Introduction
Gaucher disease (GD) is an autosomal recessive
lysosomal storage disorder caused by glucocerebrosidase (GBA)
enzyme deficiency and is characterized by hepatosplenomegaly,
anemia, thrombocytopenia, bone pain and growth retardation
(1,2). GD may be categorized into three
subtypes depending on clinical symptoms: Non-neuronopathic (type
1), acute neuronopathic (type 2) and subacute neuronopathic (type
3) (3). Over 300 mutations of the
GBA gene have been identified and there appear to be certain
genotype-phenotype correlations (4). In Japan, the use of recombinant GBA
(imiglucerase) was approved in 1998(5). With respect to treatment, the
development of enzyme replacement therapy (ERT) has markedly
improved patient prognosis (6).
The prevalence associated with GD in Japan is lower
than that in Western countries (1 per 330,000 vs. 1.16 per 100,000
live births, respectively) (7,8). In
Japan, only ~150 patients were estimated to have GD and the
proportion of GD type 1 is rarer than that in other countries.
Therefore, data on Japanese pediatric patients with GD type 1 are
limited. The present study reported the case of a Japanese
pediatric patient diagnosed with GD type 1, who presented with
hepatosplenomegaly and thrombocytopenia at the age of 2 years, and
was subsequently treated with ERT, which proved to be significantly
effective for treating this condition.
Case report
A 2-year and 10-month-old female patient suffering
from hepatosplenomegaly was referred to Dokkyo Medical University
Saitama Medical Center (Saitama, Japan) in June 2009. The patient's
medical history revealed no past episodes of fractures or
convulsions and there were no signs of developmental delay. There
was no family history of GD or Parkinson's disease. At 1-year and
0-month old, the patient started experiencing occasional petechiae
with a bleeding tendency. On admission, the patient's body weight
and length were 12.0 kg [-0.9 standard deviation (SD); normal
range, -2.0 SD to +2.0 SD] and 87.0 cm (-2.1 SD), respectively. On
physical examination, the patient was revealed to have severe
