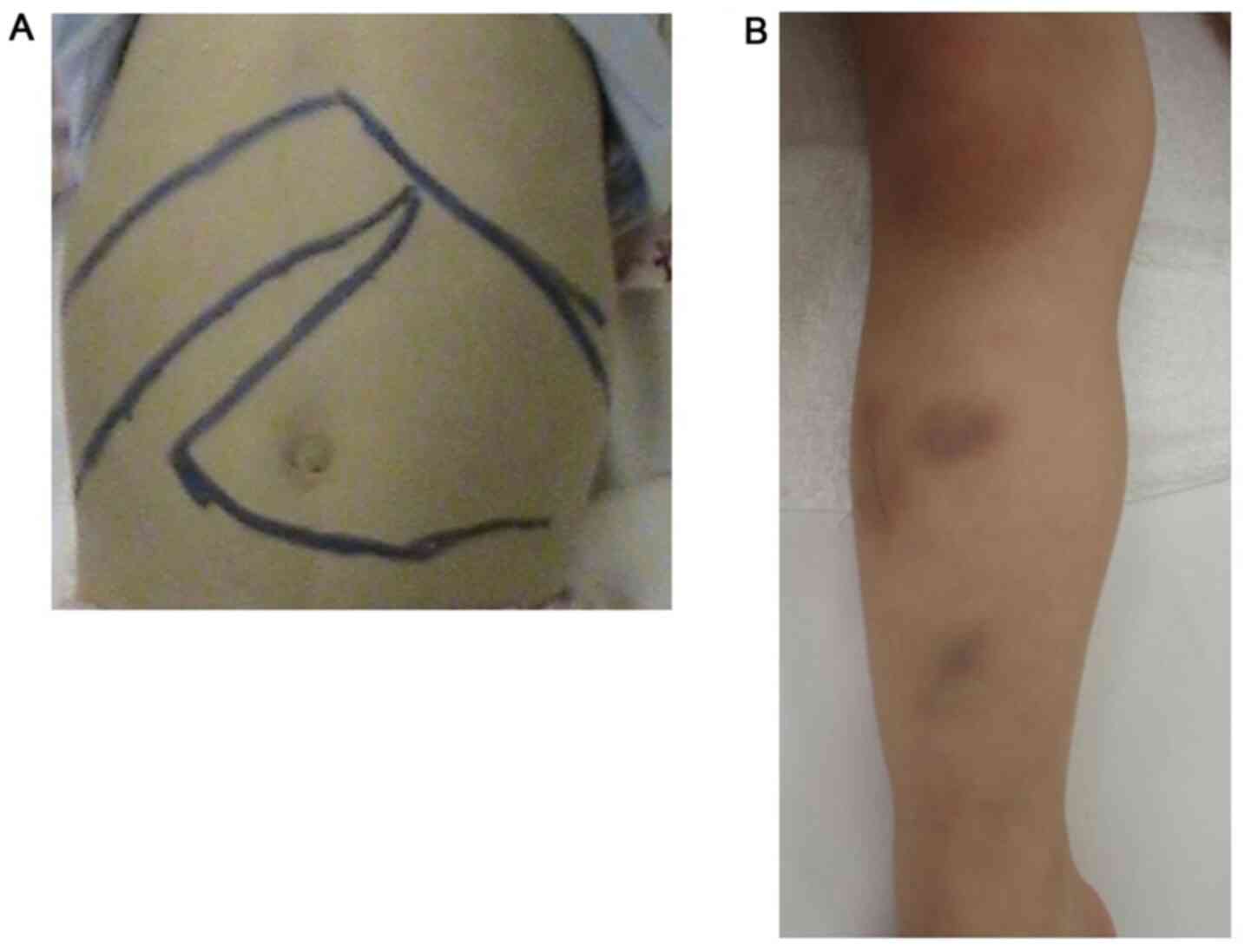
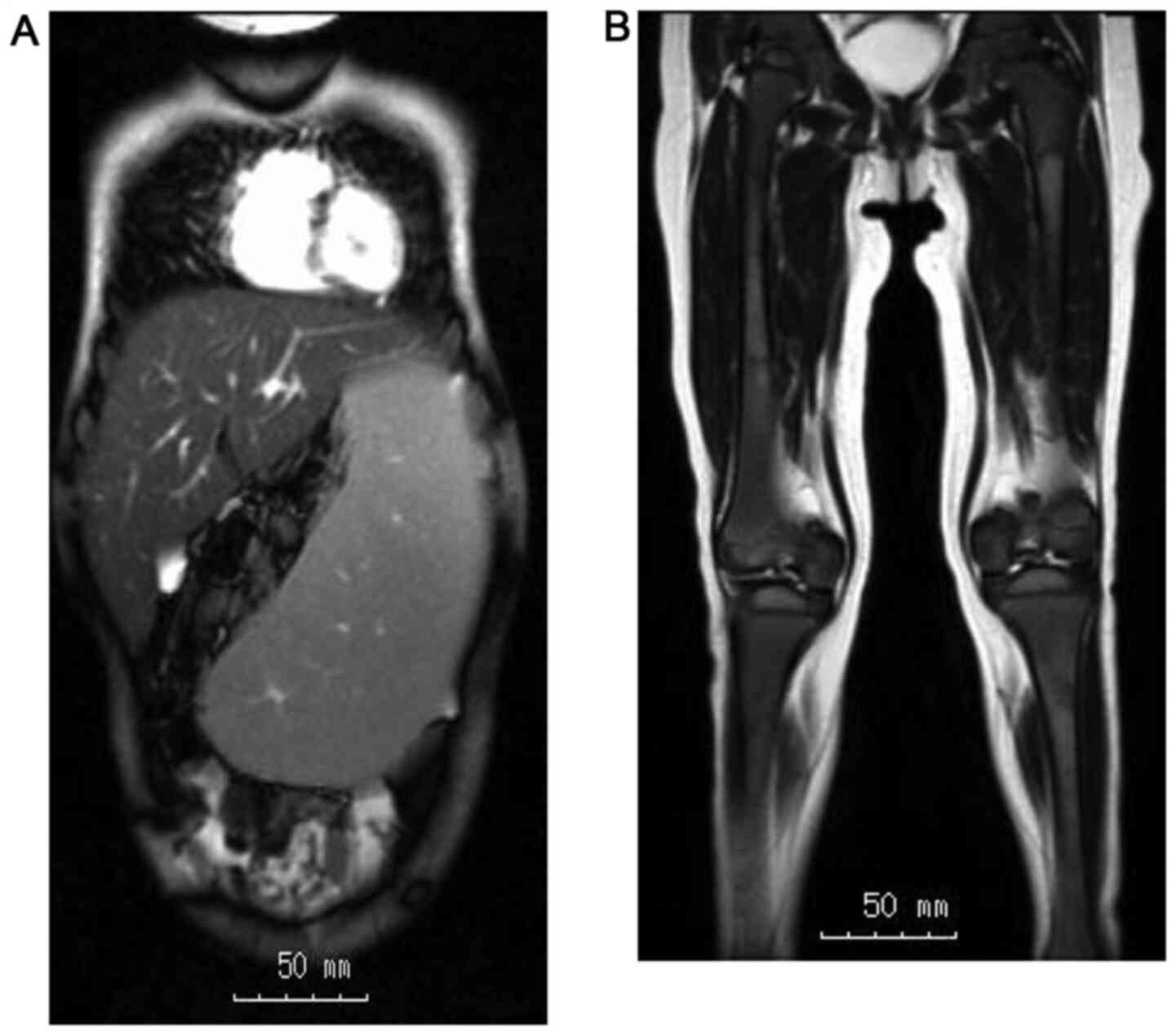
