Introduction
Trauma is the most common cause of death for all age
groups below the age of 44 and the single largest cause for years
of life lost in the United States (1-3).
Acute trauma care, therefore, is not only of the utmost importance
from a clinical point of view but also from a public health
perspective (4). One of the most
dire consequences of severe trauma, and a leading cause of
post-injury death, is hemorrhagic shock as it may result in
ischemia-reperfusion injury (IRI), systemic inflammation, and
multi-organ failure (5). Despite
enormous amounts of research, the underlying pathomechanisms are
still poorly understood, hindering a target-oriented therapy.
Hemorrhagic shock is known to cause a mismatch
between oxygen supply and demand. The tissue hypoxia that occurs
results in pathophysiological disturbances of the cellular
machinery. Protein maturation and folding in the endoplasmic
reticulum (ER) is a highly energy-dependent cellular process
(6). Perturbations in the ER
homeostasis result in an impaired ER function and an accumulation
of unfolded proteins in the ER lumen-a condition defined as ER
stress (7). Consequently, the cell
activates specific signaling pathways, which are collectively known
as the unfolded protein response (UPR) consisting of three primary
branches: Protein kinase RNA-like endoplasmic reticulum kinase
(PERK), activating transcription factor 6 (ATF6), and
inositol-requiring enzyme 1 (IRE1). Under physiological conditions,
binding immunoglobulin protein (BiP), a molecular chaperone and
master regulator of ER function, binds to the luminal domains of
PERK, ATF6, and IRE1(8). Upon ER
stress, BiP dissociates from each stress sensor and facilitates
their activation. Whereas PERK and IRE1 undergo oligomerization and
trans-autophosphorylation (9), ATF6
is activated by proteolytic cleavage in the Golgi compartment
(10). After its translocation to
the nucleus, ATF6 promotes the transcription of genes coding for
adaptive proteins, such as chaperones, and of X-box binding protein
1 (XBP1) mRNA (11). Before its
translation, XBP1 mRNA is spliced by activated IRE1
endoribonuclease (12). XBP1 codes
for an active transcription factor, which amplifies the synthesis
of components of the ER-associated protein degradation machinery
(13). Additionally, the activation
of the PERK pathway results in a global attenuation of protein
synthesis by phosphorylation of eukaryotic initiation factor 2α
(eIF2α) (14). In brief, the UPR is
primarily a pro-survival cellular response aiming to restore
protein homeostasis in the ER by facilitating protein folding,
reducing protein synthesis, and increasing protein degradation.
However, if the UPR fails to reestablish protein homeostasis and ER
stress persists, cell death may occur (15).
As reported in previous studies, the above-described
cellular mechanisms influence the extent of liver damage following
hemorrhagic shock and reperfusion (HS/R) (16-18).
Furthermore, the UPR mediates the protective effects of stress
preconditioning, an established concept to mitigate subsequent IRI.
Previous studies have demonstrated that ischemia-reperfusion
associated hepatocellular, myocardial, and neuronal cell damage can
be alleviated by stress preconditioning (e.g. by remote ischemic
preconditioning or lipopolysaccharide pretreatment) (19,20).
From a clinical point of view, the therapeutic potential of this
method has already been demonstrated by the bench-to-bedside
transfer of remote ischemic preconditioning in patients undergoing
coronary artery bypass surgery (21,22).
Even though IRI and HS/R signaling pathways have been studied for
decades, some of the underlying mechanisms remain elusive. However,
developing novel therapeutic approaches requires a deeper
understanding of the pathophysiology of IRI and HS/R. Based on the
above-described findings and our previous results, we hypothesized
that ER stress preconditioning alleviates liver damage following
HS/R and may thereby reveal a target with potential therapeutic
relevance. To investigate this hypothesis and to identify the
underlying protective mechanisms, we injected mice with the
pharmacological ER stress inducer tunicamycin (TM) and subjected
them to HS/R 48 h later.
Materials and methods
Animal care
C57BL/6 mice, 10 weeks of age, were purchased from
Charles River Laboratories (Sulzfeld, Germany). Due to the
influence of female sex steroids on post-hemorrhagic organ damage,
only male mice were included (23).
The animals were housed in the animal facility of the University of
Heidelberg at a temperature of 21˚C and a 12 h light/dark cycle.
The mice had one to two weeks for acclimatization and had access to
water and chow ad libitum. During acclimatization the
animals were housed in group cages and with start of the experiment
animals were placed individually. All study protocols were reviewed
and approved by the section for agriculture and veterinary services
of the Regional Council, Karlsruhe, Germany
(35-9185.81/G-65/13).
