Introduction
The mevalonate (MVA) pathway is indispensable for
de novo synthesis of cholesterol as well as other molecules
essential for various cellular functions, including cell
proliferation and apoptosis (1).
MVA is irreversibly synthesized from
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA), and further
metabolized into farnesyl diphosphate, which is also called
farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate
(2). These isoprenoids are
precursors for a number of important metabolites, including
sterols, dolichols, ubiquinones (also known as coenzyme Q) and
carotenoids (3).
HMG-CoA reductase (HMGCR) is the rate-limiting
enzyme in cholesterol biosynthesis, which catalyzes the conversion
of HMG-CoA to MVA (1). Statin drugs
are capable of inhibiting the synthesis of endogenous cholesterol
by competitive inhibition of HMGCR (4). Statins were originally used to treat
hypercholesterolemia (5), but have
since been found to exert many pleiotropic effects (6), including immunomodulatory,
anti-inflammatory (7), and
neuroprotective (8) effects.
Clinical studies have demonstrated that statins can be used to
treat diseases that are not clearly related to low-density
lipoprotein cholesterol (LDL-C) or cholesterol (9,10).
This may be due to the inhibitory effect of statins on the
production of isoprenoid intermediates in the cholesterol
biosynthesis pathway (9,11). Post-translational prenylation of
small guanosine triphosphate binding proteins, such as Rho and Rac,
and their downstream effectors, such as Rho kinase and nicotinamide
adenine dinucleotide phosphate oxidase, is also inhibited by
statins (6). However, the use of
statins in the treatment of heart failure remains controversial
(12-14).
In a previous study, it was demonstrated that rats
given a high-salt diet developed high blood pressure, diastolic
heart dysfunction and increased rates of myocardial fibrosis
(15). Transcriptome analysis
demonstrated that the MVA pathway served an important role in the
pathophysiology of myocardial fibrosis (15,16).
Other studies have also indicated the relevance of key enzymes and
downstream GTPases, including Rho and Ras, in the MVA pathway to
myocardial fibrosis (17,18). Inhibition of Rho and Ras can reduce
myocardial hypertrophy and fibrosis (17). The present study used multiple
drugs, including rosuvastatin, alendronate, and fasudil, which
regulate the MVA pathway at different points. Rosuvastatin, one of
the most commonly used drugs in secondary prevention of
cardiovascular disease, is a potent, effective, and safe HMGCR
inhibitor (19,20). Alendronate is a farnesyl
pyrophosphate synthase (FDPS) inhibitor that improved vascular
endothelial function in a hypertensive rat model (21,22).
Fasudil is a clinically used Rho/Rho associated coiled-coil
containing protein kinase (ROCK) inhibitor that has a good
therapeutic effect in cardiovascular diseases including angina,
hypertension, and coronary spasms (23,24).
By observing the effects of these drugs on myocardial fibroblasts,
the present study explored the mechanism by which the MVA pathway
is involved in myocardial fibrosis. The present study could provide
new directions and theoretical basis for the development of novel
targeted drugs, as well as for the clinical treatment and
prevention of diastolic heart failure and myocardial fibrosis.
Materials and methods
Isolation and primary culture of
cardiac fibroblasts
A total of 10 C57BL/6 male mice (6 weeks; weight,
20±2 g) were obtained from the Shanghai Experimental Animal Center
of the Chinese Academy of Sciences and kept in a climate-controlled
room (temperature, 25±1˚C; relative humidity, 50-60%; free access
to food and water; and 12-h light/dark cycle.) for 1 week of
acclimatization. All experimental protocols involving live animals
were reviewed and approved by the Institutional Animal Care and Use
Committee of the Tongji Hospital of Tongji University (approval no.
2019-DW-008) (Shanghai, China). Murine cardiac fibroblasts were
isolated as previously described (25,26).
In short, C57BL/6 mice were anaesthetized with 3% isoflurane until
loss of limb reflexes.
Hearts were isolated from C57 mice at room
temperature (RT) and subjected to 5 min perfusion with DMEM/F12
supplemented with 1% penicillin, 1% streptomycin, and 1%
amphotericin B solution (all Gibco; Thermo Fisher Scientific, Inc.)
at RT. The hearts were perfused with collagenase type II (Gibco;
Thermo Fisher Scientific, Inc.) at 37˚C for 20 min before
dissociation of the tissue at RT and incubation with dilute
collagenase (0.05% w/v) at 37˚C for 10 min. The collagenase was
neutralized by adding 2X the existing volume of DMEM/F12 complete
medium supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.). The cell suspension was then passed through a 40-μm sterile
cell strainer to remove undigested tissue. The filtered suspension
was collected and centrifuged at 500 x g for 10 min at room
temperature. After centrifugation, the supernatant was discarded
and the cell pellet was resuspended in 10 ml growth medium
(DMEM/F12 with 10% FBS) and then diluted to a final volume of 30
ml. Cells were seeded in cell culture dishes and allowed to adhere
for 2 h in 5% CO2 at 37˚C. The cell cultures were moved
into T-25 flasks (4 ml cell suspension) and incubated overnight at
37˚C. The following day, the cultures were washed twice with
phosphate-buffered saline (PBS) and the growth medium was replaced.
The culture medium was replaced once per day in this way until
harvesting after 3 days.
Flow cytometry
Third-generation cardiac fibroblasts were prepared
into a single cell suspension with 0.25% trypsin and the cell
density was adjusted to 1x106 cells/ml. The cells were
fixed and permeabilized with FIX & PERM Cell Permeabilization
Kit (cat. no. GAS003; Thermo Fisher Scientific, Inc.) at 37˚C for
30 min. The cells were then incubated at 37˚C for 30 min with PBS
and anti-vimentin antibody (1:100, cat. no. ab92547; Abcam), before
being incubated with FITC-labeled secondary antibody (1:1000, cat.
no. ab6717) at 37˚C for 30 min. Cells were then analyzed using a BD
FACSCanto™ II flow cytometer (BD Biosciences) with FlowJo software
(Version 7.6, BD Biosciences).
Drug intervention experiment and cell
staining
A total of five treatment groups were established
for the fibroblasts: i) negative control (NC); ii) angiotensin II
(Ang II) model, iii) Ang II + rosuvastatin (ROS); iv) Ang II +
alendronate (ALE), and v) Ang II + fasudil (FAS). The drug
concentrations were based on preliminary experiments: Ang II,
1x10-5 mol/l; rosuvastatin, 1x10-5 mol/l;
alendronate, 1x10-6 mol/l and fasudil, 10 µg/ml. The
fibroblasts were treated with the drugs for 48 h at 37˚C. No drugs
were added to the NC group. For crystal violet staining, the medium
was discarded and the cells were fixed with 10% methanol for 30 sec
at RT, followed by staining with 0.5% crystal violet staining
solution for 20 min at RT. Cell morphology was observed by light
microscopy (magnification, x100).