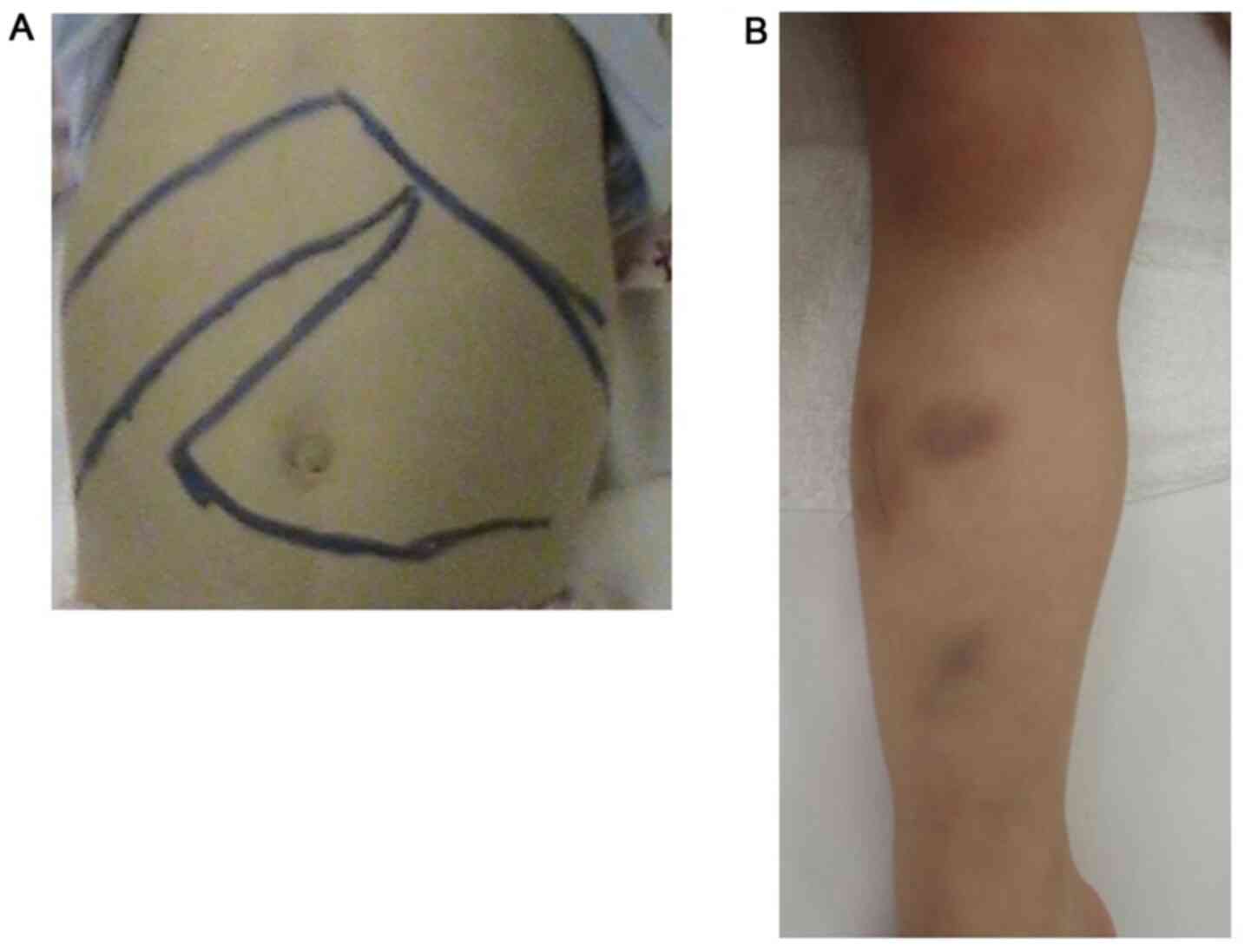
hepatosplenomegaly, petechiae and purpura (Fig. 1). Neurologic examinations revealed
no abnormalities. The patient's laboratory data are summarized in
Table I. The patient had
thrombocytopenia with slight anemia (platelets,
3.5x104/µl; hemoglobin, 10.4 g/dl). Blood analysis
revealed high levels of acid phosphatase (ACP) and
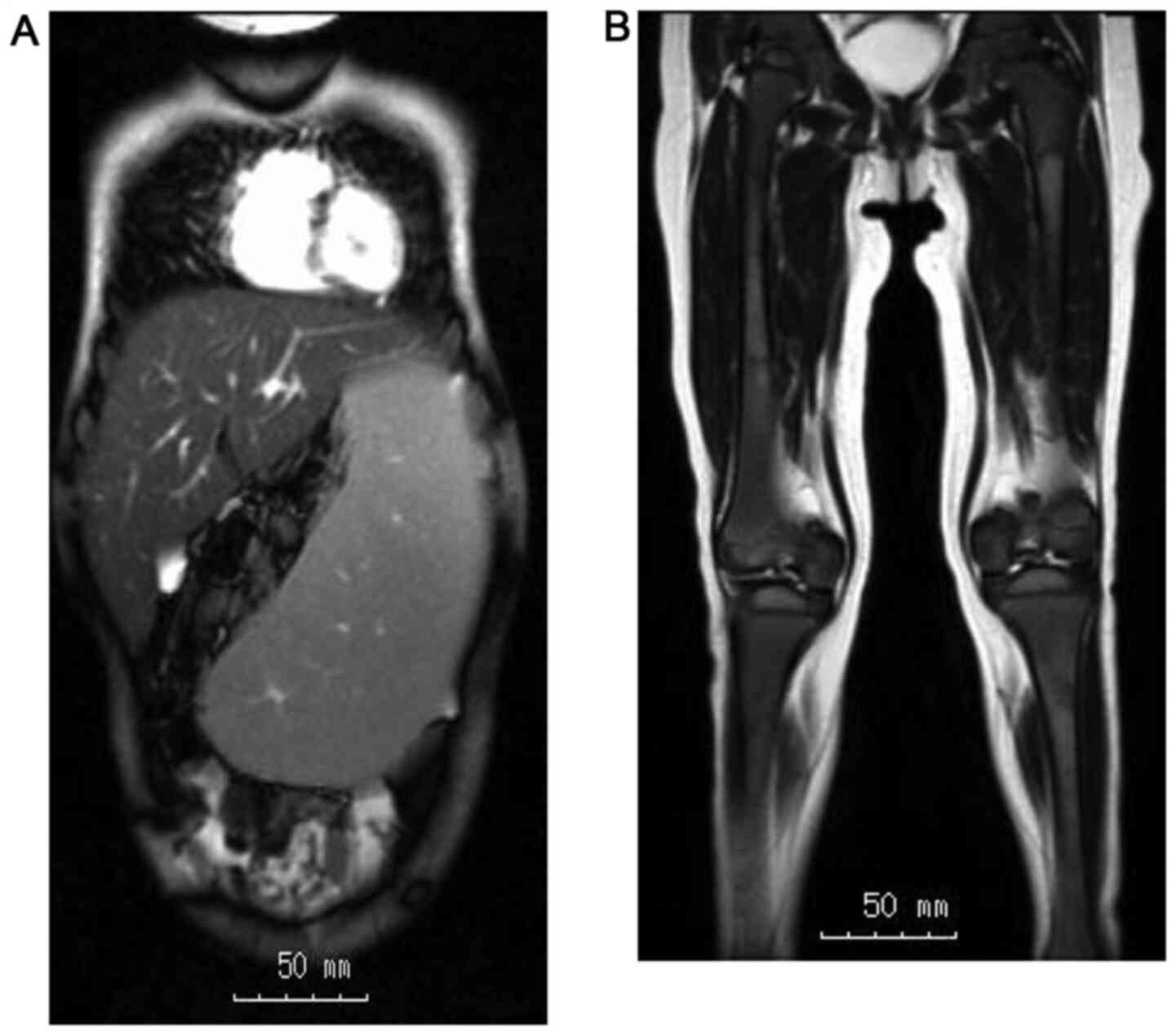
angiotensin-converting enzyme (ACE). Abdominal MRI revealed huge
hepatosplenomegaly (liver, 13.6x14.0 cm; spleen, 14.2x6.7 cm;
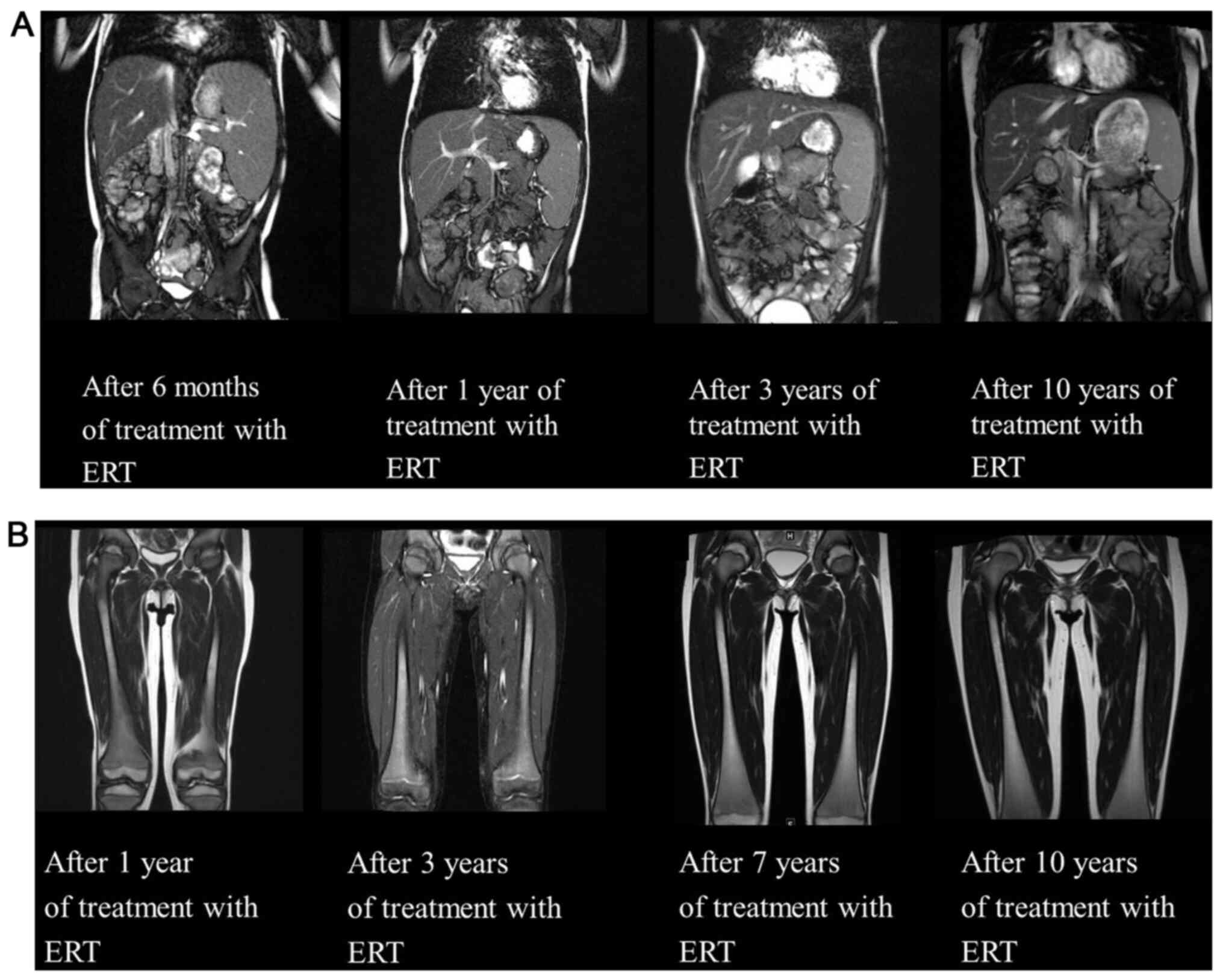
Fig. 2). An Erlenmeyer flask
deformity was seen on X-ray examination. MRI of the femora featured
a high-intensity area within the diaphysis region (Fig. 2). Each MRI was collected on a
Siemens Magnetom Avanto version VE11 (Siemens AG) and the software
used to analyze the images was SYNAPSE VINCENT version 2.8.0
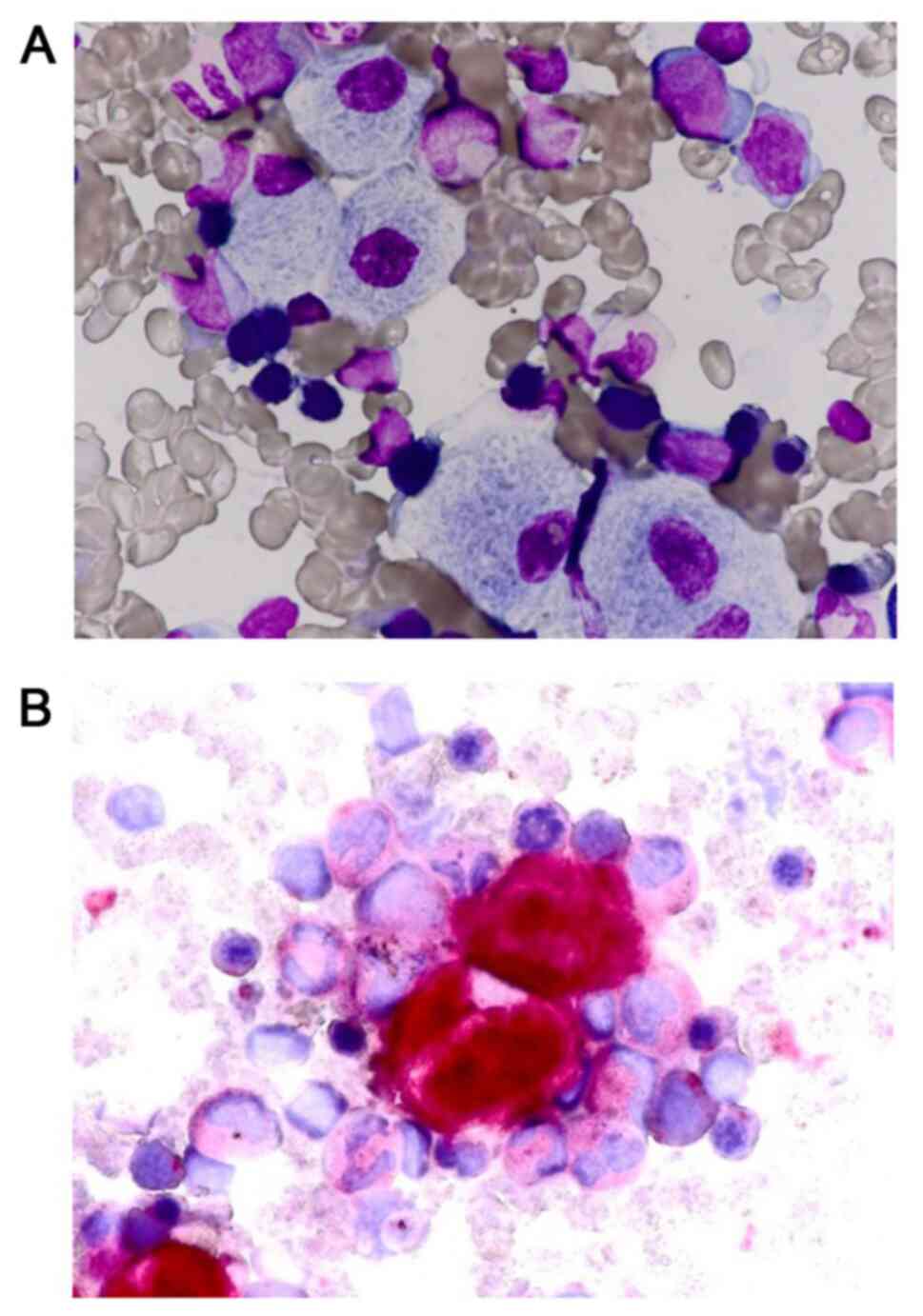
(Fujifilm Medical Co., Ltd). A bone marrow biopsy revealed the
presence of Gaucher cells (Fig. 3).