Experimental model
The shock protocol was performed as previously
published (17,18). Briefly, anesthesia was induced via
inhalation of 4% isoflurane (Abbott Laboratories Ltd.) in an
acrylic glass chamber. After loss of righting reflex the animals
were placed in a supine position on a heating cushion and
anesthesia was maintained by administering ~1.2% isoflurane via a
face mask. For temperature control (37.0±0.5˚C) a rectal probe was
inserted. Before the bilateral dissection of the groins, 25 µl (~5
mg/kg body weight) of 0.5% Bupivacaine hydrochloride (AstraZeneca
Gmbh) was applied for local anesthesia intraincisionally.
Subsequently, the femoral arteries were cannulated with a
polyethylene tubing, previously flushed with a heparin solution.
The right catheter was connected to a blood pressure analyzer
(BPA-400, Micro-med Inc.), the left catheter was used to withdraw
blood and induce a hemorrhagic shock. The mean arterial pressure
(MAP) was maintained for 90 min at 30±5 mmHg. Afterwards, Ringer's
solution, three times the shed blood volume, was injected for
resuscitation. Subsequently, the catheters were removed, the
vessels were ligated, and the skin was closed. After the
discontinuation of isoflurane inhalation, the mouse was placed in
its cage and observed till emergence.
Mice were randomly assigned to five different
groups. HS/R groups were treated as outlined above. Depending on
the group assignment mice received either TM (0.75 mg/kg BW in
solution, Merck KGaA) or its drug vehicle (DV) dimethyl sulfoxide
(DMSO), dissolved in 100 µl Ringer's solution, which was given
intraperitoneally 48 h before shock induction. Sham controls (SC)
were created for each HS/R group. SC groups received the same
treatment as the corresponding HS/R group but did not undergo
hemorrhagic shock. For the evaluation of physiologic baseline
values, euthanasia was performed under anesthesia without any prior
treatment given to the mice. This baseline control (BC) group as
well as the HS/R+TM group consisted of six animals whereas all
other groups (SC+DV, SC+TM, HS/R+DV) contained three animals.
Experimental group size calculation was based on our previous
study, which compared mice undergoing HS/R procedure and receiving
the drug vehicle (DV) DMSO during reperfusion with mice undergoing
HS/R procedure without any pharmaceutical intervention (18). Each group included 6-7 animals. The
same comparison was performed for mice undergoing sham procedure
with 5 animals per group. Using Mann-Whitney U test we could not
find any significant differences in transaminase levels or
percentage of cell death areas (HS/R groups). Therefore, we
concluded that the applied dosage of our solvent DMSO does not
influence our main outcome parameters and decided to limit the
number to three animals per control group in the present study.
Tissue harvesting and plasma
analysis
A period of 14 h after shock induction, anesthesia
was induced and maintained via inhalation of 4% isoflurane. After
loss of the paw withdrawal reflex and observation of agonal
breathing, a laparotomy and thoracotomy were performed. For
euthanasia the right heart ventricle was punctured using a
heparinized 1-ml syringe. Death was confirmed by observation of
cardiac and respiratory arrest. The collected blood was centrifuged
(10 and 5 min at 2,000 x g) and 50 µl of the plasma was used to
measure aspartate aminotransferase (ASAT) as well as alanine
aminotransferase (ALAT) concentrations (Fuji Dri-Chem NX500i;
FujiFilm Europe GmbH). Subsequently, the body was flushed with a
heparin solution through puncture of the left ventricle. The liver
was then harvested and halved. One half was snap-frozen by
submerging the sample tubes into liquid nitrogen and the other half
was placed in 4% paraformaldehyde (PFA).
Histology
After fixation in 4% PFA for at least 24 h the
livers were dehydrated using a series of alcohols with increasing
concentrations and acetone. Hereafter, the organs were embedded in
paraffin. The tissues were then cut into 5 µm sections. For
deparaffinization the slides were placed in xylene and afterwards
immersed in a series of alcohols with decreasing concentrations for
rehydration. The sections were then either processed for
immunohistochemistry or stained with hematoxylin and eosin
(H&E) using a standard protocol. To quantify liver damage, the
H&E-stained liver tissue sections were assessed for dead cells
assessed by at least two investigators experienced in analyzing
histological slides. We first measured the percentage of vessels
and dead cells using ImageJ (Version: 1.51f; Wayne Rasband,
National Institutes of Health). In the following, the number of
pixels covered by vessels were subtracted from the total pixel
amount and the percentage of irreversibly damaged tissue was
calculated. Corresponding morphological features were a rupture of
the nuclear envelope or chromatin condensation, loss of cell
borders with irregular fragmentation and/or washed-out image of
cytoplasm (24,25). Since cell swelling per se is a
reversible state, swollen cells were not considered as dead cells
(24). Six-eight representative
visual fields (100 x) per animal of the HS/R+DV and 1-5
representative visual fields per animal of the HS/R+TM group were
analyzed. In total, we evaluated 20 representative visual fields
for each HS/R group. The varying numbers of analyzed visual fields
per animal resulted from the different group sizes. Subsequently,
the median percentage of damaged tissue per animal was calculated
and used for further statistical analysis.