For collagen staining, the medium was discarded
following drug intervention and the cells were fixed with 4%
paraformaldehyde for 30 min at RT, and stained using Masson's
Trichrome Stain Kit according to the manufacturer's instructions
(cat. no. G1340, Beijing Solarbio Science & Technology Co.,
Ltd.). Cell collagen staining was observed by light microscopy
(magnification, x100).
Cell proliferation assay
Cell proliferation was assessed using the Cell
Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies Inc.).
Briefly, cells were seeded in 96-well plates at 1x104
cells/well and treated with drugs for 0, 24 and 48 h. CCK-8
solution (5 µl) was added to each well and incubated at 37˚C for an
additional 2 h. Optical density (OD) was determined at 450 nm.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA of cells was extracted with
TRIzol® (Invitrogen; Thermo Fisher Scientific Inc.).
cDNA synthesis was carried out using a TOYOBO ReverTra Ace qPCR RT
kit (Toyobo Life Science) and qPCR was performed using the SYBR
Supermix PCR kit (Kapa Biosystems; Roche Diagnostics), according to
the manufacturer's protocols. Primers were obtained from Sangon
Biotech Co., Ltd. and the sequences are listed in Table I. The reverse transcription reaction
step as follows: 37˚C for 15 min and 95˚C for 5 min. The
thermocycling conditions were as follows: Initial denaturation at
95˚C for 5 min, followed by 40 cycles of 95˚C for 30 sec, 61˚C for
30 sec and 72˚C for 30 sec. Gene expression levels were measured
using cycle threshold (CQ) values and the 2-ΔΔCq
calculation (27) and normalized to
GAPDH.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene name | Primer sequence (5'
to 3') | Amplicon size
(bp) |
|---|
| GAPDH | F:
GCAAGTTCAACGGCACAGTCA | |
| | R:
ACGACATACTCAGCACCAGCAT | 127 |
| HMGCR | F:
CTTGACGCTCTGGTGGAATG | |
| | R:
GTTGGCAAGCACGGACATAC | 105 |
| FDPS | F:
AGCAGAATTTCATCCAGCACT | |
| | R:
GTCAGACCCCGATTGTACT | 148 |
| RhoA | F:
ATGTGGCAGATATTGAAGTGGA | |
| | R:
GTGTCTGGGTAGGAGAGAGG | 106 |
| FGF-2 | F:
ACCCACACGTCAAACTACAG | |
| | R:
GGCGTTCAAAGAAGAAACACTC | 150 |
| TGF-β1 | F:
CCTGAGTGGCTGTCTTTTGA | |
| | R:
CGTGGAGTACATTATCTTTGCTG | 124 |
| VEGF | F:
ACTGGACCCTGGCTTTACT | |
| | R:
ATTGGACGGCAATAGCTGC | 136 |
| COL1A1 | F:
GACGCATGGCCAAGAAGAC | |
| | R:
ACTTCTGCGTCTGGTGATAC | 234 |
| HSP47 | F:
GTCCATCAACGAGTGGGC | |
| | R:
CAGTGCGGCTTAAAGAACATG | 117 |
| MVD | F:
GTCAACATCGCGGTTATCAA | |
| | R:
CCTCTGTGAAGTCCTTGCTAA | 142 |
| MVK | F:
CCAAACGTCGGTATTAAGCA | |
| | R:
GCCTTCGTTGCCTACACA | 159 |
Western blotting
RIPA buffer (cat. no. 9806; Cell Signaling
Technologies Inc.) was used for cells protein extraction and
protein concentration was determined using bicinchoninic acid
protein assay reagent (cat. no. 7780; Cell Signaling Technologies
Inc.). A total of 50-100 µg protein/lane were subjected to SDS-PAGE
(10% gel) and transferred onto a PVDF membrane. The membrane was
blocked by 5% non-fat milk powder for 1 h at room temperature
before incubation with the following primary antibodies: TGF-β1
(1:1,000; cat. no. ab92486; Abcam), collagen I (1:1,000; cat. no.
ab34710; Abcam), heat shock protein 47 (HSP47; 1:1,000; cat. no.
ab109117; Abcam) or GAPDH (1:4,000; cat. no. E12-042; EnoGene
Biotech Co., Ltd.) at 4˚C overnight. Goat-anti-rabbit antibody
(1:2,000; cat. no. ab205718; Abcam) was used as the secondary
antibody at RT for 1 h. Chemiluminescent visualization was
performed using ECL reagent (cat. no. 32106; Pierce; Thermo Fisher
Scientific Inc.). The protein loading control was GAPDH. Optical
density analysis was performed by ImageJ software (version 1.8,
National Institutes of Health).
