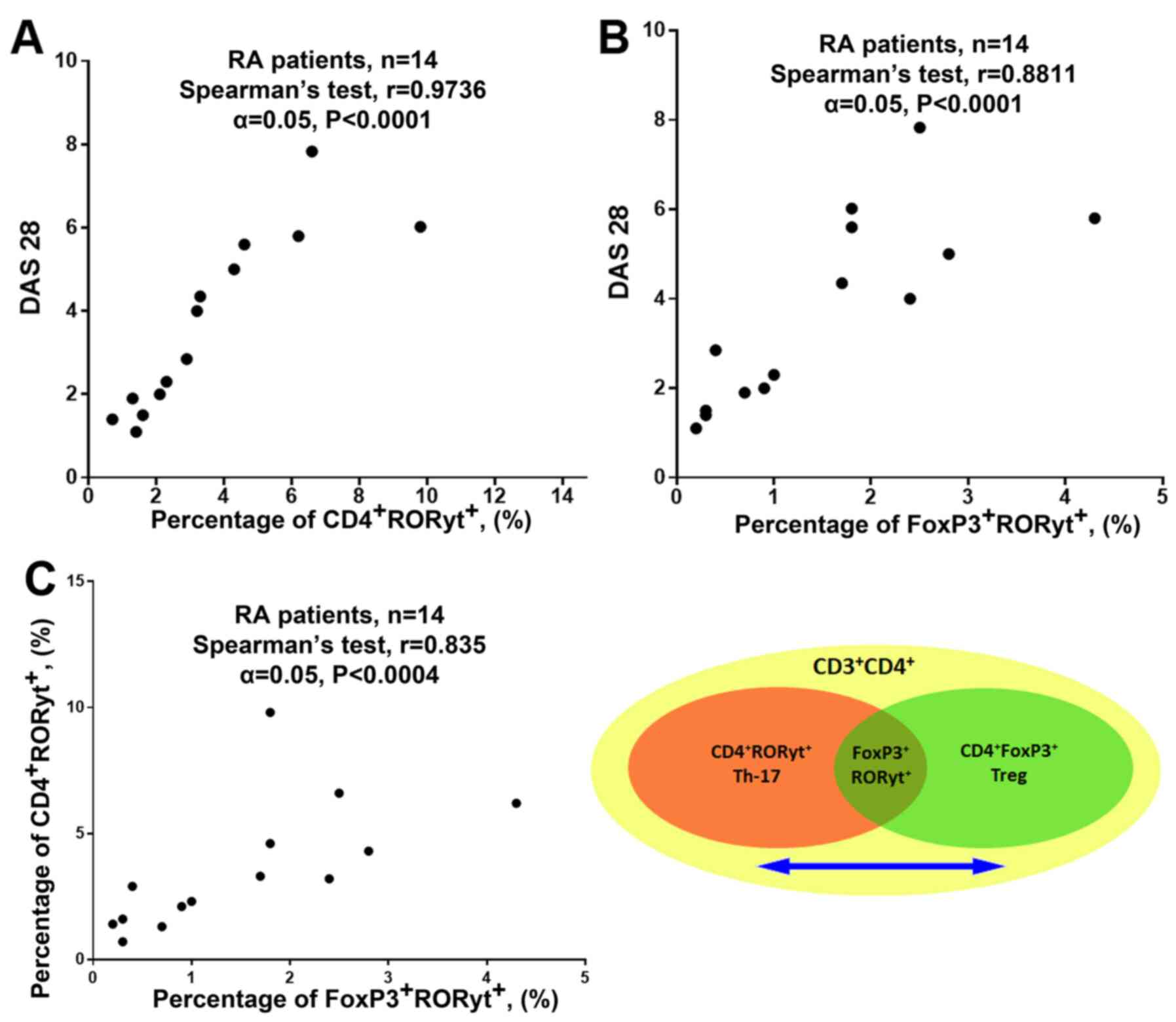
more prone to differentiate into Th17(29). Furthermore, a strong correlation was
found between the percentage of CD4+RORyt+
cells and DAS-28, and between the percentage of
FoxP3+RORyt+ cells and DAS-28, which
suggested that the process of transdifferentiation from Treg to
Th17 could be involved in the pathogenesis of RA (Fig. 4A and B; RA patients from the general group).
Additionally, a strong correlation was found between the percentage
of FoxP3+RORyt+ and
CD4+RORyt+ cells, which indicates
transdifferentiation between Treg cells and Th-17 lymphocytes.
(Fig. 4C; RA patients from the
general group) (30).
Thus, it was revealed that features of Treg cells in
patients with RA could contribute to the pathogenesis of the
disease by promotion and maintenance of inflammation. It is
important to note that some studies did not find IL-17 expression
in different Treg cell subsets, while Th17 cells expressing IL-17
were present in the blood (10,31).
This may be due to the expression of RORyt being initiated at the
early stage of Th17 cell differentiation and IL-17 being
undetectable in the transient stage of
FoxP3+RORyt+ cells (32).