Enzymatic activity of leukocyte β-glucosidase, the measurement of
which is necessary for the definitive diagnosis of Gaucher disease
(2), had decreased to 186.7
nmol/h/mg (reference range, 1,424.0-2,338.0 nmol/h/mg). Based on
these results, the patient was clinically diagnosed with GD. GBA
gene was analyzed using GBA sequence analysis. The analysis
revealed the compound heterozygote mutation of F213I (c.754T>A)
on exon 7 and L444P (c.1448T>C) on exon 11 by using Sanger
sequencing (9).
 | Table ILaboratory findings prior to enzyme
treatment therapy. |
Table I
Laboratory findings prior to enzyme
treatment therapy.
| Parameter | Value | Normal reference
range/limit |
|---|
| Hematology | | |
|
White blood
cells (x103/µl) | 6.2 | 4.0-12.0 |
|
Hemoglobin
(g/dl) | 10.4 | 11.5-14.5 |
|
Hematocrit
(%) | 32.9 | 33.0-45.0 |
|
Platelets
(x103/µl) | 3.5 | 15.0-40.0 |
| Leukocyte count | | |
|
Neutrophils
(%) | 49.0 | 38.0-68.0 |
|
Lymphocytes
(%) | 40.0 | 27.0-47.0 |
|
Monocytes
(%) | 9.0 | 3.0-7.0 |
| Blood chemistry | | |
|
Total
bilirubin (mg/dl) | 0.84 | 0.40-1.00 |
|
Aspartate
aminotransferase (U/l) | 45 | 20-60 |
|
Alanine
aminotransferase (U/l) | 13 | 5-45 |
|
Alkaline
phosphatase (U/l) | 253 | 410-1,150 |
|
Gamma-glutamyl
transpeptidase (U/l) | 14 | 5-32 |
|
Total
protein (g/dl) | 6.7 | 6.1-7.9 |
|
Albumin
(g/dl) | 4.39 | 3.40-5.10 |
|
Blood urea
nitrogen (mg/dl) | 11.0 | 5.0-18.0 |
|
Creatinine
(mg/dl) | 0.20 | 0.17-0.37 |
|
Sodium
(mEq/l) | 139 | 134-143 |
|
Potassium
(mEq/l) | 4.3 | 3.3-4.6 |
|
Chloride
(mEq/l) | 107 | 98-106 |
|
Immunoglobulin