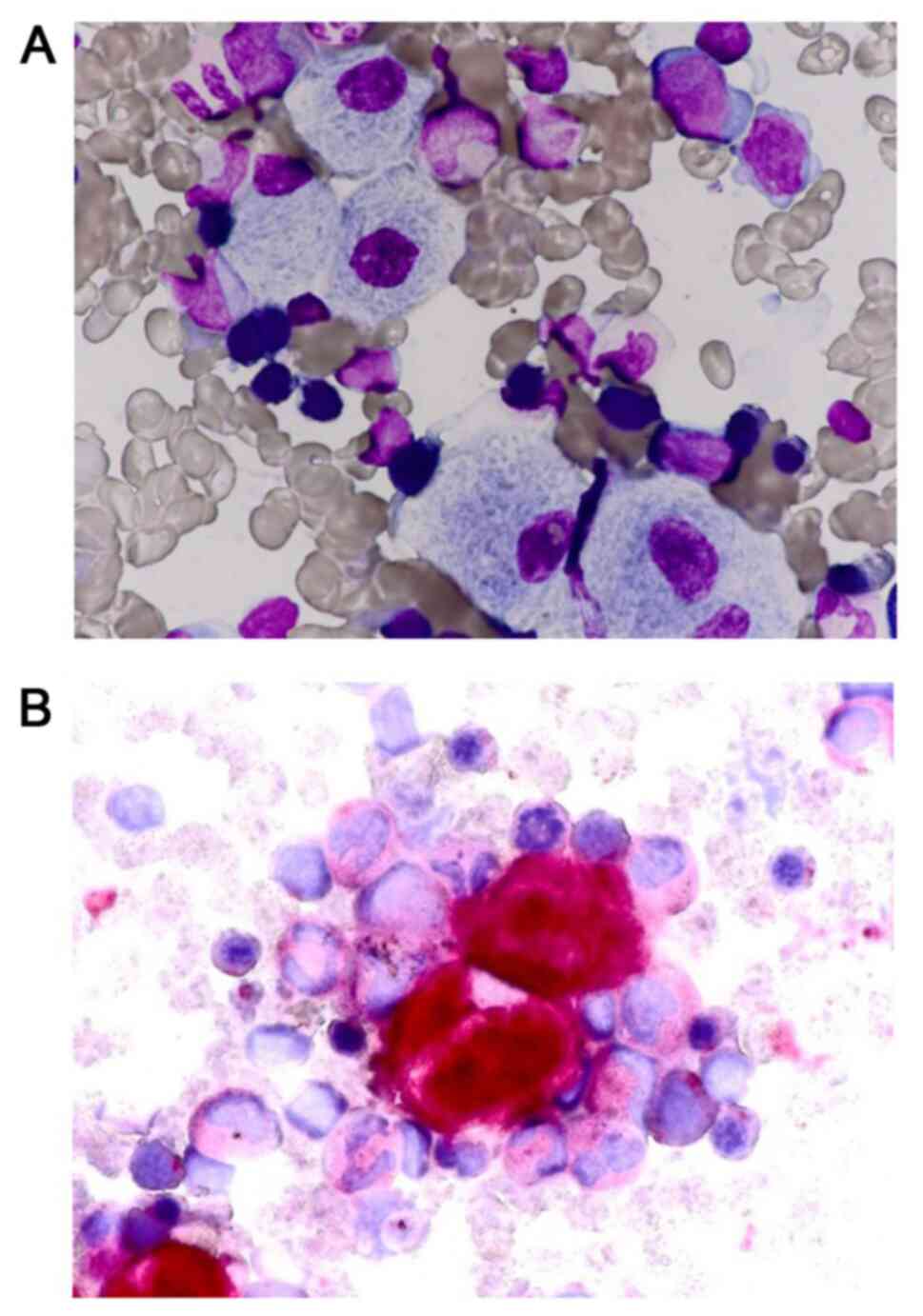