Expression of functional molecules and
activation molecules on Treg-cells in RA
For the following experiments, 12 donors and 12
patients with RA (DAS-28 >3.2) were randomly selected from the
general group, who were comparable by sex and age, and the
expression of CD86, CTLA-4, PD-L1, HLA-DR and CCR4 (the gating
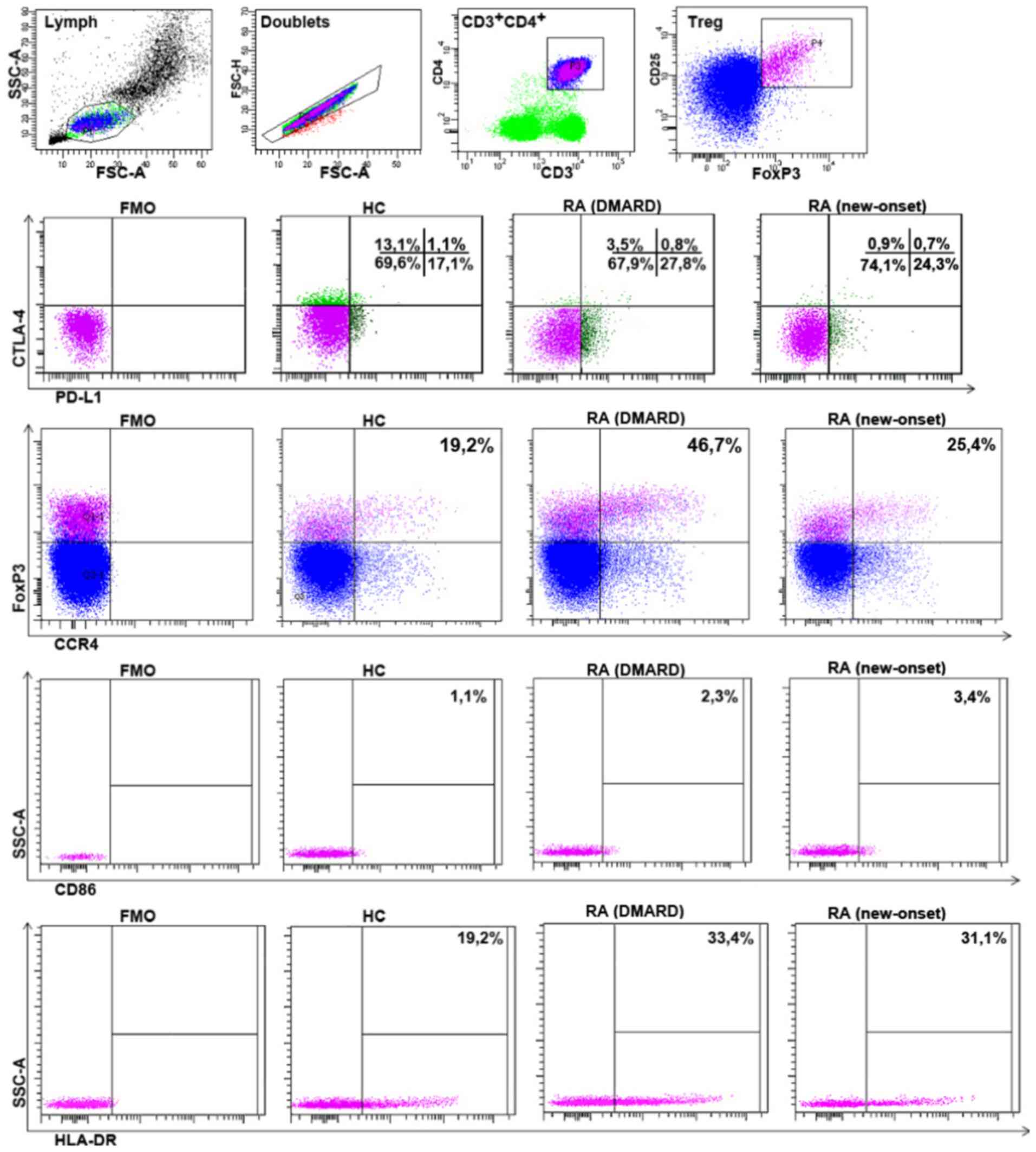
strategy is shown in Fig. 5), which
are involved in the functional activity of Treg cells (14), was investigated. A total of 6
patients had new-onset RA and six were receiving DMARD therapy.
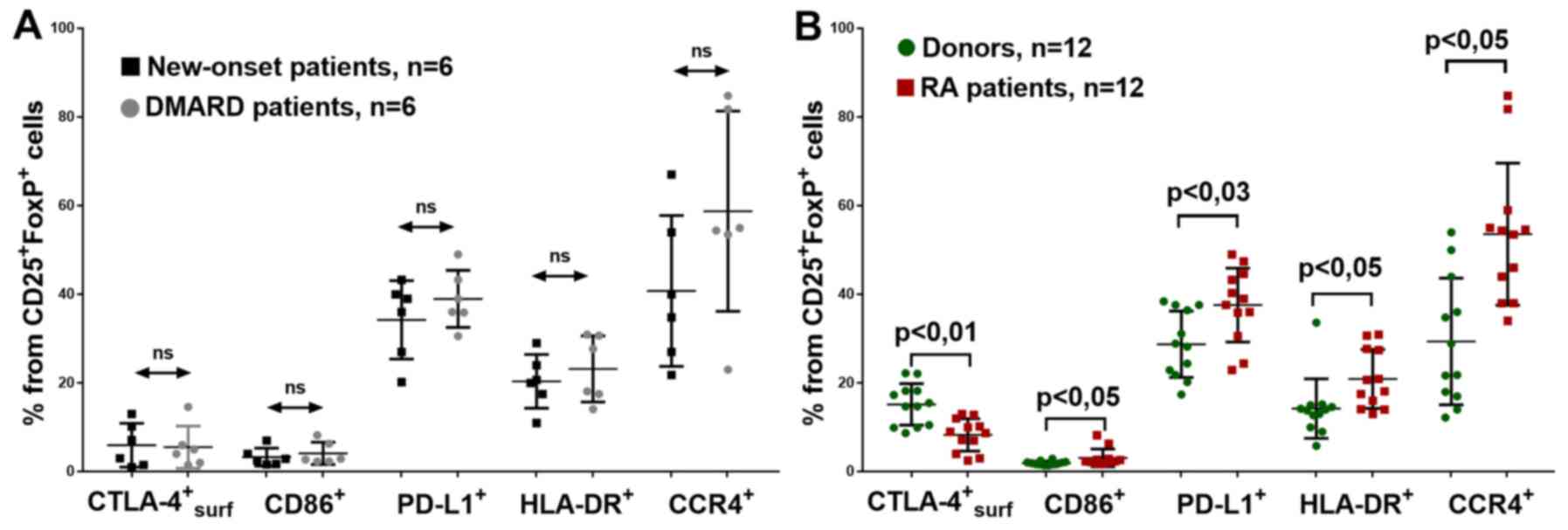
However, no significant differences were found between these RA
groups using expression density (MFI) of these molecules or the
percentage of Treg cells, as determined by the positive expression
of CD86, CTLA-4, PD-L1, HLA-DR and CCR4 (Fig. 6A). Notably, with the patients with
RA receiving DMARD therapy, the percentage of CCR4+Treg
cells was slightly higher compared with that in the patients with
new-onset RA. This may be due to the different average durations of
the disease in these groups (6.2±4.5 years and <2 weeks
respectively).
Some differences were also found between donors and
patients with RA from the common group. Investigating CD86
expression on the surface of Treg, it was found that in patients
with RA, the percentage of
CD86+CD25+FoxP3+ cells was
significantly higher (Fig. 6B).
The percentage of Treg cells with surface CTLA-4
expression was significantly lower in the all patients with RA
compared with that in the healthy donors (Fig. 6B). The mean fluorescence, which is
indicative of the CTLA-4 expression density on cell surface, was
also significantly lower for the general patient group (data not
shown). However, at the same time, neither donors nor patients
differed significantly, with respect to total (surficial +
intracellular) CTLA-4 expression: 94.5 (range, 94-98%) vs. 96.8%
(range, 97-98%), respectively (data not shown). This may be
explained by the higher intensity of internalization of the CTLA-4
molecule, in complex with CD86, in patients with RA, and by the
exhaustion of the CTLA-4 mediated mechanism of Treg-suppression
(15).