G (mg/dl) | 631 | 345-1,236 |
| Coagulation | | |
|
Prothrombin
time activity (%) | 102.0 | 70.0-120.0 |
|
Activated
partial thromboplastin time (sec) | 39.1 | |
|
Fibrinogen
(mg/dl) | 417 | 130-350 |
| Acid phosphatase
(U/l) | 134.2 | <14.4 |
|
Angiotensin-converting-enzyme (U/µl) | 103.2 | 9.3-21.4 |
| Leukocyte
β-glucosidase (nmol/h/mg) | 186.7 | 1,424.0-2,338 |
At the age of 3.0 years, ERT was initiated for the
patient, along with an intravenous infusion of 60 U/kg imiglucerase
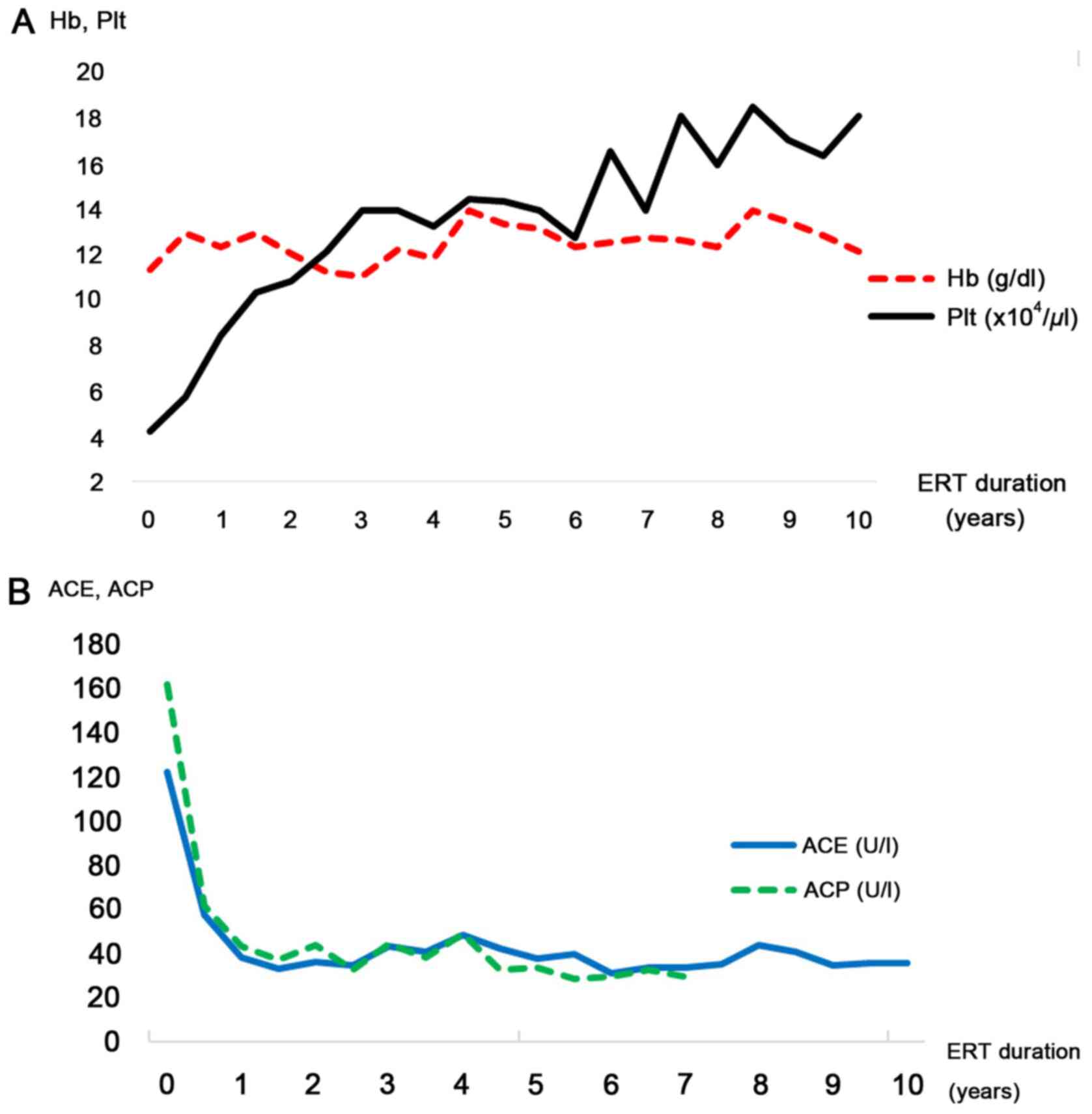
every other week. Hemoglobin levels and the platelet count were
observed to improve gradually, and they normalized after two years
(Fig. 4A). ACP and ACE levels,
biomarkers of GD progression, were also indicated to improve
(Fig. 4B). Abdominal MRI at six
months after the initiation of ERT revealed a decrease in the size
of the liver and spleen (liver: From 13.6x14.0 to 12.8x8.6 cm;
spleen, from 14.2x6.7 to 13.5x5.1 cm), which also normalized after
1 year (Fig. 5A). Since then,
follow-up abdominal MRIs were performed yearly to ensure early
recognition of any changes (Fig.
5). Conversely, MRI of the femora indicated no improvement of
the high-intensity area within the diaphysis region over 10 years
(Fig. 5B). Although there was a
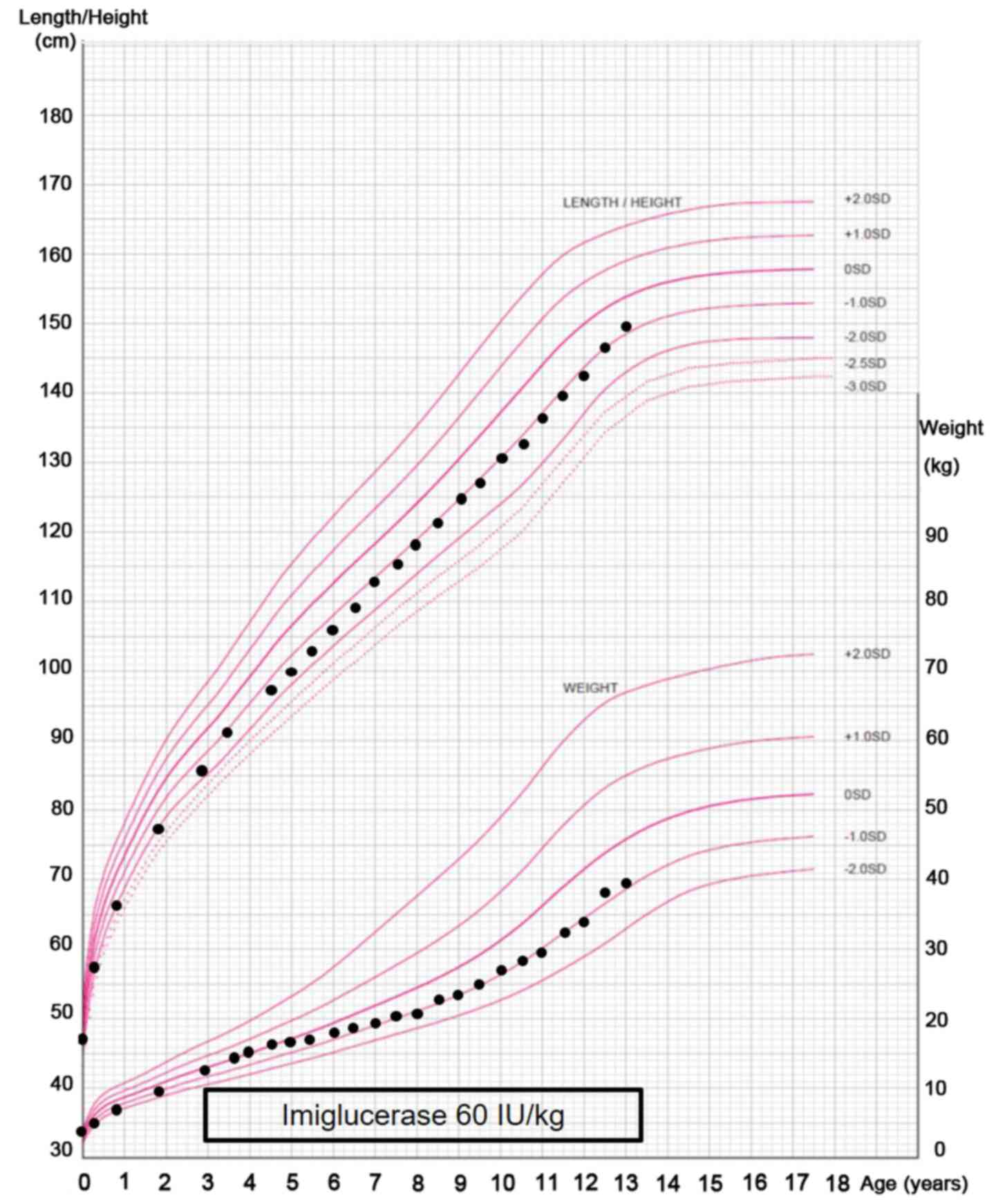
certain degree of growth retardation [body weight 12.0 kg (-0.9 SD)
and length 87.0 cm (-2.1 SD), normal body weight, 12.8±1.5 kg;
normal length, 90.9±3.3 cm)] at the initiation of ERT, this was
observed to improve relatively early following initiation of ERT
(Fig. 6).