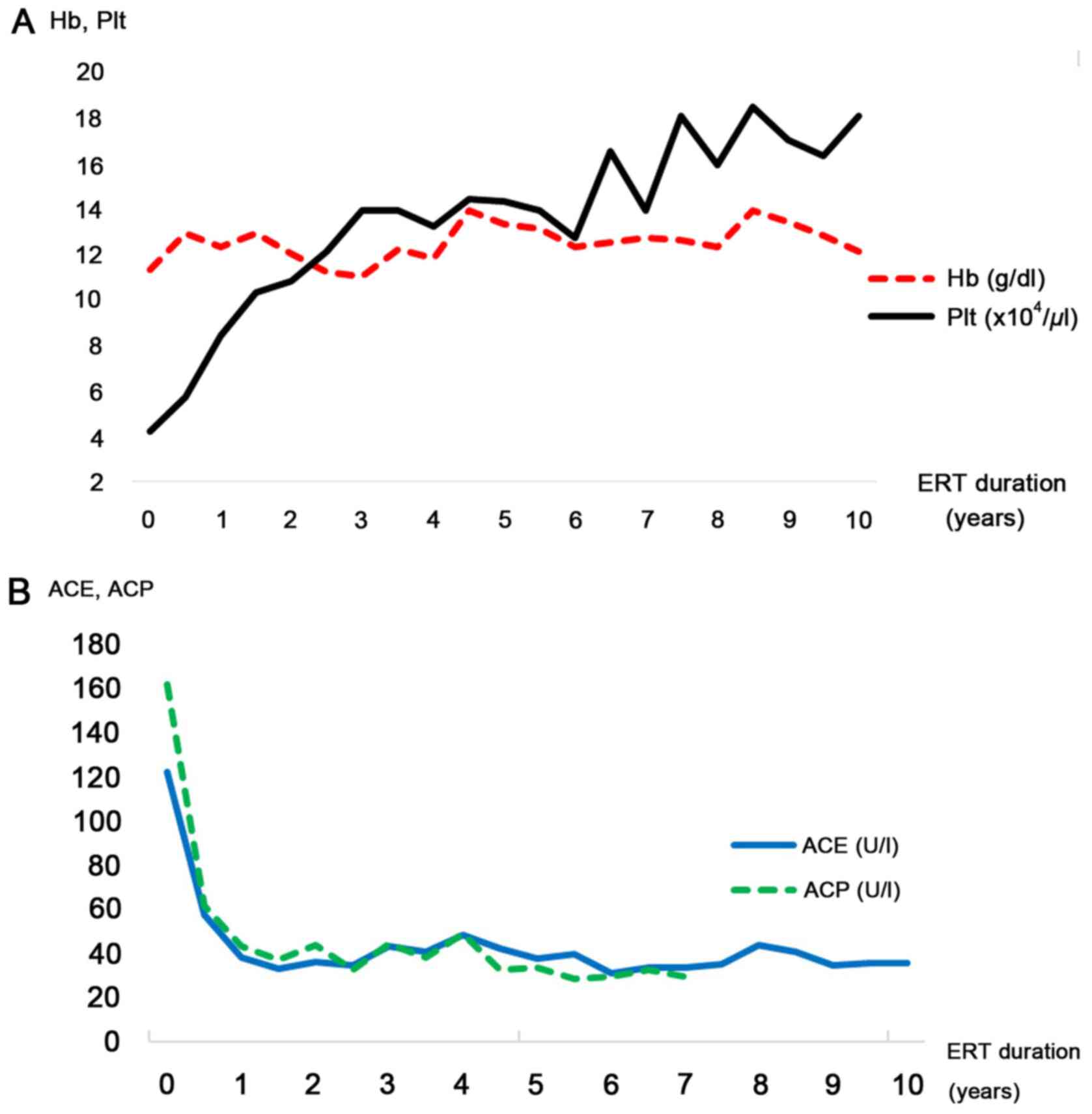
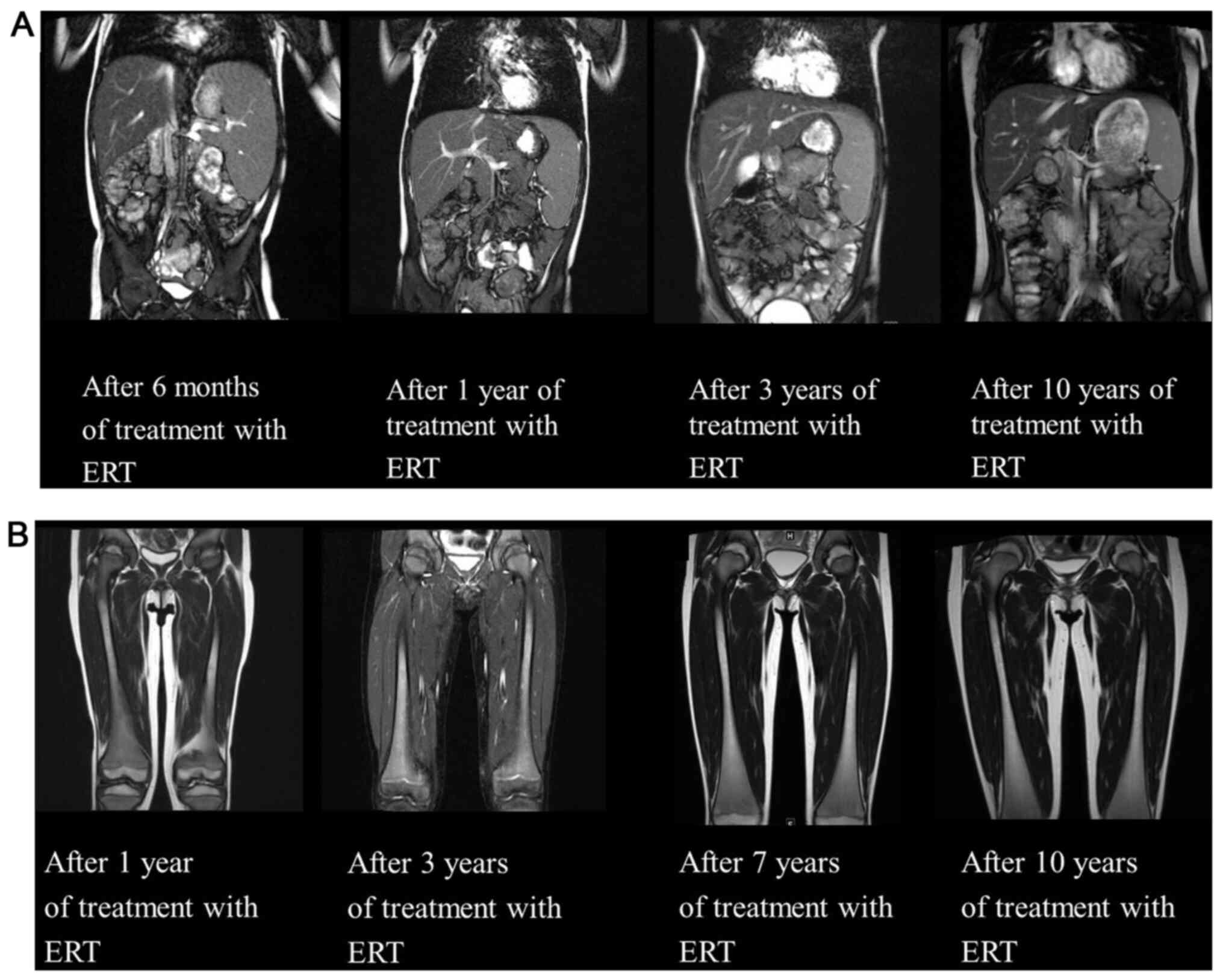