Patients with RA also had significantly more
PD-L1-expressing Treg cells in peripheral blood (Fig. 6B). This could be due to the higher
CD86+Treg concentrations and was possibly associated
with the Treg activation in RA. To further investigate this issue,
the HLA-DR activation marker expression on Treg lymphocytes was
determined. The expression of this molecule was previously found to
be associated with elevated FoxP3 expression (14). HLA-DR+Treg lymphocytes
are activated inflammation-associated Treg cells predominant in
inflammation foci in RA, and they can recirculate and emerge in
peripheral blood. Such Treg cells have strong suppressive effects
against effector lymphocytes (13,33).
The present study identified an increased HLA-DR+Treg
percentage in all patients with RA (Fig. 6B).
To investigate the migration of Treg cells, the
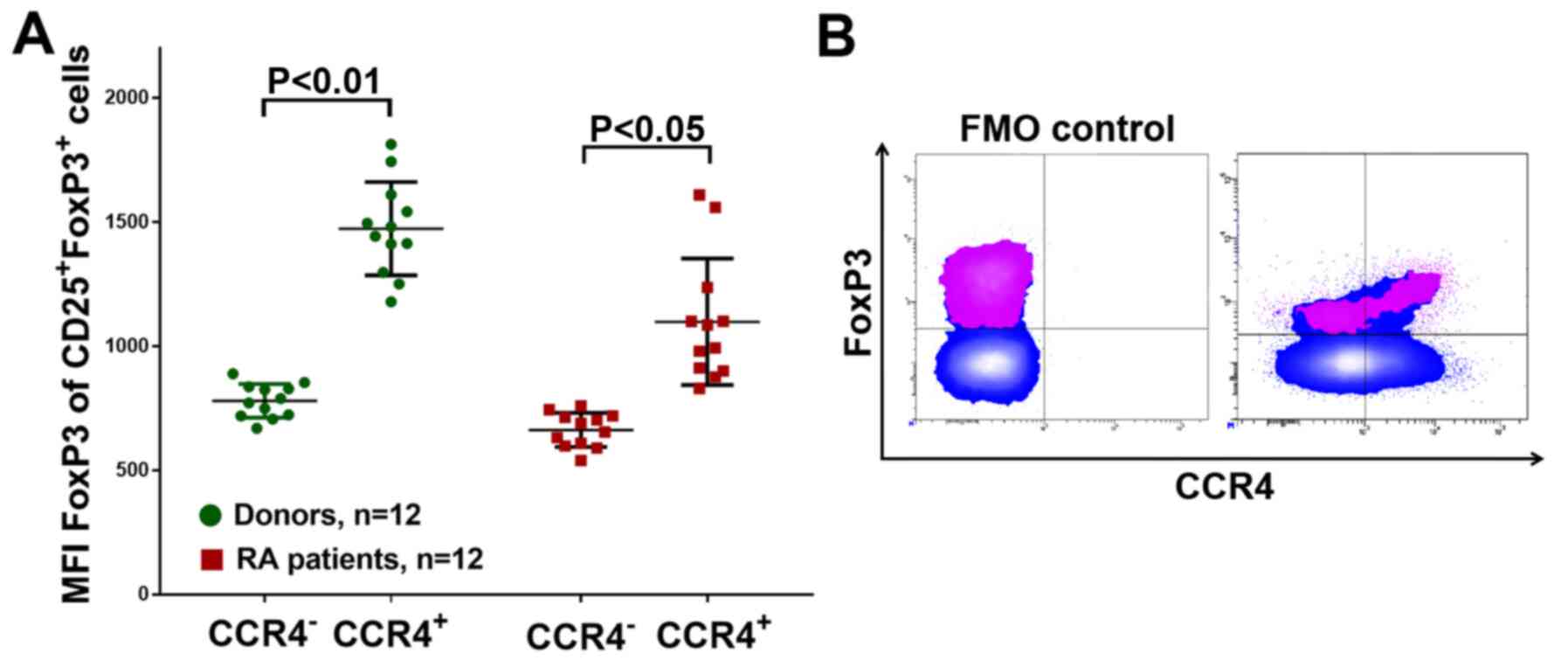
expression of CCR4 was investigated. Patients with RA had a higher
CCR4+Treg percentage (Fig.
6B), In addition, CCR4+Treg cells had a higher FoxP3
expression in either group, as determined by the mean of the
FoxP3-PE channel fluorescence intensity (Fig. 7), which might also indicate the
activation and greater functional activity of CCR4+Treg
in RA in addition to HLA-DR+Treg.
Thus, patients with RA had Treg cells with a lower
CTLA-4 expression; however, they had higher PD-L1, CD86, HLA-DR and
CCR4 expression.
Functional activity of Treg-cells in
RA
To investigate the Treg functionality and the key
mechanisms involved, the Treg ability to inhibit the proliferation
of CD4+ and CD8+ lymphocytes, the TGF-β and
IL-10 concentrations in the supernatant, and the CTLA-4/PD-L1
expression on the Treg membrane were determined. Patients with RA
were exactly matched to age- and sex-corresponded healthy donors
for this analysis. In addition, in the Treg suppression assay,
DMARD and new-onset RA patients were calculated as two separate
groups.