Statistical analysis
GraphPad Prism v.7.0 (Graph Pad Inc.) was used for
statistical analysis. The data is expressed as the means ± SD from
experimental repeats. The comparison of multiple groups was
performed using one-way analysis of variance followed by the post
hoc Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
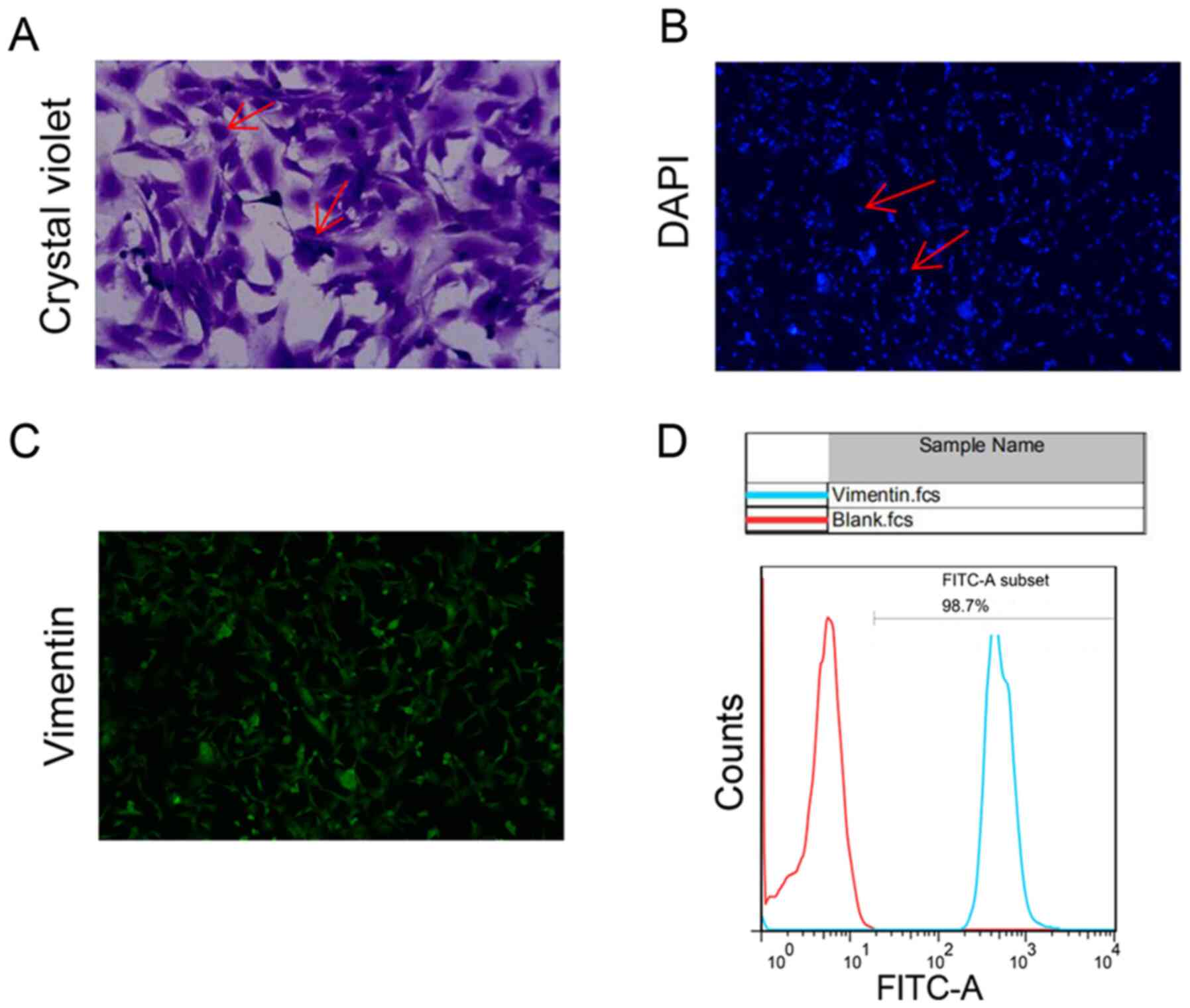
Fibroblast cell identification
We performed primary cultures of cardiac fibroblasts
and identified by flow staining. Microscopically, the fibroblast
cell bodies were large and spindle-shaped or stellate and flat with
multiple spindles (Fig. 1A). DAPI
staining shows regular oval nuclei (Fig. 1B). Anti-vimentin FITC was detected
on the surface of 98.7% of cultured cells (Fig. 1C and D).
Drug therapy alleviate the process of
Ang II-promoted cell fibrosis
Cells were large and mostly spindle-shaped in the NC
group (Fig. 2A). Cells were
proliferative and growth was dense and radial in the Ang II group
(Fig. 2A). There was no significant
difference in cell morphology between the ALE and Ang II groups
(Fig. 2A). The FAS and ROS groups
had fewer cells, more cytoplasm, and some cells were round compared
with those in the Ang II group (Fig.
2A). Collagen fiber staining in the Ang II group was
significantly more intense compared with that in the NC group
(Fig. 2A). Collagen staining in the
ALE, FAS, and ROS groups was weak compared with the Ang II group
(Fig. 2A). These results suggested
that drug intervention can significantly inhibit the Ang II-induced
increase in collagen level in myocardial cells. Ang II can
significantly promote proliferation compared with that in the NC
group, whilst the rates of proliferation in the ROS, ALE and FAS
treatment groups were lower compared with those in the Ang II group
at both 24 and 48 h (Fig. 2B). This
indicated that drug therapy can alleviate the process of Ang
II-promoted cell fibrosis.
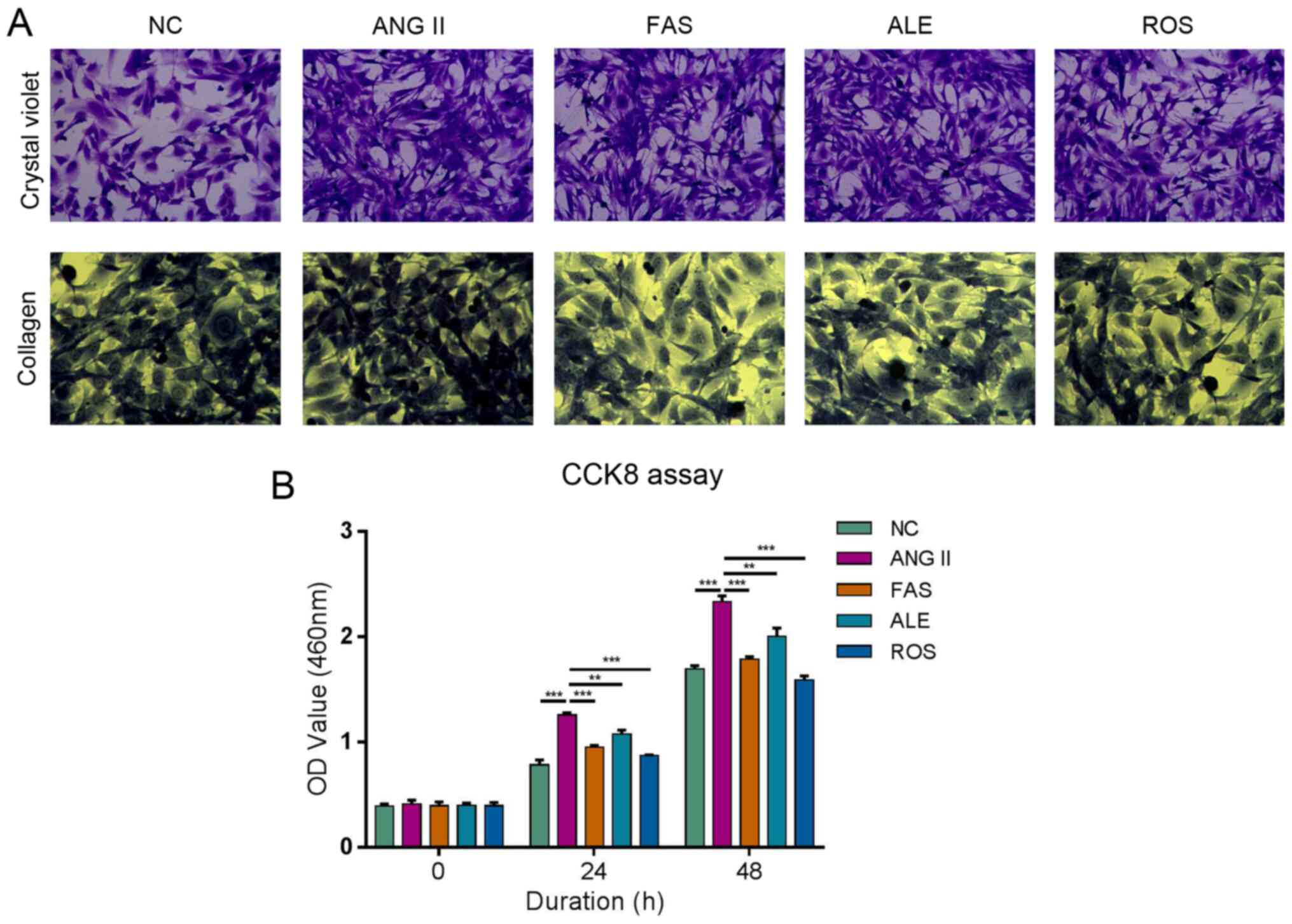 | Figure 2ALE, FAS and ROS therapy alleviate
the process of Ang II-promoted cell fibrosis and viability of
cardiac fibroblasts. (A) Crystal violet staining (magnification,
x200) shows cells were proliferative and growth was dense in the
Ang II group. There was no difference in cell morphology between
the ALE and Ang II groups. The FAS and ROS groups had fewer cells,
more cytoplasm, and some cells were round compared with those in
Ang II group. Collagen staining (magnification, x200) in the Ang II
group was more intense compared with that in the NC group. ALE,
FAS, and ROS groups was weak compared with the Ang II group. (B)
Cell proliferation assay shows Ang II significantly promote
proliferation compared with NC group, whilst the rates of