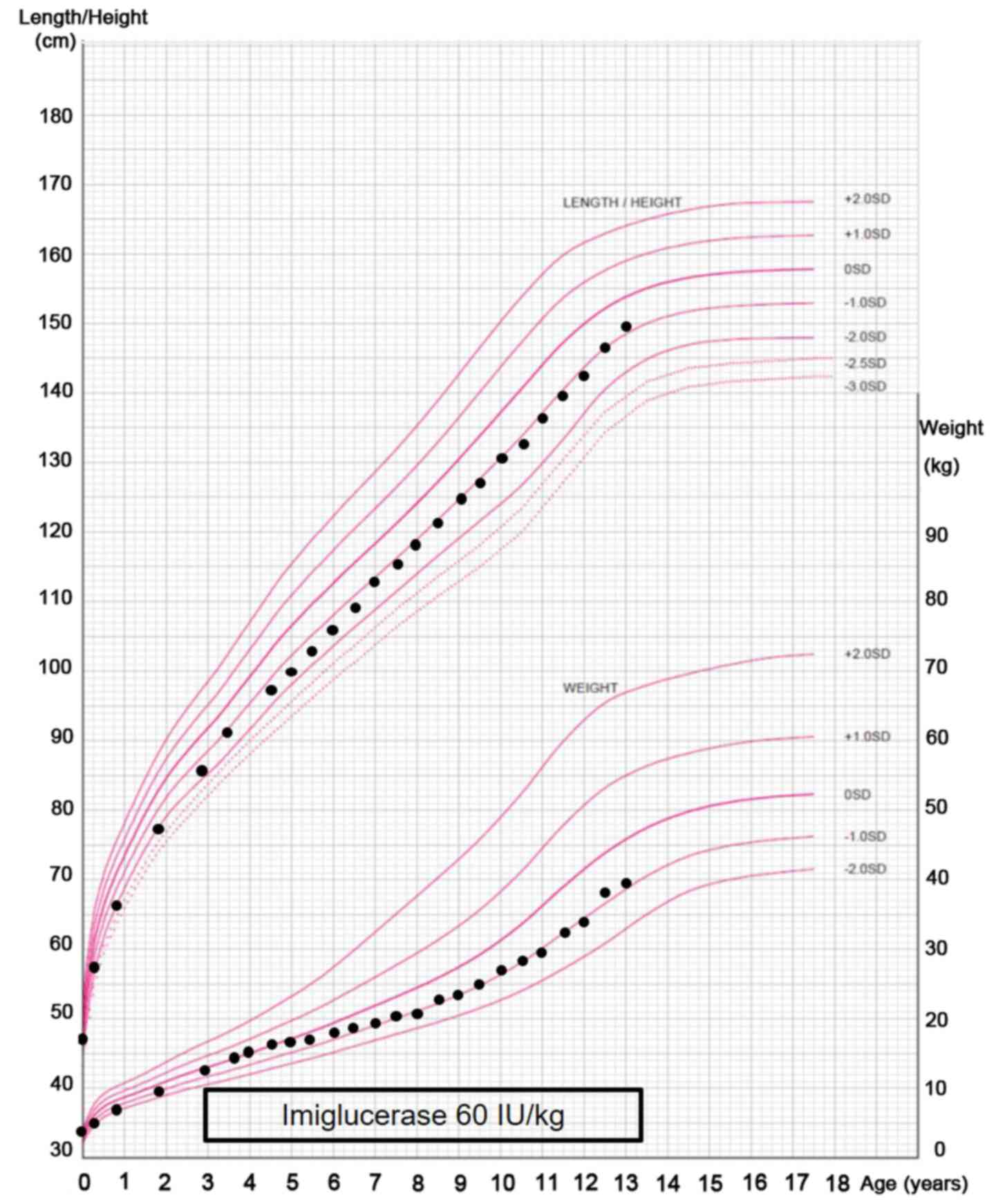
|
1
|
Baris HN, Cohen IJ and Mistry PK: Gaucher
disease: The metabolic defect, pathophysiology, phenotypes and
natural history. Pediatr Endocrinol Rev. 12 (Suppl 1):72–81.