Discussion
The present study described the case of a Japanese
pediatric patient diagnosed with GD type 1 who significantly
improved on ERT. In Japan, the prevalence of GD is lower compared
with that in Western countries (8),
and thus, limited data on Japanese pediatric patients with GD are
currently available.
At 6 months following initiation of ERT, the liver
and spleen of the patient had significantly decreased in size and
had normalized after 1 year of ERT. Hemoglobin levels and the
platelet count improved gradually and had normalized after 2 years.
ACP and ACE, which are commonly used for clinical diagnosis and
assessment of treatment effectiveness, decreased gradually after
initiation of ERT. Andersson et al (10) reported that long-term ERT for
pediatric patients with GD type 1 was efficient. Anemia and
thrombocytopenia, which were present in >50% of patients at
baseline, had resolved in >95% of patients after 8 years of
treatment. The response in the present case was consistent with
this. Conversely, in the present case, chitotriosidase, chemokine
C-C motif ligand 18 (CCL18) and glucosylsphingosine could not be
evaluated as good markers of disease progression prior to and
during ERT, as the quantification of these markers was not
commercially available in Japan. CCL18 and chitotriosidase are
secreted by Gaucher cells and are markers for disease burden;
consequently, plasma levels of these biomarkers are significantly
increased in untreated patients with GD (11,12).
Glucosylsphingosine is also a sensitive and specific biomarker for
primary diagnosis and follow-up monitoring of patients with GD
(13). Increasing the accuracy of
these biomarkers may spare patients from invasive examinations such
as bone marrow biopsies to diagnose GD. A previous study indicated
that long-term high-dose ERT (60 U/kg every 2 weeks) was required
to obtain sufficient improvement to maintain health among pediatric
patients with severe GD and ERT reduction (from 60 to 30 or 15 U/kg
every 2 weeks) was associated with insufficient improvement of
hemoglobin levels and the platelet count (14). ERT with imiglucerase was reported to
achieve a dose-dependent improvement in hematological and visceral
parameters in GD type 1(15).
Therefore, in the present case, long-term high-dose ERT (60 U/kg
every 2 weeks) was maintained without any dose reduction.
Early manifestation of GD during childhood leads to
growth retardation and a pathological growth pattern with
tubulation of the metaphyseal area of the long bone, in particular,
metaphysics of the distal femur and proximal tibia (16). Erlenmeyer flask deformity results
from enlargement of the metaphyseal area and consequent absence of
the typical concave diametaphyseal curve. Although it is generally
held that ERT accelerates the growth rate (10), Mendelsohn et al (17) reported that ERT did not
significantly impact the mean final height SD score. In their
study, the effect of delayed puberty on stature was highlighted:
Short stature during childhood is compensated by a long time of
linear growth usually up to 15 years old, thereby enabling the
patients to reach a normal body height in adulthood. In the present
case, growth retardation was present before treatment, but it
improved ~2 years following initiation of ERT. With respect to MRI,
as bone fractures were never observed and bone pain was not
reported (no lameness or complaints observed), it is unclear
whether the high-intensity area within the diaphysis region of the
femora on MRI had any pathological significance. The MRI of the
femora did not improve in terms of intensity level within the
diaphysis region 10 years after initiation of ERT. The MRI of the
femora was evaluated by using the bone marrow signal intensity
scores for the femurs (18,19). The scores were 5 points prior to ERT
and they did not improve for 3 years. At 4 years after initiation,
the scores decreased to 4 points but did not change anymore
thereafter. Certain studies suggested that ERT is effective for
bone lesions (20-22),
while others demonstrated limited effectiveness (23). The response to ERT for bone disease
in adult patients with GD is considered much slower compared with
pediatric patients. In the study by Sims et al (23), patients who received long-term
high-dose ERT exhibited improvement in bone pain and bone crisis.
However, bone pain was present in 73% of patients at baseline and
39% of patients still reported bone pain after 48 months of ERT.
Furthermore, compared with the rapid reduction of bone pain and
bone crisis, associated morphological changes were observed only
after longer periods of therapy. According to one study,
improvements on MRI were observed at the earliest after 2-3 years
of treatment (24), whereas in
another study, a decrease in bone marrow accumulation of
glucocerebroside was apparent only after 3-4 years of ERT (25). The latter study indicated that
generalized bone pain may be easily ameliorated by ERT, whereas
pain attributable to focal, irreversible lesions tends to be more
resistant (25). Furthermore,
evidence from other lysosomal disorders may also support the role
of inflammatory mediators in osteoarticular disease. In a similar
manner to that in GD, bone complications in patients with
mucopolysaccharidosis type 1 are less significantly modified by ERT
with α-L-iduronidase compared to that in other organs such as the
liver and spleen (26). In general,
the efficacy of ERT in bone and heart tissue is inferior in terms
of uptake compared to that in the liver and spleen. Therefore, the
effect on bone lesions may be delayed compared to that in the liver
and spleen.