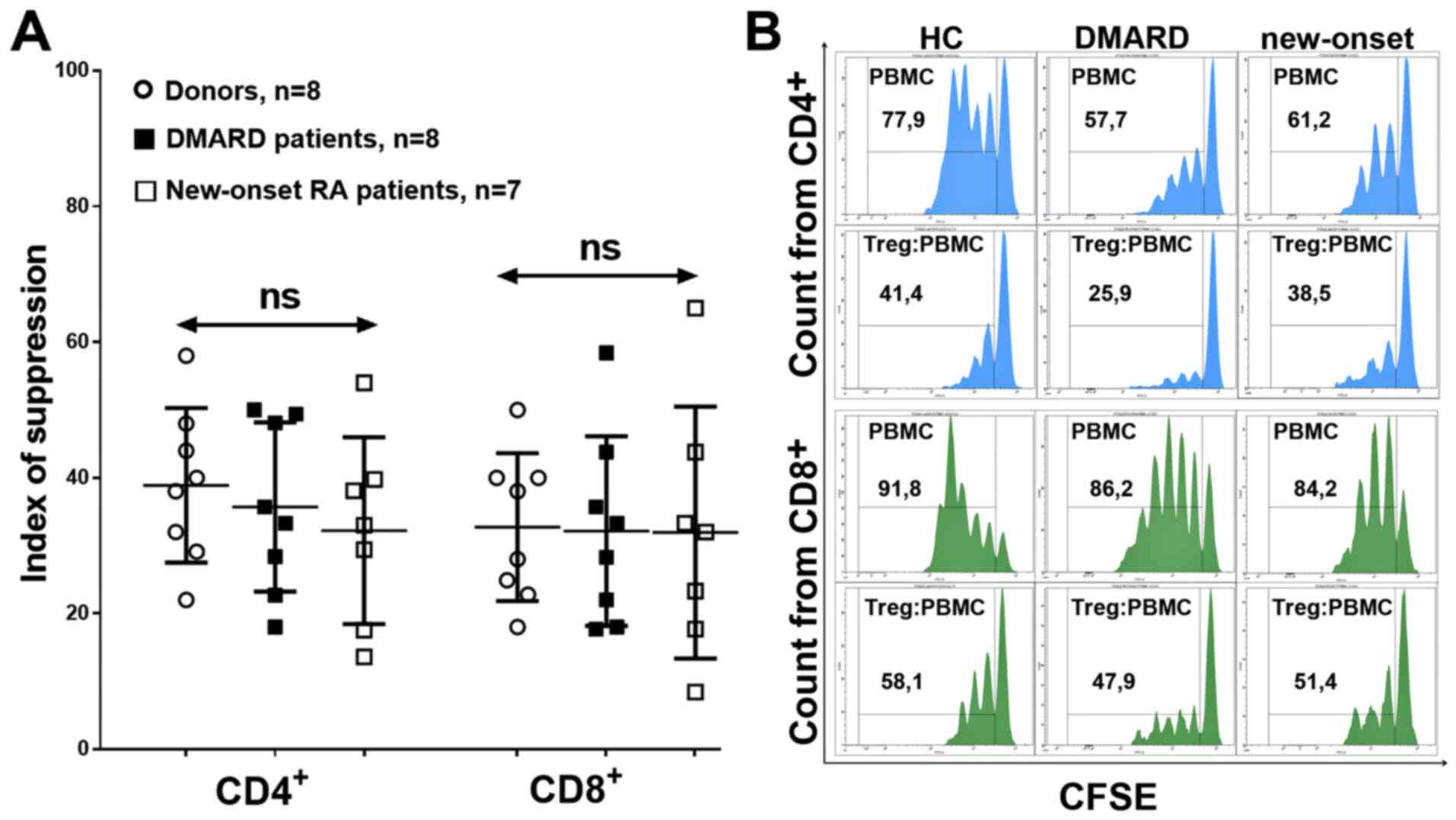
Suppression of proliferation was determined using
the suppressor index, which did not differ in donors or different
groups of patients, which indicated preserved suppressor activity
of the general Treg cell pool in RA, with respect to both
CD4+ and CD8+ (Fig. 8).
As the suppression activity of Treg cells did not
differ between patients with RA and receiving DMARD therapy and
patients with new-onset RA, the expression of CTLA-4/PD-L1 and the
secretion of IL-10 and TGF-β were determined in a common group of
patients. After 4 days of cultivation, surface CTLA-4 and PD-L1
expression did not differ between donors and patients (Fig. 9A). In addition, in donors and
patients with RA, the percentage of CTLA-4+Treg
increased during stimulation. The percentage of
PD-L1+Treg was significantly increased under
anti-CD3+IL-2 stimulation only in donors.
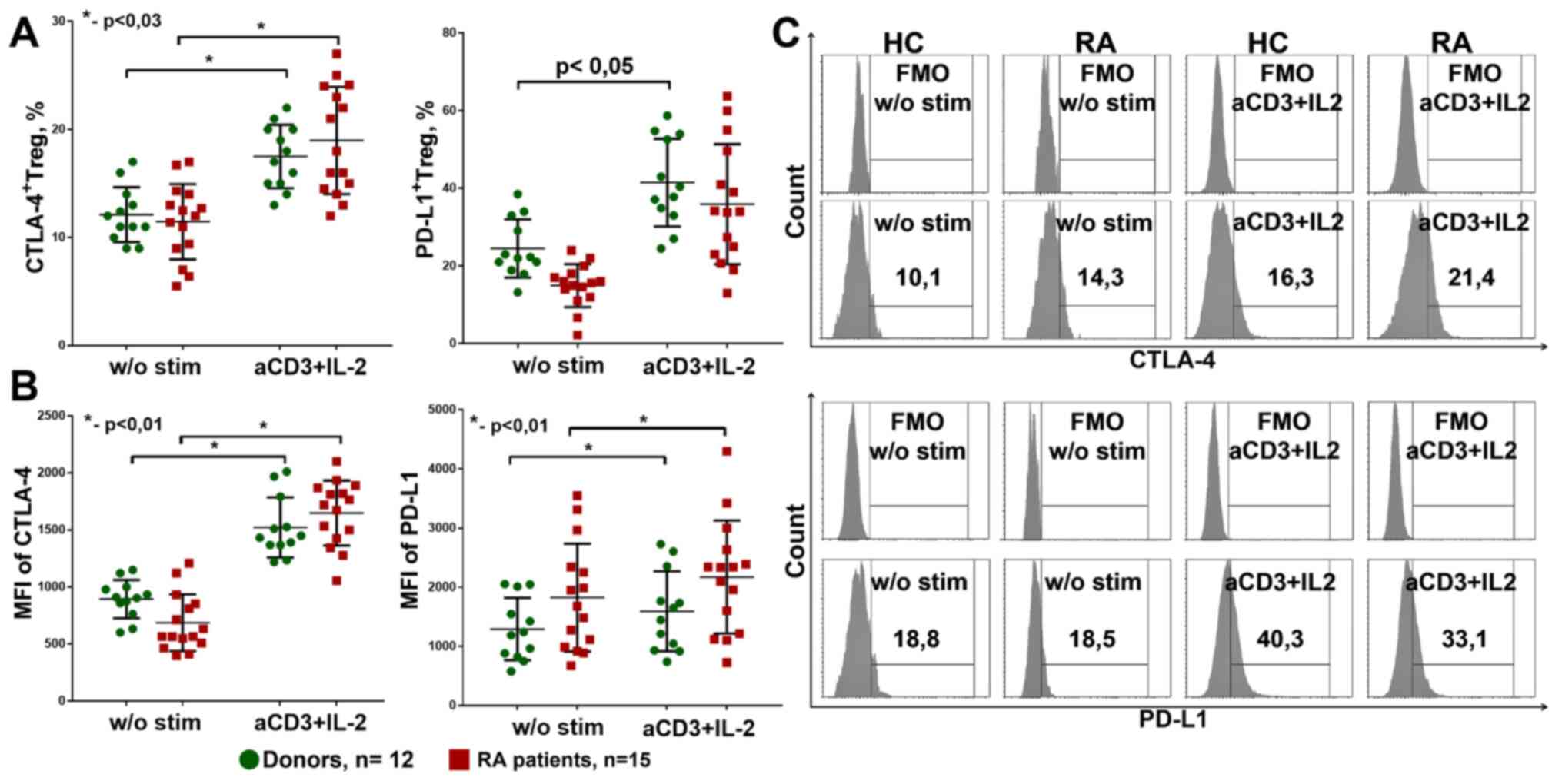 | Figure 9CTLA4+ and
PD-L1+ expression on Treg. (A) CTLA4+ and
PD-L1+Treg percentage from culture. Healthy donors,
n=12, RA patients, n=15. (B) MFI of CTLA-4 and PD-L1 expression on
Treg in culture. Healthy donors, n=12, RA patients, n=15. (C)
Gating strategy for CTLA-4 and PD-L1. The data are presented as the
median and 25-75% interquartile range, and was analyzed using the
Mann-Whitney test for independent groups and with the Wilcoxon test
for related groups. *P<0.01. w/o, without; a, anti;
stim, stimulated; Treg, regulatory T cells; HC, healthy controls;
RA, rheumatoid arthritis; MFI, mean fluorescence intensity; FMO,
fluorescence minus one. |
The densities of CTLA-4 and PD-L1 expression on Treg
cells also increased during stimulation with anti-CD3+IL-2 in both
the patients with RA and the donor groups, but did not differ
between these groups (Fig. 9B).