2014.PubMed/NCBI
|
|
2
|
Cox TM and Schofield JP: Gaucher's
disease: Clinical features and natural history. Baillieres Clin
Haematol. 10:657–689. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Thomas AS, Mehta A and Hughes DA: Gaucher
disease: Haematological presentations and complications. Br J
Haematol. 165:427–440. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hruska KS, La Marca ME, Scott CR and
Sidransky E: Gaucher disease: mutation and polymorphism spectrum in
the glucocerebrosidase gene (GBA). Hum Mutat. 29:567–583.
2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Eto Y, Ida H, Ohashi T, Okuyama T, Sakai
N, Takayanagi M, Narita A and Nanba E (eds): Gaucher Disease
Update. 1st edition. Shindantochiryousha, pp91-92, 2016 (In
Japanese).
|
|
6
|
Starzyk K, Richards S, Yee J, Smith SE and
Kingma W: The long-term international safety experience of
imiglucerase therapy for Gaucher disease. Mol Genet Metab.
90:157–163. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Poorthuis BJ, Wevers RA, Kleijer WJ,
Groener JE, de Jong JG, van Weely S, Niezen-Koning KE and van
Diggelen OP: The frequency of lysosomal storage diseases in The
Netherlands. Hum Genet. 105:151–156. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tajima A, Yokoi T, Ariga M, Ito T,
Kaneshiro E, Eto Y and Ida H: Clinical and genetic study of
Japanese patients with type 3 Gaucher disease. Mol Genet Metab.
97:272–277. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ida H, Rennert OM, Kawame H, Maekawa K and
Eto Y: Mutation prevalence among 47 unrelated Japanese patients
with Gaucher disease: Identification of four novel mutations. J
Inherit Metab Dis. 20:67–73. 1997.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Andersson H, Kaplan P, Kacena K and Yee J:
Eight-year clinical outcomes of long-term enzyme replacement
therapy for 884 children with Gaucher disease type 1. Pediatrics.
122:1182–1190. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Boot RG, Verhoek M, de Fost M, Hollak CE,
Maas M, Bleijlevens B, van Breemen MJ, van Meurs M, Boven LA, Laman
JD, et al: Marked elevation of the chemokine CCL18/PARC in Gaucher
disease: A novel surrogate marker for assessing therapeutic
intervention. Blood. 103:33–39. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hollak CE, van Weely S, van Oers MH and
Aerts JM: Marked elevation of plasma chitotriosidase activity. A
novel hallmark of Gaucher disease. J Clin Invest. 93:1288–1292.
1994.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Arkadir D, Dinur T, Revel-Vilk S, Becker
Cohen M, Cozma C, Hovakimyan M, Eichler S, Rolfs A and Zimran A:
Glucosylsphingosine is a reliable response biomarker in Gaucher
disease. Am J Hematol. 93:E140–E142. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ida H, Rennert OM, Kobayashi M and Eto Y:
Effects of enzyme replacement therapy in thirteen Japanese
paediatric patients with Gaucher disease. Eur J Pediatr. 160:21–25.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Grabowski GA, Kacena K, Cole JA, Hollak
CE, Zhang L, Yee J, Mistry PK, Zimran A, Charrow J and vom Dahl S:
Dose-response relationships for enzyme replacement therapy with
imiglucerase/alglucerase in patients with Gaucher disease type 1.