proliferation in the ROS, ALE, and FAS treatment groups were lower
compared Ang II group at both 24 and 48 h. **P<0.01
and ***P<0.001. NC, negative control; Ang II,
angiotensin II; ROS, Ang II + rosuvastatin; ALE, Ang II +
alendronate; FAS, Ang II + fasudil. |
mRNA expression levels of MVA pathway
and fibrosis-related genes under different drug interventions
Subsequently, effects of rosuvastatin, fasudil and
alendronate on gene expression in cultured primary cardiac
fibroblasts were investigated by RT-qPCR. After 72 h treatment with
the different drug interventions, relative mRNA abundance of genes
including TGF-β1, HSP47, collagen type I α1
(COL1A1), HMGCR, FDPS, ras homolog family
member A (RhoA), mevalonate diphosphate decarboxylase
(MVD), mevalonate kinase (MVK), fibroblast growth
factor-2 (FGF-2), and vascular endothelial growth factor
(VEGF) was examined. TGF-β1, HSP47, COL1A1, VEGF and
FGF2 expression was significantly reduced in the FAS and ROS
groups compared with the Ang II group (Fig. 3). HMGCR expression was
significantly lower in the drug intervention groups compared with
that in the Ang II group. FDPS expression was downregulated
in the ALE group and elevated in the FAS and ROS groups compared
with the Ang II group (Fig. 3).
RhoA expression was downregulated in the FAS and ROS groups
compared with the Ang II group and the expression of MVK was
decreased in the ROS group compared with the other groups (Fig. 3). The results indicated
rosuvastatin, fasudil and alendronate can target different target
genes that regulate the MVA pathway and interfere with the
expression of fibrosis-related genes.
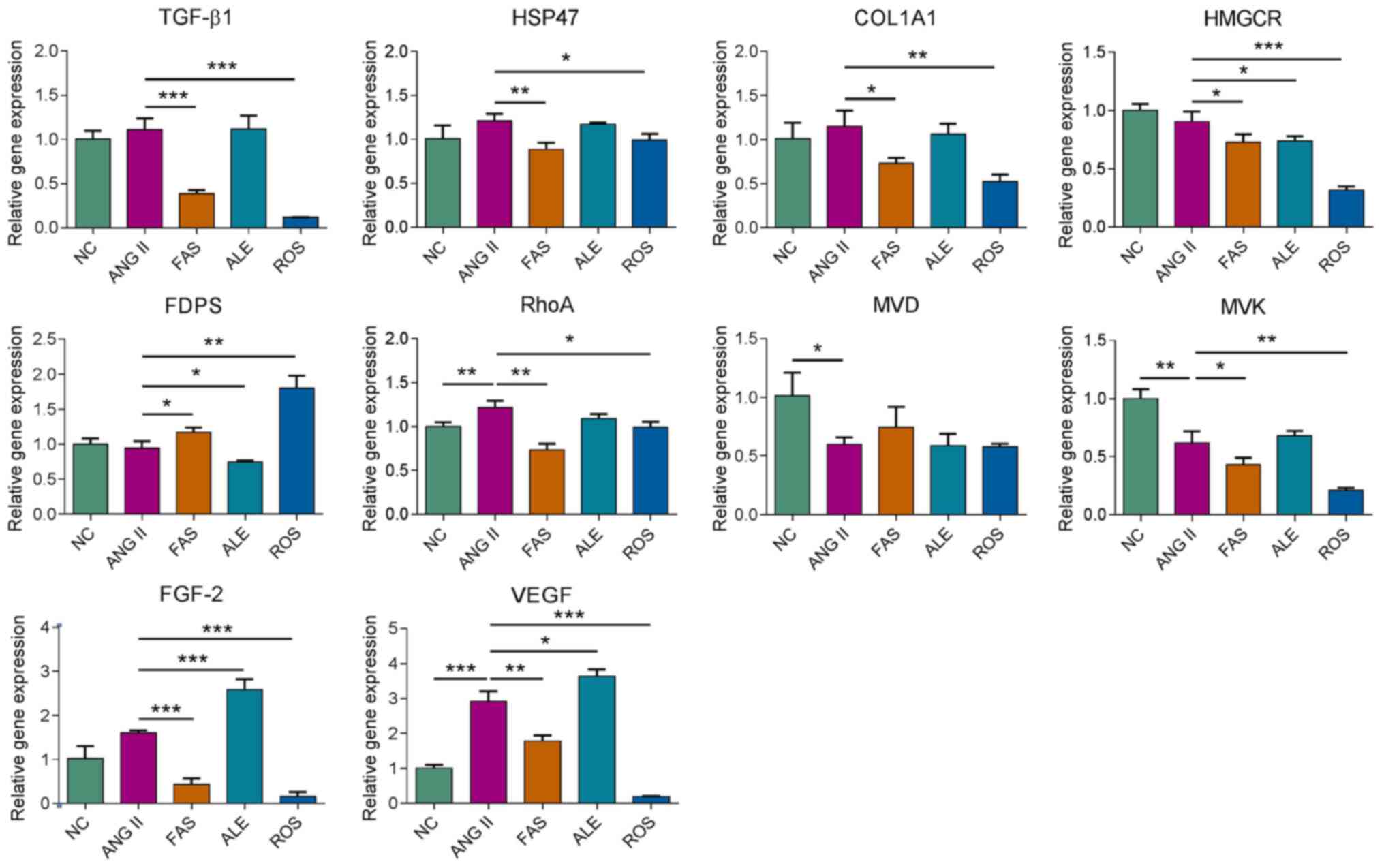 | Figure 3Reverse transcription-quantitative
polymerase chain reaction analysis of the genetic regulatory
effects of drug treatment. *P<0.05,
**P<0.01, ***P<0.001. NC, negative
control; ROS, Ang II + rosuvastatin; ALE, Ang II + alendronate;
FAS, Ang II + fasudil; HSP47, heat shock protein 47; COL1A1,
collagen type I α1; HMGCR, HMG-CoA reductase; FDPS, farnesyl
pyrophosphate synthase; MVD, mevalonate diphosphate decarboxylase;
MVK, mevalonate kinase; Ang II, angiotensin II; FGF-2, fibroblast
growth factor-2; VEGF, vascular endothelial growth factor. |
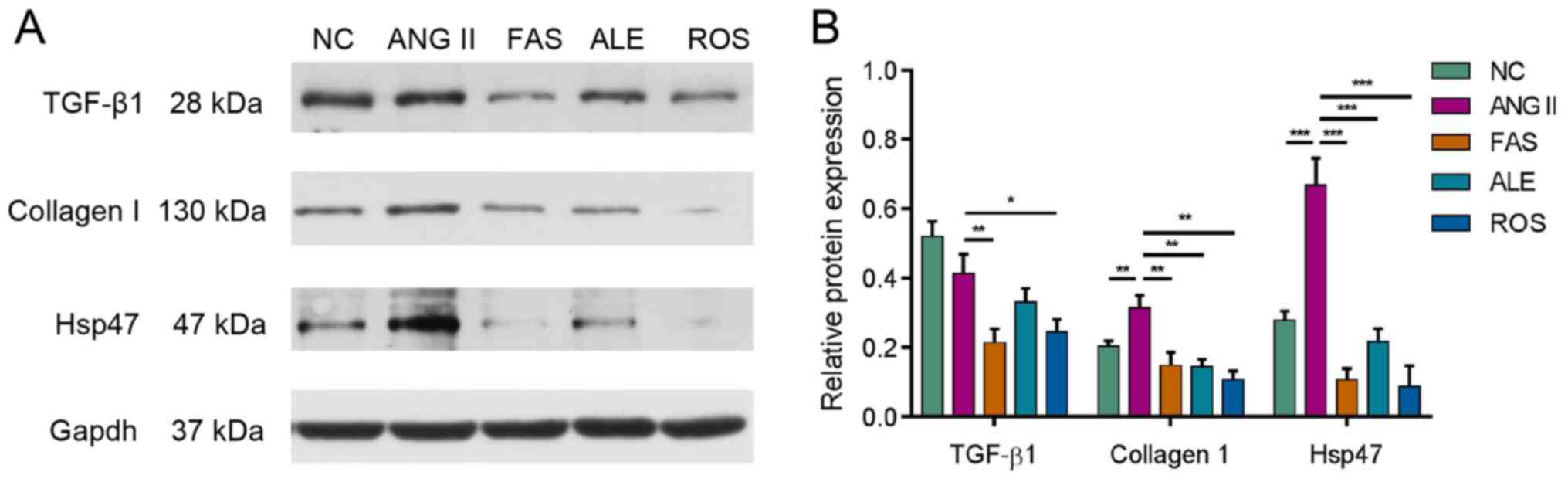
Fibrosis-related protein
expression
Western blotting was used to analyze expression of
key signaling molecules involved in myocardial fibrosis, including
TGF-β1, collagen I and HSP47. Ang II stimulation resulted in a
significant increase in collagen I and HSP47 expression (Fig. 4). Protein expression of TGF-β1,
collagen I and HSP47 all decreased following drug treatment