Thus, anti-CD3+IL-2 stimulation could increase Treg suppressive
ability by promoting PD-L1 and CTLA-4 expression.
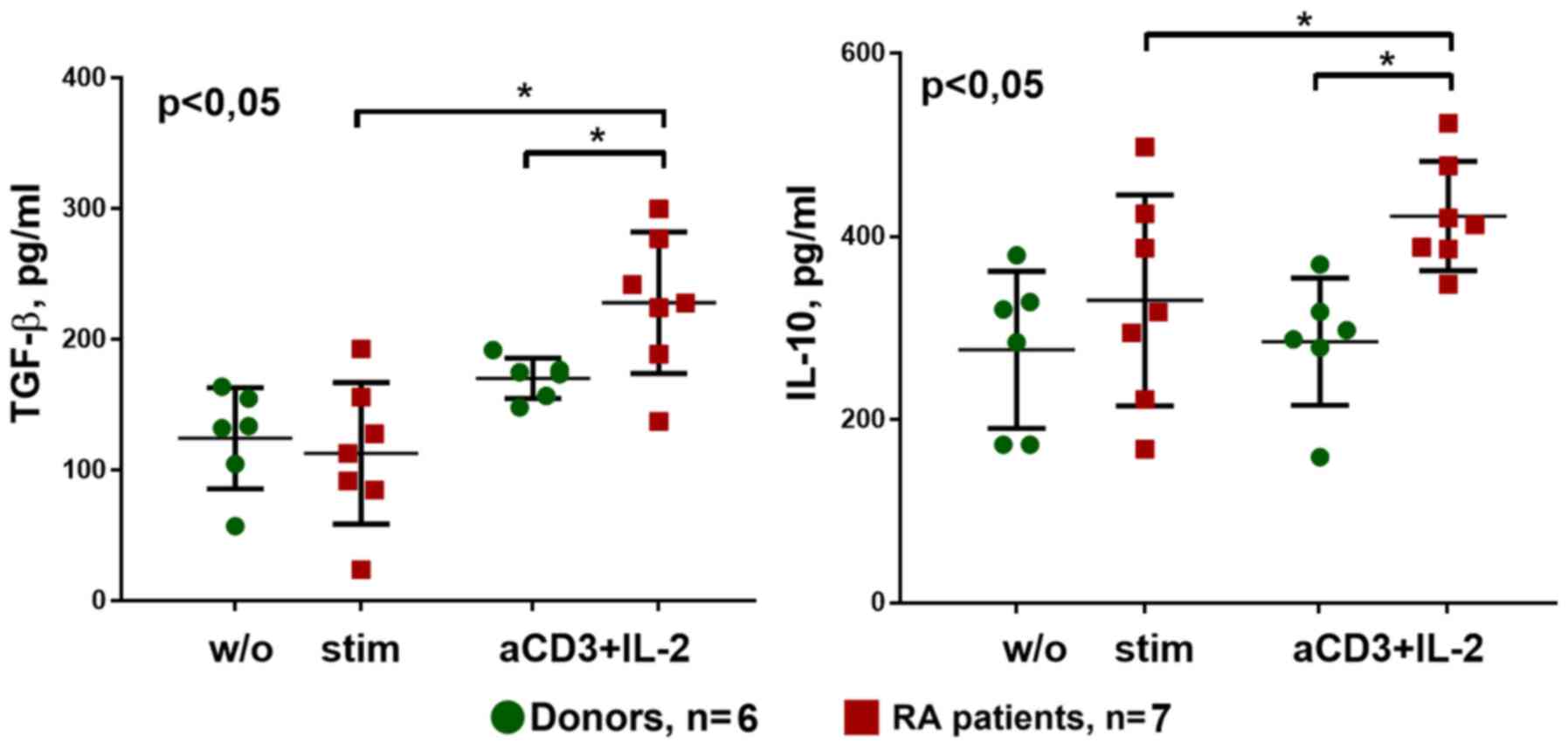
With respect to the secretion of cytokines during
cultivation with anti-CD3 + IL-2 stimulation, TGF-β and IL-10
production was significantly higher in patients with RA compared
with that in donors. In addition, anti-CD3+IL-2 stimulation
increased secretion of these cytokines, particularly in the patient
group (Fig. 10).
Discussion
Previous studies have revealed that autotolerance
disorders are associated with autoimmune conditions, for example,
RA develops from the imbalance of regulatory and effector
mechanisms, and the imbalance itself was associated, not only with
the excess activation of effectors, but also with defects in
regulatory T cells (34-36).
However, in the present study it was found that the Treg pool in
patients with RA has the same quantitative and qualitative
characteristics compared with that in the healthy donors. Namely,
that the general Treg pool retains healthy suppressor activity,
with respect to CD4+ and CD8+ lymphocytes.
Furthermore, there were no significant associations between
percentage of Treg and clinical parameters, such as ESR, CRP and RF
(data not shown). This may be due to the preservation of Treg
numbers in the peripheral blood in RA, which is consistent with the
ROC analysis of Treg percentage.