Genet Med. 11:92–100. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wenstrup RJ, Roca-Espiau M, Weinreb NJ and
Bembi B: Skeletal aspects of Gaucher disease: A review. Br J
Radiol. 75 (Suppl 1):A2–A12. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mendelsohn E, Meir A, Abrahamov A, Elstein
D, Zimran A and Levy-Khademi F: Growth and final height of children
with Gaucher disease: A 15-year follow-up at an Israeli Gaucher
center. Blood Cells Mol Dis. 68:97–99. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Robertson PL, Maas M and Goldblatt J:
Semiquantitative assessment of skeletal response to enzyme
replacement therapy for Gaucher's disease using the bone marrow
burden score. AJR Am J Roentgenol. 188:1521–1528. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vom Dahl S, Poll L, Di Rocco M, Ciana G,
Denes C, Mariani G and Maas M: Evidence-based recommendations for
monitoring bone disease and the response to enzyme replacement
therapy in Gaucher patients. Curr Med Res Opin. 22:1045–1064.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hughes D, Mikosch P, Belmatoug N, Carubbi
F, Cox T, Goker-Alpan O, Kindmark A, Mistry P, Poll L, Weinreb N,
et al: Gaucher Disease in Bone: From Pathophysiology to Practice. J
Bone Miner Res. 34:996–1013. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Weinreb NJ, Goldblatt J, Villalobos J,
Charrow J, Cole JA, Kerstenetzky M, vom Dahl S and Hollak C:
Long-term clinical outcomes in type 1 Gaucher disease following 10
years of imiglucerase treatment. J Inherit Metab Dis. 36:543–553.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Charrow J, Dulisse B, Grabowski GA and
Weinreb NJ: The effect of enzyme replacement therapy on bone crisis
and bone pain in patients with type 1 Gaucher disease. Clin Genet.
71:205–211. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sims KB, Pastores GM, Weinreb NJ,
Barranger J, Rosenbloom BE, Packman S, Kaplan P, Mankin H, Xavier
R, Angell J, et al: Improvement of bone disease by imiglucerase
(Cerezyme) therapy in patients with skeletal manifestations of type
1 Gaucher disease: Results of a 48-month longitudinal cohort study.
Clin Genet. 73:430–440. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Poll LW, Koch JA, vom Dahl S, Willers R,
Scherer A, Boerner D, Niederau C, Häussinger D and Mödder U:
Magnetic resonance imaging of bone marrow changes in Gaucher
disease during enzyme replacement therapy: First German long-term
results. Skeletal Radiol. 30:496–503. 2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Poll LW, Maas M, Terk MR, Roca-Espiau M,
Bembi B, Ciana G and Weinreb NJ: Response of Gaucher bone disease
to enzyme replacement therapy. Br J Radiol. 75 (Suppl 1):A25–A36.
2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Simonaro CM, Haskins ME and Schuchman EH:
Articular chondrocytes from animals with a dermatan sulfate storage
disease undergo a high rate of apoptosis and release nitric oxide
and inflammatory cytokines: A possible mechanism underlying
degenerative joint disease in the mucopolysaccharidoses. Lab
Invest. 81:1319–1328. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Horowitz M, Tzuri G, Eyal N, Berebi A,
Kolodny EH, Brady RO, Barton NW, Abrahamov A and Zimran A:
Prevalence of nine mutations among Jewish and non-Jewish Gaucher
disease patients. Am J Hum Genet. 53:921–930. 1993.PubMed/NCBI
|
|
28
|
Beutler E, Gelbart T and West C:
Identification of six new Gaucher disease mutations. Genomics.
15:203–205. 1993.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ida H, Rennert OM, Ito T, Maekawa K and
Eto Y: Type 1 Gaucher disease: Phenotypic expression and natural
history in Japanese patients. Blood Cells Mol Dis. 24:73–81.
1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ida H, Owen MR, Iwasawa K, Kobayashi M and
Eto Y: Clinical and genetic studies of Japanese homozygotes for the
Gaucher disease L444P mutation. J Hum Genet. 105:120–126.
1999.PubMed/NCBI View Article : Google Scholar
|