compared with the Ang II group (Fig.
4).
Discussion
Statins have been used for several decades to
prevent hypercholesterolemia and cardiovascular diseases, including
hyperlipidemia and atherosclerosis (19,20).
The primary mechanism of action of statin drugs is the lowering of
serum cholesterol (28). However,
statins may exert cardiovascular protective effects that are
independent of LDL-C lowering, such as pleiotropic effects
(29). The mechanism of these
effects may be related to suppression of the MVA pathway and
downstream GTPase activity (30,31).
Some cell culture and animal studies have suggested
that statins can prevent the development of cardiac hypertrophy and
fibrosis (32-35),
which may reduce ventricular compliance and is a major cause of
cardiac diastolic dysfunction (36). A meta-analysis of 11 eligible
studies, including 17,985 patients with heart failure with a
preserved ejection fraction demonstrated that statin therapy may be
associated with improved survival rates in these patients (37). However, 2 large-scale randomized
controlled studies of patients with chronic heart failure (the
CORONA and GISSI-HF trials) demonstrated that the majority of
patients exhibited heart failure with a reduced ejection fraction.
The aforementioned studies demonstrated that compared with the
control groups, the use of statins did not reduce the primary
outcome (death from any cause) (12,14).
The present study demonstrated that rosuvastatin can
inhibit the increased collagen levels in myocardial fibroblasts
caused by Ang II, reduce the proliferation of myocardial
fibroblasts induced by Ang II, and significantly decrease the gene
expression of FGF-2, TGF-β1, VEGF and COL1A1, and the
protein expression of TGF-β1, collagen I, and HSP47. HSP47 is a
collagen-specific molecular chaperone residing in the endoplasmic
reticulum (38). HSP47 is essential
for collagen synthesis in vertebrates (39). Previous studies have shown that
inhibition of HSP47 can improve the phenotype of collagen-related
diseases, such as pulmonary artery fibrosis, peritoneal fibrosis,
and liver fibrosis (40-42).
In addition, HSP47 has been found to be associated with fibrosis
level after myocardial infarction (43). Another study suggested that the
beneficial effect of atorvastatin on cardiac fibrosis may be due to
lowered HSP47 levels (34). The
results of the present study are consistent with the aforementioned
studies, with statins having the strongest effect among the tested
drugs. The results of the present study suggested that statins have
antimyocardial fibrosis effects and may have clinical therapeutic
potency for the treatment of diastolic heart failure.