Stimulation with anti-CD3+IL-2 promoted PD-L1 and
CTLA-4 expression on Treg cells in healthy donors and RA patients
since IL-2 is one of the most important cytokines for Treg cell
homeostasis, which, when combined with a strong signal using TCR,
by anti-CD3 stimulation, can lead to activation and increasing
suppressor capacity of Treg cells (3).
In addition, the cultures of RA patients had higher
IL-10 and TGF-β production levels, under anti-CD3+IL-2 stimulation,
which is a sign of increased Treg activation. It can be
hypothesized that regulatory T cells require activation to maintain
the immunity balance in RA, i.e. to compensate for the excess
activation of effector cells. This hypothesis was supported by the
raised concentrations of CCR4+ -a chemokine receptor
that enables homing to inflammation foci, and
HLA-DR+Treg cells with higher FoxP3 expression, as well
as by the elevated expression of PD-L1 in patients with RA-a
finding consistent with previous studies (37-39).
Lower CTLA-4 expression on the surface of Tregs in RA has been
hypothesized to be associated with a reduction in suppressor
activity (40,41); however, it might also indicate a
greater Treg involvement in contact with APCs. CTLA-4 molecules are
constitutively expressed by Treg cells intracellularly, near the
membrane in vesicles (42). These
vesicles merge with the membrane from time to time, which causes
CTLA-4 molecules to emerge on the lymphocyte surface. Such
recirculation of CTLA-4 is amplified when stimulating Treg TCRs by
interacting with the peptide part of MHC-II on APCs (43). Thus, when a Treg contacts APCs,
surface CTLA-4 molecules bind to CD80/86 co-stimulation molecules
and disrupt antigen presentation (44). Then, a segment of the membrane near
the CTLA-4-CD80/86 complex submerges into the Treg and recreates
the vesicles, which merge with lysosomes, causing co-stimulation
molecules to degrade. However, not all B7 molecules undergo lysis
as some might emerge on the Treg surface (15). This leads to the hypothesis that
lower CTLA-4 expression on Treg cells, as well as a greater
CD86+Treg concentration in RA, may be due to these cells
being more active and involved in contact suppression with APCs.
With respect to the CTLA-4/CD86 interaction during strong TCR
signaling, which leads to an increase in trans-endocytosis, that
may suggest that Treg lymphocytes are involved in contacting APC to
a greater extent in RA compared with that in healthy individuals
(15). Apparently, clones with a
high affinity to self-antigens better represent in the
CD86+Treg subset. However, this assumption requires
further investigation. Therefore, it can be hypothesized that the
expression of B7 molecules on the Treg surface demonstrates the
higher functional activity of some antigen-specific clones of
Tregs; however subsequent effects of this expression are
understudied and require additional research.
The increased activation and persistent suppressor
activity of Treg cells in RA indicated that cells had adapted to
chronic inflammation; however, RA still disrupted the immune
balance. Earlier study proved that Treg cells were unable to
effectively suppress CD161+Th proliferation, while still
being able to suppress the proliferation of other
CD161-Th lymphocytes (16). CD161+Th lymphocytes are a
pool of CD4+RORyt+ cells producing IL-17A,
IL-17F, IL-22, TNFα and other pro-inflammatory factors (45,46).
This leads to the assumption that it is not a lower suppressor
activity of the general Treg pool that disrupts the immune balance
in RA; rather, it is the insufficient suppressor activity against
specific CD161+RORyt+ lymphocytes, the
peripheral-blood and synovial concentration, which rises in RA
(17,47). The present study also showed that
patients with RA had more FoxP3+RORyt+ and
CD4+RORyt+ cells, and their percentage was
highly correlated with disease activity; however, these cells are
still only a small part of the Th pool and could not have
significantly affected the Treg suppressor activity estimates. It
is worth noting that IL-17 expression determined using flow
cytometry requires previous stimulation and may not reflect the
initial expression of IL-17 in lymphocytes from PBMCs (48,49).
Therefore, the transcription factor, RORyt, was used as a marker of
differentiation for Th-17 cells, which reflects the initial stage
of transition when the IL-17 expression may not be detected
(19,29).
Another immune imbalance mechanism triggered by RA
is the effector resistance to Treg suppression in inflammation. The
resistance to Treg-issued inhibitory signals has been shown to be
unrelated to APC activation or the effector cell memory phenotype;
rather, it depends on the activation of protein kinase B
(PKB)/c-akt in effector cells exposed to TNFα and IL-6 at
inflammation sites (50).
Inhibiting this kinase restores the effector cell response to Treg
suppression. Another study showed that when removed from the
inflammatory environment, Tregs effectively suppressed the
production of proinflammatory cytokines and the proliferation of
effector T cells (51). Taken
together, the general Treg pool does not lose its function in
RA.
Balancing effector and regulatory immunity are
active processes where multiple mechanisms are involved to attain a
dynamic balance (3). This may be
why the general Treg pool retains its suppressor activity in RA,
while Treg cells have an activated phenotype due to greater tension
imposed on the regulatory mechanisms in chronic inflammation
(33). Evidently, the inflammatory
microenvironment causes some Treg cells to continuously convert
into pathogenic exFoxP3-Th-17 lymphocytes (17), that resist Treg inhibition and cause
self-sustained inflammation (16).
Rossetti et al (33) found
that some activated Treg cells, pertaining to synovial
inflammation, share the TCR antigenic specificity with effector T
lymphocytes involved in sustaining articular inflammation; these
Treg cells express HLA-DR. This means that Treg cells have some
antigen-specific effects. It therefore seems important to consider,
not only the Treg subpopulation in terms of the expression of
different molecules, but also the heterogeneity of this population,
as determined by the antigen specificity of TCRs. Perhaps the
regulatory defects in RA are not population-wide, but pertain to a
limited set of Treg clones highly specific to particular antigens,
the tolerance to which has been compromised (3,52).
In conclusion, intolerance to body-produced antigens
in RA could be due to excessive activation of effector cells, as
well as defects in regulatory T cells (1,11). The
present study showed that Treg cells were activated in RA and
retained suppressor activity against CD4+ and
CD8+ in both patients with new-onset RA and those
receiving DMARD treatment. Transitional
FoxP3+RORyt+ forms and increased
CD4+RORyt+ concentrations indicated the
pathological propensity of Treg cells to convert into Th17 in RA,
which could to be the most important factor of sustaining a chronic
autoimmune process, particularly in light of a previous report that
the inability of Treg cells to effectively suppress exFoxP3-Th17
proliferation and proinflammatory cytokine production (16).
Acknowledgements
Not applicable.