The mutation spectrum of GBA in the Japanese
population is quite different from that in Western countries. For
example, the L444P, N307S, 84GG, D409H, IVS2+1 and R463C mutations
are commonly observed in Ashkenazi Jewish and non-Jewish
populations in Israel (27). These
six mutations account for ~90 and ~75% of the total mutated alleles
observed among Jewish and non-Jewish patients with GD, respectively
(28). In particular, N307S is the
most common mutation associated with GD type 1.In contrast, the
N370S mutation have not been identified among Japanese patients.
where the L444P and F213I alleles are the most prevalent (29). These two mutations account for 54%
of all mutations. The prevalence of L444P and F213I among Japanese
patients is ~43.6 and 14.9%, respectively, whereas in Ashkenazi
Jewish patients, these mutations are relatively rare (9). Tajima et al (8) reported that L444P/L444P accounted for
61% of all genotypes in patients thought to have GD type 1 at
diagnosis but who later developed GD type 3, followed by
L444P/F213I (22%) and F213I/F213I (11%). L444P was associated with
more severe disease and Japanese patients with GD tended to
experience an earlier onset and progression with greater
neurological involvement (30). In
Japanese patients, the L444P/L444P genotype was more highly
associated with GD type 3 as in Caucasian patients, compared with
the association with GD type 1. F213I was also associated with
neurologic symptoms, which was a unique mutation among patients
with GD from Asia and it remains to be clarified how the F213I
mutation affected the clinical course (8). In the case of the present study,
symptoms were already present since the age of 1 year and the
severity appeared to be high. However, the present case was
clinically diagnosed with GD type 1 as there were no neurological
complications. It may be reasoned that as the patient was young,
neurological symptoms may not have developed and early initiation
of ERT may have contributed to the prevention of these symptoms.
Long-term neurological assessments as part of a follow-up are
required to enable the evaluation of prognosis.
The prevalence of GD in Japan is much less compared
to that in Western countries (7,8), and
it is not widely known among Japanese pediatricians and
hematologists. Although ~150 patients with GD have been identified,
there may be numerous undiagnosed patients with GD in Japan. In
addition, the genetic distribution of Japanese patients with GD is
completely different from that of Jewish patients and the frequency
of unidentified mutations remains higher than that of Jewish
patients with GD (28). Long-term
follow-up studies and advancing technologies such as next-genome
sequencing may determine the association between GD severity and
novel mutations. Therefore, it is necessary to raise the awareness
of GD among more general pediatricians and hematologists in
Japan.
In conclusion, the present study reported the case
of a Japanese pediatric patient with GD type 1 who, despite having
a high disease severity, exhibited significant improvements on ERT
over a 10-year duration.
Acknowledgements
The authors would like to thank Dr Toshiro Nagai
(Department of Pediatrics, Nakagawa-no-sato Ryouiku Center,
Saitama, Japan), Dr Yoshiko Abe (Department of Pediatrics, Abe
Clinic, Saitama, Japan), Dr Takayoshi Tsuchiya (Department of
Pediatrics, Sakurayama Pediatric Clinic, Kanagawa, Japan), Dr
Yuriko Tanaka (Department of Pediatrics, Tanaka Clinic, Shizuoka,
Japan) and Dr Kazuo Obata (Department of Pediatrics, Maple Lane
Pediatrics, Hyogo, Japan) for their useful discussions.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YO was the patient's doctor and wrote the
manuscript. TI and SN made substantial contributions to the
conception of the study and the acquisition of data. ST, HI, MS,
AN, NM and TM made substantial contributions to the analysis and
interpretation of data. HI analyzed enzymatic activity and the GBA
gene. All of the authors were involved in writing the manuscript
and revised the manuscript critically for important intellectual
content, and read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Dokkyo Medical University Saitama Medical Center (Koshigaya, Japan;
no. 2025). Formal written informed consent was obtained from the
patient and the parents of the patient.
Patient consent for publication
The authors received formal written informed consent
from the patient and the parents of the patient approving the
publication of these data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Baris HN, Cohen IJ and Mistry PK: Gaucher
disease: The metabolic defect, pathophysiology, phenotypes and
natural history. Pediatr Endocrinol Rev. 12 (Suppl 1):72–81.
2014.PubMed/NCBI
|
|
2
|
Cox TM and Schofield JP: Gaucher's
disease: Clinical features and natural history. Baillieres Clin
Haematol. 10:657–689. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Thomas AS, Mehta A and Hughes DA: Gaucher
disease: Haematological presentations and complications. Br J
Haematol. 165:427–440. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hruska KS, La Marca ME, Scott CR and
Sidransky E: Gaucher disease: mutation and polymorphism spectrum in
the glucocerebrosidase gene (GBA). Hum Mutat. 29:567–583.
2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Eto Y, Ida H, Ohashi T, Okuyama T, Sakai
N, Takayanagi M, Narita A and Nanba E (eds): Gaucher Disease
Update. 1st edition. Shindantochiryousha, pp91-92, 2016 (In
Japanese).
|
|
6
|
Starzyk K, Richards S, Yee J, Smith SE and
Kingma W: The long-term international safety experience of
imiglucerase therapy for Gaucher disease. Mol Genet Metab.
90:157–163. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Poorthuis BJ, Wevers RA, Kleijer WJ,
Groener JE, de Jong JG, van Weely S, Niezen-Koning KE and van
Diggelen OP: The frequency of lysosomal storage diseases in The
Netherlands. Hum Genet. 105:151–156. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tajima A, Yokoi T, Ariga M, Ito T,
Kaneshiro E, Eto Y and Ida H: Clinical and genetic study of
Japanese patients with type 3 Gaucher disease. Mol Genet Metab.