FDPS is a key enzyme in the MVA pathway (44). This chain-elongation enzyme
catalyzes head-to-tail condensation of 2 molecules of isopentenyl
diphosphate with dimethylallyl diphosphate to form FPP (45). FPP is an important intermediate in
cholesterol and sterol biosynthesis, a substrate for protein
farnesylation and geranylgeranylation and a ligand for certain
hormone receptors and growth receptors, such as thyroid hormone
nuclear receptor-β (TR-β) and farnesoid X receptor (46). Prenylation of small GTP-binding
proteins by FPP is critical for enabling such proteins to be
incorporated into membranes, hence determining their localization
and function, and serving a crucial role in signal transduction
(47). Previous studies have
demonstrated that compared with Wistar Kyoto rats, the expression
of FDPS is significantly increased in aortic smooth muscle cells
and in the left ventricles of spontaneously hypertensive rats (SHR)
(48). Upregulation of FDPS has
also been detected in the heart and abdominal aorta in rat models
of overload-induced cardiac hypertrophy and associated diastolic
dysfunction (49). Additionally,
cardiac-specific overexpression of FDPS has been demonstrated to
induce cardiac hypertrophy, fibrosis, and heart failure (18). These pathological changes were
associated with the activation of RhoA and other known kinases,
including ERK1/2 and p38 MAPK in the hypertrophic signaling pathway
(18). FDPS inhibitors, namely
nitrogen-containing bisphosphonate (N-BP), are commonly used to
treat osteoporosis and osteolytic bone lesions (50). Previous studies have demonstrated
that N-BP may have effects on the cardiovascular system (21,22,51).
Ibandronate, a potent N-BP has been demonstrated to reduce reactive
oxygen species production in the vascular smooth muscle of SHR rats
(21), improve vascular endothelial
function (21) and prevent
noradrenaline-induced fibrosis of vascular smooth muscle (22). Another previous study also suggested
that chronic treatment with alendronate can reduce myocardial
hypertrophy and fibrosis whilst improving aortic remodeling through
a pathway involving inhibition of the geranylgeranylation and
activation of RhoA (52).
The present study demonstrated that alendronate
reversed the proliferation of myocardial fibroblasts induced by Ang
II. Collagen staining suggested that aledronate can inhibit the
increase of collagen levels of myocardial fibroblasts induced by
Ang II. However, alendronate had the weakest effect among the three
chosen drugs in the present study. In addition, alendronate did not
decrease the mRNA expression of TGF-β1, COL1A1, VEGF or
FGF-2 in the present study, which suggested that alendronate
has limited preventative effects in myocardial fibrosis and
collagen synthesis. This result is inconsistent with those of
previous studies (21,22,53).
This may be because the doses of N-BP were much larger in the
animal models used (50-100 times the doses used in humans)
(53). The effect in these previous
studies was positively associated with the duration of drug
administration (21,22,53).
The present study was an in vitro one in which the optimal
concentration and duration of alendronate treatment remain to be
identified. There are 2 types of signaling pathways involved in Ang
II activation: G protein-dependent and -independent pathways.
Alendronate only affects the G protein-dependent pathway allowing
Ang II to continue to promote fibrosis via other pathways,
including the epidermal growth factor receptor and insulin receptor
signaling pathways (21,22).
The small GTPase, RhoA, belongs to the Rho GTPase
family, part of the Ras-like GTP-binding protein superfamily, whose
members serve important roles in the regulation of cytoskeletal
dynamics, cell adhesion and migration (54). RhoA controls actin stress fiber
formation and acto-myosin contraction at the rear of the cell
(54). The RhoA/ROCK signaling
pathway is closely associated with a number of cardiovascular and
cerebrovascular diseases, including coronary atherosclerotic heart
disease, stroke, and pulmonary vascular disease (55-57).
Evidence has suggested that RhoA is intricately involved in the
pathophysiology of cardiac diseases (58). Constitutive overexpression of RhoA
in cardiomyocytes leads to the spontaneous development of dilated
cardiomyopathy, heart failure, and bradycardia (59). RhoA/ROCK activation is required for
the development of fibrosis in animal models of cardiac fibrosis as
well as fibrosis of other organs, such as the lung, liver, and
kidney (60). Fasudil is a
clinically applied Rho-kinase-inhibitor, which has demonstrated
therapeutic effects against cerebral vasospasm after subarachnoid
hemorrhage (61). Fasudil has also
been demonstrated to have various biological roles in the
cardiovascular system and cardioprotective functions in myocardial
ischemia/reperfusion (I/R) injury animal models. These examples
include improvements in coronary vasodilation, inhibition of
apoptosis and oxidative stress, relieving inflammation and
reduction in endoplasmic reticulum stress and metabolism (24). However, the mechanism of fasudil
action remains unclear. In the present study, fasudil increased
collagen levels of myocardial fibroblasts induced by Ang II,
prevented their proliferation, significantly reduced the mRNA
expression of FGF-2, TGF-β1, VEGF, COL1A1, and HSP47,
and the protein expression of TGF-β1, HSP47, and collagen I. The
efficacy of fasudil was similar to that of rosuvastatin and
slightly lower compared with that of the statin. Considering that
statins act on HMGCR, the rate-limiting enzyme of the MVA pathway
(4), while fasudil acts on
downstream small GTPases (23), it
is speculated that the antimyocardial fibrosis effect induced by
inhibition of the MVA pathway is largely due to the inhibition of
small GTPases.
The findings of the present study suggested that 3
drugs acting at different points in the MVA pathway (rosuvastatin,
aledronate, and fasudil) can all inhibit the proliferation and
collagen synthesis of myocardial fibroblasts induced by Ang II,
with the strongest effect induced by rosuvastatin and the weakest
by aledronate. Results from the present study could provide new
directions and theoretical basis for the clinical treatment and
prevention of diastolic heart failure and the regulation of
myocardial fibrosis.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81700316).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request
Authors' contributions
HX and ML designed the present study. HX, YS, CL, HW
and JH performed the experiments. HX, ML and PX analyzed the data.
HX and ML are responsible for confirming this authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental protocols involving live animals
were reviewed and approved by the Institutional Animal Care and Use
Committee of the Tongji Hospital of Tongji University (approval no.
2019-DW-008; Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Bathaie SZ, Ashrafi M, Azizian M and
Tamanoi F: Mevalonate Pathway and Human Cancers. Curr Mol
Pharmacol. 10:77–85. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yeganeh B, Wiechec E, Ande SR, Sharma P,
Moghadam AR, Post M, Freed DH, Hashemi M, Shojaei S, Zeki AA, et
al: Targeting the mevalonate cascade as a new therapeutic approach
in heart disease, cancer and pulmonary disease. Pharmacol Ther.