Funding
RFBR and Novosibirsk Region funded this work
according to the research project № 17-44-540167
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DS performed experimental work, contributed to the
conception, drafting of the manuscript, and design. VT contributed
to the conception and revision of the manuscript. VK contributed to
the final approval of the manuscript. AS, OS, and VK contributed to
patients recruitment and clinical evaluation of parameters of RA
patients. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Local Ethics
Committee's Approval (protocol no. 110: October 11, 2018; RIFCI
Ethics Committee).
Patient consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Smolen JS, Aletaha D, Barton A, Burmester
GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH,
Strand V and Yamamoto K: Rheumatoid arthritis. Nat Rev Dis Primers.
4(18001)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fang Q, Zhou C and Nandakumar KS:
Molecular and cellular pathways contributing to joint damage in
rheumatoid arthritis. Mediators Inflamm.
2020(3830212)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Shevyrev D and Tereshchenko V: Treg
heterogeneity, function, and homeostasis. Front Immunol.
10(3100)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ehrenstein MR, Evans JG, Singh A, Moore S,
Warnes G, Isenberg DA and Mauri C: Compromised function of
regulatory T cells in rheumatoid arthritis and reversal by
anti-TNFalpha therapy. J Exp Med. 200:277–285. 2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Morita T, Shima Y, Wing JB, Sakaguchi S,
Ogata A and Kumanogoh A: The proportion of regulatory T cells in
patients with rheumatoid arthritis: A Meta-analysis. PLoS One.
11(e0162306)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang X, Zhang X, Zhuang L, Xu C, Li T,
Zhang G and Liu Y: Decreased regulatory T-cell frequency and
interleukin-35 levels in patients with rheumatoid arthritis. Exp
Ther Med. 16:5366–5372. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Abazaa N, EL-kabarityb RH and Abo-Shadyb
RA: Deficient or abundant but unable to fight? Estimation of
circulating FoxP3+ T regulatory cells and their counteracting
FoxP3-in rheumatoid arthritis and correlation with disease
activity. Egypt Rheum. 35:185–192. 2013.
|
|
8
|
Walter GJ, Evans HG, Menon B, Gullick NJ,
Kirkham BW, Cope AP, Geissmann F and Taams LS: Interaction with
activated monocytes enhances cytokine expression and suppressive
activity of human
CD4+CD45ro+CD25+CD127low
regulatory T cells. Arthritis Rheum. 65:627–638. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kanjana K, Paisooksantivatana K,
Matangkasombut P, Chevaisrakul P and Lumjiaktase P: Efficient
short-term expansion of human peripheral blood regulatory T cells
for co-culture suppression assay. J Immunoassay Immunochem.
40:573–589. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Walter GJ, Fleskens V, Frederiksen KS,
Rajasekhar M, Menon B, Gerwien JG, Evans HG and Taams LS:
Phenotypic, functional, and gene expression profiling of peripheral
CD45RA+ and CD45RO+
CD4+CD25+CD127low treg cells in
patients with chronic rheumatoid arthritis. Arthritis Rheumatol.
68:103–116. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nie H, Zheng Y, Li R, Guo TB, He D, Fang
L, Liu X, Xiao L, Chen X, Wan B, et al: Phosphorylation of FOXP3
controls regulatory T cell function and is inhibited by TNF-α in
rheumatoid arthritis. Nat Med. 19:322–328. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang N, Schröppel B, Lal G, Jakubzick C,
Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y and Bromberg
JS: Regulatory T cells sequentially migrate from inflamed tissues
to draining lymph nodes to suppress the alloimmune response.
Immunity. 30:458–469. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baecher-Allan C, Wolf E and Hafler DA: MHC
class II expression identifies functionally distinct human
regulatory T cells. J Immunol. 176:4622–4631. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shevyrev DV, Blinova EA and Kozlov VA: The
influence of humoral factors of homeostatic proliferation on
t-regulatory cells in vitro. Bull Siberian Med. 18:286–293.
2019.
|
|
15
|
Qureshi OS, Zheng Y, Nakamura K, Attridge
K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z,
et al: Trans-endocytosis of CD80 and CD86: A molecular basis for
the cell-extrinsic function of CTLA-4. Science. 332:600–603.
2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Basdeo SA, Moran B, Cluxton D, Canavan M,
McCormick J, Connolly M, Orr C, Mills KH, Veale DJ, Fearon U and
Fletcher JM: Polyfunctional, pathogenic CD161+ Th17
lineage cells are resistant to regulatory T cell-mediated
suppression in the context of autoimmunity. J Immunol. 195:528–540.
2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Komatsu N, Okamoto K, Sawa S, Nakashima T,
Oh-hora M, Kodama T, Tanaka S, Bluestone JA and Takayanagi H:
Pathogenic conversion of Foxp3+ T-cells into TH17 cells in
autoimmune arthritis. Nat Med. 20:62–70. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Korn T, Bettelli E, Oukka M and Kuchroo
VK: IL-17 and Th17 cells. Annu Rev Immunol. 27:485–517.
2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ivanov II, McKenzie BS, Zhou L, Tadokoro
CE, Lepelley A, Lafaille JJ, Cua DJ and Littman DR: The orphan
nuclear receptor RORγt directs the differentiation program of
proinflammatory IL-17+ T helper cells. Cell.
126:1121–1133. 2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Aletaha D, Ward MM, Machold KP, Nell VP,
Stamm T and Smolen JS: Remission and active disease in rheumatoid
arthritis: Defining criteria for disease activity states. Arthritis
Rheum. 52:2625–2636. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
van der Heijde DM, van't Hof M, van Riel
PL and van de Putte LB: Development of a disease activity score
based on judgment in clinical practice by rheumatologists. J
Rheumatol. 20:579–581. 1993.PubMed/NCBI
|
|
22
|
Scott PJ and Huskisson EC: Measurement of
functional capacity with visual analogue scales. Rheumatol Rehabil.
16:257–259. 1977.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 Rheumatoid arthritis classification criteria:
An American College of Rheumatology/European League Against
Rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Böyum A: Isolation of leucocytes from
human blood. Further observations. Methylcellulose, dextran, and
ficoll as erythrocyteaggregating agents. Scand J Clin Lab Invest
Suppl. 97:31–50. 1968.PubMed/NCBI
|
|
25
|
Böyum A: Isolation of mononuclear cells
and granulocytes from human blood. Isolation of monuclear cells by
one centrifugation, and of granulocytes by combining centrifugation
and sedimentation at 1 g. Scand J Clin Lab Invest Suppl. 97:77–89.