97:272–277. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ida H, Rennert OM, Kawame H, Maekawa K and
Eto Y: Mutation prevalence among 47 unrelated Japanese patients
with Gaucher disease: Identification of four novel mutations. J
Inherit Metab Dis. 20:67–73. 1997.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Andersson H, Kaplan P, Kacena K and Yee J:
Eight-year clinical outcomes of long-term enzyme replacement
therapy for 884 children with Gaucher disease type 1. Pediatrics.
122:1182–1190. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Boot RG, Verhoek M, de Fost M, Hollak CE,
Maas M, Bleijlevens B, van Breemen MJ, van Meurs M, Boven LA, Laman
JD, et al: Marked elevation of the chemokine CCL18/PARC in Gaucher
disease: A novel surrogate marker for assessing therapeutic
intervention. Blood. 103:33–39. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hollak CE, van Weely S, van Oers MH and
Aerts JM: Marked elevation of plasma chitotriosidase activity. A
novel hallmark of Gaucher disease. J Clin Invest. 93:1288–1292.
1994.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Arkadir D, Dinur T, Revel-Vilk S, Becker
Cohen M, Cozma C, Hovakimyan M, Eichler S, Rolfs A and Zimran A:
Glucosylsphingosine is a reliable response biomarker in Gaucher
disease. Am J Hematol. 93:E140–E142. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ida H, Rennert OM, Kobayashi M and Eto Y:
Effects of enzyme replacement therapy in thirteen Japanese
paediatric patients with Gaucher disease. Eur J Pediatr. 160:21–25.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Grabowski GA, Kacena K, Cole JA, Hollak
CE, Zhang L, Yee J, Mistry PK, Zimran A, Charrow J and vom Dahl S:
Dose-response relationships for enzyme replacement therapy with
imiglucerase/alglucerase in patients with Gaucher disease type 1.
Genet Med. 11:92–100. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wenstrup RJ, Roca-Espiau M, Weinreb NJ and
Bembi B: Skeletal aspects of Gaucher disease: A review. Br J
Radiol. 75 (Suppl 1):A2–A12. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mendelsohn E, Meir A, Abrahamov A, Elstein
D, Zimran A and Levy-Khademi F: Growth and final height of children
with Gaucher disease: A 15-year follow-up at an Israeli Gaucher
center. Blood Cells Mol Dis. 68:97–99. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Robertson PL, Maas M and Goldblatt J:
Semiquantitative assessment of skeletal response to enzyme
replacement therapy for Gaucher's disease using the bone marrow
burden score. AJR Am J Roentgenol. 188:1521–1528. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vom Dahl S, Poll L, Di Rocco M, Ciana G,
Denes C, Mariani G and Maas M: Evidence-based recommendations for
monitoring bone disease and the response to enzyme replacement
therapy in Gaucher patients. Curr Med Res Opin. 22:1045–1064.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hughes D, Mikosch P, Belmatoug N, Carubbi
F, Cox T, Goker-Alpan O, Kindmark A, Mistry P, Poll L, Weinreb N,
et al: Gaucher Disease in Bone: From Pathophysiology to Practice. J
Bone Miner Res. 34:996–1013. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Weinreb NJ, Goldblatt J, Villalobos J,
Charrow J, Cole JA, Kerstenetzky M, vom Dahl S and Hollak C:
Long-term clinical outcomes in type 1 Gaucher disease following 10
years of imiglucerase treatment. J Inherit Metab Dis. 36:543–553.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Charrow J, Dulisse B, Grabowski GA and
Weinreb NJ: The effect of enzyme replacement therapy on bone crisis
and bone pain in patients with type 1 Gaucher disease. Clin Genet.
71:205–211. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sims KB, Pastores GM, Weinreb NJ,
Barranger J, Rosenbloom BE, Packman S, Kaplan P, Mankin H, Xavier
R, Angell J, et al: Improvement of bone disease by imiglucerase
(Cerezyme) therapy in patients with skeletal manifestations of type
1 Gaucher disease: Results of a 48-month longitudinal cohort study.
Clin Genet. 73:430–440. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Poll LW, Koch JA, vom Dahl S, Willers R,
Scherer A, Boerner D, Niederau C, Häussinger D and Mödder U:
Magnetic resonance imaging of bone marrow changes in Gaucher
disease during enzyme replacement therapy: First German long-term
results. Skeletal Radiol. 30:496–503. 2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Poll LW, Maas M, Terk MR, Roca-Espiau M,
Bembi B, Ciana G and Weinreb NJ: Response of Gaucher bone disease
to enzyme replacement therapy. Br J Radiol. 75 (Suppl 1):A25–A36.
2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Simonaro CM, Haskins ME and Schuchman EH:
Articular chondrocytes from animals with a dermatan sulfate storage
disease undergo a high rate of apoptosis and release nitric oxide
and inflammatory cytokines: A possible mechanism underlying
degenerative joint disease in the mucopolysaccharidoses. Lab
Invest. 81:1319–1328. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Horowitz M, Tzuri G, Eyal N, Berebi A,
Kolodny EH, Brady RO, Barton NW, Abrahamov A and Zimran A:
Prevalence of nine mutations among Jewish and non-Jewish Gaucher
disease patients. Am J Hum Genet. 53:921–930. 1993.PubMed/NCBI
|
|
28
|
Beutler E, Gelbart T and West C:
Identification of six new Gaucher disease mutations. Genomics.
15:203–205. 1993.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ida H, Rennert OM, Ito T, Maekawa K and
Eto Y: Type 1 Gaucher disease: Phenotypic expression and natural
history in Japanese patients. Blood Cells Mol Dis. 24:73–81.
1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ida H, Owen MR, Iwasawa K, Kobayashi M and
Eto Y: Clinical and genetic studies of Japanese homozygotes for the
Gaucher disease L444P mutation. J Hum Genet. 105:120–126.
1999.PubMed/NCBI View Article : Google Scholar
|