143:87–110. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ference BA, Ray KK, Catapano AL, Ference
TB, Burgess S, Neff DR, Oliver-Williams C, Wood AM, Butterworth AS,
Di Angelantonio E, et al: Mendelian Randomization Study of ACLY and
Cardiovascular Disease. N Engl J Med. 380:1033–1042.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Raper A, Kolansky DM and Cuchel M:
Treatment of familial hypercholesterolemia: Is there a need beyond
statin therapy? Curr Atheroscler Rep. 14:11–16. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Oesterle A, Laufs U and Liao JK:
Pleiotropic effects of statins on the cardiovascular system. Circ
Res. 120:229–243. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Steffens S and Mach F: Anti-inflammatory
properties of statins. Semin Vasc Med. 4:417–422. 2004.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Greenwood J, Steinman L and Zamvil SS:
Statin therapy and autoimmune disease: From protein prenylation to
immunomodulation. Nat Rev Immunol. 6:358–370. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Ward NC, Watts GF and Eckel RH: Statin
toxicity. Circ Res. 124:328–350. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Schleyer T, Hui S, Wang J, Zhang Z, Knapp
K, Baker J, Chase M, Boggs R and Simpson RJ Jr: Quantifying unmet
need in statin-treated hyperlipidemia patients and the potential
benefit of further LDL-C reduction through an EHR-based
retrospective cohort study. J Manag Care Spec Pharm. 25:544–554.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Toth PP and Banach M: Statins: Then and
now. Methodist Debakey Cardiovasc J. 15:23–31. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Nochioka K, Sakata Y, Miyata S, Miura M,
Takada T, Tadaki S, Ushigome R, Yamauchi T, Takahashi J and
Shimokawa H: CHART-2 Investigators. Prognostic impact of statin use
in patients with heart failure and preserved ejection fraction.
Circ J. 79:574–582. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kjekshus J, Apetrei E, Barrios V, Böhm M,
Cleland JG, Cornel JH, Dunselman P, Fonseca C, Goudev A, Grande P,
et al: CORONA Group: Rosuvastatin in older patients with systolic
heart failure. N Engl J Med. 357:2248–2261. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu G, Zheng XX, Xu YL, Ru J, Hui RT and
Huang XH: Meta-analysis of the effect of statins on mortality in
patients with preserved ejection fraction. Am J Cardiol.
113:1198–1204. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xu H, Qing T, Shen Y, Huang J, Liu Y, Li
J, Zhen T, Xing K, Zhu S and Luo M: RNA-seq analyses the effect of
high-salt diet in hypertension. Gene. 677:245–250. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li X, Han J, Li L, Wang KJ and Hu SJ:
Effect of farnesyltransferase inhibition on cardiac remodeling in
spontaneously hypertensive rats. Int J Cardiol. 168:3340–3347.
2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhao CZ, Zhao XM, Yang J, Mou Y, Chen B,
Wu HD, Dai DP, Ding J and Hu SJ: Inhibition of farnesyl
pyrophosphate synthase improves pressure overload induced chronic
cardiac remodeling. Sci Rep. 6(39186)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang J, Mou Y, Wu T, Ye Y, Jiang JC, Zhao
CZ, Zhu HH, Du CQ, Zhou L and Hu SJ: Cardiac-specific
overexpression of farnesyl pyrophosphate synthase induces cardiac
hypertrophy and dysfunction in mice. Cardiovasc Res. 97:490–499.
2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Perez-Calahorra S, Laclaustra M,
Marco-Benedi V, Pinto X, Sanchez-Hernandez RM, Plana N, Ortega E,
Fuentes F and Civeira F: Comparative efficacy between atorvastatin
and rosuvastatin in the prevention of cardiovascular disease
recurrence. Lipids Health Dis. 18(216)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wander GS, Hukkeri MYK, Yalagudri S,
Mahajan B and Panda AT: Rosuvastatin: Role in Secondary Prevention
of Cardiovascular Disease. J Assoc Physicians India. 66:70–74.
2018.PubMed/NCBI
|
|
21
|
Han J, Jiang DM, Ye Y, Du CQ, Yang J and
Hu SJ: Farnesyl pyrophosphate synthase inhibitor, ibandronate,
improves endothelial function in spontaneously hypertensive rats.
Mol Med Rep. 13:3787–3796. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Du CQ, Yang L, Yang J, Han J, Hu XS, Wu T
and Hu SJ: Inhibition of farnesyl pyrophosphate synthase prevents
norepinephrine-induced fibrotic responses in vascular smooth muscle
cells from spontaneously hypertensive rats. Hypertens Res.
37:26–34. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Oh KS, Oh BK, Park CH, Seo HW, Kang NS,
Lee JH, Lee JS and Ho Lee B: Cardiovascular effects of a novel
selective Rho kinase inhibitor,
2-(1H-indazole-5-yl)amino-4-methoxy-6-piperazino triazine (DW1865).
Eur J Pharmacol. 702:218–226. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Huang YY, Wu JM, Su T, Zhang SY and Lin
XJ: Fasudil, a Rho-kinase inhibitor, exerts cardioprotective
function in animal models of myocardial ischemia/reperfusion
injury: a meta-analysis and review of preclinical evidence and
possible mechanisms. Front Pharmacol. 9(1083)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Landry NM, Rattan SG and Dixon IMC: An
improved method of maintaining primary murine cardiac fibroblasts
in two-dimensional cell culture. Sci Rep. 9(12889)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ackers-Johnson M, Li PY, Holmes AP,
O'Brien SM, Pavlovic D and Foo RS: A simplified, Langendorff-free
method for concomitant isolation of viable cardiac myocytes and
nonmyocytes from the adult mouse heart. Circ Res. 119:909–920.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) μethod. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Alonso R, Cuevas A and Cafferata A:
Diagnosis and management of statin intolerance. J Atheroscler
Thromb. 26:207–215. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Krähenbühl S, Pavik-Mezzour I and von
Eckardstein A: Unmet νeeds in LDL-C lowering: when statins won't
do! Drugs. 76:1175–1190. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sethunath V, Hu H, De Angelis C,
Veeraraghavan J, Qin L, Wang N, Simon LM, Wang T, Fu X, Nardone A,
et al: Targeting the mevalonate pathway to overcome acquired
anti-HER2 treatment resistance in breast cancer. Mol Cancer Res.
17:2318–2330. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Longo J, Mullen PJ, Yu R, van Leeuwen JE,
Masoomian M, Woon DTS, Wang Y, Chen EX, Hamilton RJ, Sweet JM, et
al: An actionable sterol-regulated feedback loop modulates statin
sensitivity in prostate cancer. Mol Metab. 25:119–130.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wu CK, Yeh CF, Chiang JY, Lin TT, Wu YF,
Chiang CK, Kao TW, Hung KY and Huang JW: Effects of atorvastatin
treatment on left ventricular diastolic function in peritoneal
dialysis patients-The ALEVENT clinical trial. J Clin Lipidol.
11:657–666. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Beck AL, Otto ME, D'Avila LB, Netto FM,
Armendaris MK and Sposito AC: Diastolic function parameters are
improved by the addition of simvastatin to enalapril-based
treatment in hypertensive individuals. Atherosclerosis.