1968.PubMed/NCBI
|
|
26
|
Collison LW and Vignali DA: In vitro Treg
suppression assays. Methods Mol Biol. 707:21–37. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mahmud SA, Manlove LS and Farrar MA:
Interleukin-2 and STAT5 in regulatory T cell development and
function. JAKSTAT. 2(e23154)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hua J, Inomata T, Chen Y, Foulsham W,
Stevenson W, Shiang T, Bluestone JA and Dana R: Pathological
conversion of regulatory T cells is associated with loss of
allotolerance. Sci Rep. 8(7059)2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ivanov II, Zhou L and Littman DR:
Transcriptional regulation of Th17 cell differentiation. Semin
Immunol. 19:409–417. 2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tada Y, Ono N, Suematsu R, Tashiro S,
Sadanaga Y, Tokuda Y, Ono Y, Nakao Y, Maruyama A, Ohta A and
Koarada S: The balance between Foxp3 and Ror-γt expression in
peripheral blood is altered by tocilizumab and abatacept in
patients with rheumatoid arthritis. BMC Musculoskelet Disord.
17(290)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kugyelka R, Kohl Z, Olasz K, Mikecz K,
Rauch TA, Glant TT and Boldizsar F: Enigma of IL-17 and Th17 cells
in rheumatoid arthritis and in autoimmune animal models of
arthritis. Mediators Inflamm. 2016(6145810)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kim BS, Lu H, Ichiyama K, Chen X, Zhang
YB, Mistry NA, Tanaka K, Lee YH, Nurieva R, Zhang L, et al:
Generation of RORγt+ Antigen-Specific T regulatory 17
cells from Foxp3+ precursors in autoimmunity. Cell Rep.
21:195–207. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rossetti M, Spreafico R, Consolaro A,
Leong JY, Chua C, Massa M, Saidin S, Magni-Manzoni S, Arkachaisri
T, Wallace CA, et al: TCR repertoire sequencing identifies synovial
Treg cell clonotypes in the bloodstream during active inflammation
in human arthritis. Ann Rheum Dis. 76:435–441. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dejaco C, Duftner C, Grubeck-Loebenstein B
and Schirmer M: Imbalance of regulatory T cells in human autoimmune
diseases. Immunology. 117:289–300. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lee GR: The Balance of Th17 versus Treg
cells in autoimmunity. Int J Mol Sci. 19(730)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Horwitz DA, Fahmy TM, Piccirillo CA and La
Cava A: Rebalancing immune homeostasis to treat autoimmune
diseases. Trends Immunol. 40:888–908. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shalini PU, Debnath T, Jvs V, Kona LK,
Kamaraju SR, Kancherla R and Chelluri LK: A study on FoxP3 and
Tregs in paired samples of peripheral blood and synovium in
rheumatoid arthritis. Cent Eur J Immunol. 40:431–436.
2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li N, Wei W, Yin F, Chen M, Ma TR, Wu Q,
Zhou JR, Zheng SG and Han J: The abnormal expression of CCR4 and
CCR6 on Tregs in rheumatoid arthritis. Int J Clin Exp Med.
8:15043–15053. 2015.PubMed/NCBI
|
|
39
|
Al-Banna NA, Vaci M, Slauenwhite D,
Johnston B and Issekutz TB: CCR4 and CXCR3 play different roles in
the migration of T cells to inflammation in skin, arthritic joints,
and lymph nodes. Eur J Immunol. 44:1633–1643. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Cribbs AP, Kennedy A, Penn H, Read JE,
Amjadi P, Green P, Syed K, Manka SW, Brennan FM, Gregory B and
Williams RO: Treg cell function in rheumatoid arthritis is
compromised by ctla-4 promoter methylation resulting in a failure
to activate the indoleamine 2,3-dioxygenase pathway. Arthritis
Rheumatol. 66:2344–2354. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Flores-Borja F, Jury EC, Mauri C and
Ehrenstein MR: Defects in CTLA-4 are associated with abnormal
regulatory T cell function in rheumatoid arthritis. Proc Natl Acad
Sci USA. 105:19396–19401. 2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Walker LS: Treg and CTLA-4: Two
intertwining pathways to immune tolerance. J Autoimmun. 45:49–57.
2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Linsley PS, Bradshaw J, Greene J, Peach R,
Bennett KL and Mittler RS: Intracellular trafficking of CTLA-4 and
focal localization towards sites of TCR engagement. Immunity.
4:535–543. 1996.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sansom DM: CD28, CTLA-4 and their ligands:
Who does what and to whom? Immunology. 101:169–177. 2000.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Pesenacker AM, Bending D, Ursu S, Wu Q,
Nistala K and Wedderburn LR: CD161 defines the subset of
FoxP3+ T cells capable of producing proinflammatory
cytokines. Blood. 121:2647–2658. 2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Afzali B, Mitchell PJ, Edozie FC, Povoleri
GA, Dowson SE, Demandt L, Walter G, Canavan JB, Scotta C, Menon B,
et al: CD161 expression characterizes a subpopulation of human
regulatory T cells that produces IL-17 in a STAT3-dependent manner.
Eur J Immunol. 43:2043–2054. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sato K, Suematsu A, Okamoto K, Yamaguchi
A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y,
et al: Th17 functions as an osteoclastogenic helper T cell subset
that links T cell activation and bone destruction. J Exp Med.
203:2673–2682. 2006.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Pappu BP and Dong C: Measurement of
interleukin-17. Curr Protoc Immunol: Chapter 6:Unit 6.25, 2007.
doi:10.1002/0471142735.im0625s79.
|
|
49
|
Zhao L, Chou Y, Jiang Y, Jiang Z and Chu
CQ: Analysis of IL-17 production by flow cytometry and ELISPOT
assays. Methods Mol Biol. 1172:243–256. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Wehrens EJ, Mijnheer G, Duurland CL, Klein
M, Meerding J, van Loosdregt J, de Jager W, Sawitzki B, Coffer PJ,
Vastert B, et al: Functional human regulatory T cells fail to
control autoimmune inflammation due to PKB/c-Akt hyperactivation in
effector cells. Blood. 118:3538–3548. 2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Herrath J, Müller M, Amoudruz P, Janson P,
Michaëlsson J, Larsson PT, Trollmo C, Raghavan S and Malmström V:
The inflammatory milieu in the rheumatic joint reduces regulatory
T-cell function. Eur J Immunol. 41:2279–2290. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zemmour D, Zilionis R, Kiner E, Klein AM,
Mathis D and Benoist C: Single-cell gene expression reveals a
landscape of regulatory T cell phenotypes shaped by the TCR. Nat
Immunol. 19:291–301. 2018.PubMed/NCBI View Article : Google Scholar
|