222:444–448. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Akahori H, Tsujino T, Naito Y, Matsumoto
M, Sasaki N, Iwasaku T, Eguchi A, Sawada H, Hirotani S and Masuyama
T: Atorvastatin ameliorates cardiac fibrosis and improves left
ventricular diastolic function in hypertensive diastolic heart
failure model rats. J Hypertens. 32:1534–1541; discussion 1541.
2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Choi SY, Park JS, Roh MS, Kim CR, Kim MH
and Serebruany V: Inhibition of angiotensin II-induced cardiac
fibrosis by atorvastatin in adiponectin knockout mice. Lipids.
52:415–422. 2017.PubMed/NCBI View Article : Google Scholar : Erratum in: Lipids
52: 1061, 2017.
|
|
36
|
Vasan RS, Benjamin EJ and Levy D:
Congestive heart failure with normal left ventricular systolic
function. Clinical approaches to the diagnosis and treatment of
diastolic heart failure. Arch Intern Med. 156:146–157.
1996.PubMed/NCBI
|
|
37
|
Fukuta H, Goto T, Wakami K and Ohte N: The
effect of statins on mortality in heart failure with preserved
ejection fraction: A meta-analysis of propensity score analyses.
Int J Cardiol. 214:301–306. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Florkowski CM, Molyneux SL and George PM:
Rosuvastatin in older patients with systolic heart failure. N Engl
J Med. 358(1301): author reply 1301. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ito S and Nagata K: Roles of the
endoplasmic reticulum-resident, collagen-specific molecular
chaperone Hsp47 in vertebrate cells and human disease. J Biol Chem.
294:2133–2141. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Brown KE, Broadhurst KA, Mathahs MM, Brunt
EM and Schmidt WN: Expression of HSP47, a collagen-specific
chaperone, in normal and diseased human liver. Lab Invest.
85:789–797. 2005.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nishino T, Miyazaki M, Abe K, Furusu A,
Mishima Y, Harada T, Ozono Y, Koji T and Kohno S: Antisense
oligonucleotides against collagen-binding stress protein HSP47
suppress peritoneal fibrosis in rats. Kidney Int. 64:887–896.
2003.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hagiwara S, Iwasaka H, Matsumoto S and
Noguchi T: Introduction of antisense oligonucleotides to heat shock
protein 47 prevents pulmonary fibrosis in
lipopolysaccharide-induced pneumopathy of the rat. Eur J Pharmacol.
564:174–180. 2007.PubMed/NCBI View Article : Google Scholar : Retraction in: Eur
J Pharmacol 792: 80, 2016.
|
|
43
|
Sauk JJ, Nikitakis N and Siavash H: Hsp47
a novel collagen binding serpin chaperone, autoantigen and
therapeutic target. Front Biosci. 10:107–118. 2005.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sun S and McKenna CE: Farnesyl
pyrophosphate synthase modulators: A patent review (2006-2010).
Expert Opin Ther Pat. 21:1433–1451. 2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dhar MK, Koul A and Kaul S: Farnesyl
pyrophosphate synthase: A key enzyme in isoprenoid biosynthetic
pathway and potential molecular target for drug development. N
Biotechnol. 30:114–123. 2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Vukelic S, Stojadinovic O, Pastar I,
Vouthounis C, Krzyzanowska A, Das S, Samuels HH and Tomic-Canic M:
Farnesyl pyrophosphate inhibits epithelialization and wound healing
through the glucocorticoid receptor. J Biol Chem. 285:1980–1988.
2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hooff GP, Wood WG, Müller WE and Eckert
GP: Isoprenoids, small GTPases and Alzheimer's disease. Biochim
Biophys Acta. 1801:896–905. 2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen GP, Yao L, Lu X, Li L and Hu SJ:
Tissue-specific effects of atorvastatin on
3-hydroxy-3-methylglutarylcoenzyme A reductase expression and
activity in spontaneously hypertensive rats. Acta Pharmacol Sin.
29:1181–1186. 2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Chen B, Zhong LY, Yang JX, Pan YY, Chen F,
Yang J, Wu T and Hu SJ: Alteration of mevalonate pathway related
enzyme expressions in pressure overload-induced cardiac hypertrophy
and associated heart failure with preserved ejection fraction. Cell
Physiol Biochem. 32:1761–1775. 2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Holstein SA: A patent review of
bisphosphonates in treating bone disease. Expert Opin Ther Pat.
29:315–325. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Sing CW, Wong AY, Kiel DP, Cheung EY, Lam
JK, Cheung TT, Chan EW, Kung AW, Wong IC and Cheung CL: Association
of alendronate and risk of cardiovascular events in patients with
hip fracture. J Bone Miner Res. 33:1422–1434. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yang J, Chen YN, Xu ZX, Mou Y and Zheng
LR: Alteration of RhoA prenylation ameliorates cardiac and vascular
remodeling in spontaneously hypertensive rats. Cell Physiol
Biochem. 39:229–241. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Susic D, Varagic J, Ahn J, Slama M and
Frohlich ED: Beneficial pleiotropic vascular effects of
rosuvastatin in two hypertensive models. J Am Coll Cardiol.
42:1091–1097. 2003.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Schaefer A, Reinhard NR and Hordijk PL:
Toward understanding RhoGTPase specificity: Structure, function and
local activation. Small GTPases. 5(6)2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Vesterinen HM, Currie GL, Carter S, Mee S,
Watzlawick R, Egan KJ, Macleod MR and Sena ES: Systematic review
and stratified meta-analysis of the efficacy of RhoA and Rho kinase
inhibitors in animal models of ischaemic stroke. Syst Rev.
2(33)2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Sun Z, Wu X, Li W, Peng H, Shen X, Ma L,
Liu H and Li H: RhoA/rock signaling mediates peroxynitrite-induced
functional impairment of Rat coronary vessels. BMC Cardiovasc
Disord. 16(193)2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Antoniu SA: Targeting RhoA/ROCK pathway in
pulmonary arterial hypertension. Expert Opin Ther Targets.
16:355–363. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Loirand G, Sauzeau V and Pacaud P: Small G
proteins in the cardiovascular system: Physiological and
pathological aspects. Physiol Rev. 93:1659–1720. 2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn
GW II, Ross J Jr, Chien KR and Brown JH: Cardiac-specific
overexpression of RhoA results in sinus and atrioventricular nodal
dysfunction and contractile failure. J Clin Invest. 103:1627–1634.
1999.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Shimizu T and Liao JK: Rho kinases and
cardiac remodeling. Circ J. 80:1491–1498. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Hasegawa S, Hasegawa Y and Miura M:
Current therapeutic drugs against cerebral vasospasm after
subarachnoid hemorrhage: a comprehensive review of basic and
clinical studies. Curr Drug Deliv. 14:843–852. 2017.PubMed/NCBI View Article : Google Scholar
|