Immunohistochemistry
Immunohistochemical staining was performed as
previously published (17,18). In the following, we describe the BiP
staining more detailed since that was our standard protocol.
Therefore, only the differences to the BiP staining process are
mentioned for the other staining procedures.
BiP
After deparaffinization and rehydration the tissue
sections were immersed in 0.45% hydrogen peroxide for 20 min to
block the endogenous peroxidase activity. Heat-induced antigen
retrieval was performed by placing the sections for 20 min in a
citrate buffer (pH 6.0, 10 mM) set to 100˚C. Following this, the
blocking agent, 1.5% donkey serum (#sc-2023, Santa Cruz
Biotechnology, Inc.) in phosphate buffered saline (PBS), was
applied. The tissue sections were then incubated overnight at 4˚C
with the primary antibody, goat anti-BiP (#sc-1050, Santa Cruz
Biotechnology, Inc.), at a dilution of 1:50. In the next step, the
secondary antibody, donkey anti-goat IgG (#sc-2023, Santa Cruz
Biotechnology, Inc.), was administered for 30 min at room
temperature, diluted at 1:200. For signal detection, alkaline
phosphatase (AP; #AK-5000, Vector Laboratories) was applied and
Fast Red was used as chromogen. To stop the reaction, the slides
were immersed in distilled water. Finally, hematoxylin was applied
for counterstaining and the slides were mounted using an aqueous
mountant.
ATF6
2.5% horse serum was used as a blocking solution
(#MP-5401, Vector Laboratories, Inc.). The tissue sections were
incubated overnight at 4˚C with the rabbit anti-ATF6 antibody
(#NBP1-77251, Novus Biologicals Europe, Cambridge, Great Britain),
diluted at 1:100. As secondary antibody we applied a horse
anti-rabbit antibody for 30 min at room temperature, which was
supplied as ready-to-use kit and already conjugated with alkaline
phosphatase by the manufacturer (#MP-5401, Vector Laboratories,
Inc.).
pPERK
To block unspecific antibody binding sites, a
solution of 5% skim milk and 1% BSA was applied. Afterwards, the
primary antibody, rabbit anti-pPERK (#ab192591, Abcam plc.,),
incubated overnight at 4˚C at a 1:50 dilution. The slides were then
covered with the secondary antibody for 30 min at room temperature,
a ready-to-use alkaline phosphatase polymer anti-rabbit reagent
(#MP-5401, Vector Laboratories, Inc.).
sXBP1
After the blocking procedure with 1.5% donkey serum
(#sc-2023, Santa Cruz Biotechnology, Inc.), the slides were
incubated overnight at 4˚C with the goat anti-XBP1 antibody
(#ab85546, Abcam plc.), diluted at 1:100. Subsequently, the
secondary antibody (#sc-2023; Santa Cruz Biotechnology, Inc.),
diluted at 1:200 in 1.5% donkey serum, was applied for 30 min at
room temperature.
Beclin 1
For deparaffinization the slides were placed in
xylene before being rehydrated by immersion in 100% isopropanol.
Afterwards, the endogenous peroxidase activity was blocked by 1.5%
methanol and antigen retrieval was performed as described above. To
block unspecific binding sites 2.5% horse serum was applied. Next,
the slides were incubated for one hour at room temperature with the
primary antibody, rabbit anti-Beclin1 IgG (#NB500-249, Novus
Biologicals Europe), at a dilution of 1:400 before the secondary
antibody (#MP-5401, Vector Laboratories, Inc.) was applied for 30
min at room temperature.
Western blot analysis
The frozen livers were thawed and homogenized using
a homogenization buffer (5 mmol/l 3-(N-morpholino) propanesulfonic
acid, 1 mmol/l ethylenediaminetetraacetic acid, 0.25 mol/l sucrose,
0.2 mmol/l dithiothreitol, 1 mmol/l ε-aminocaproic acid, 5 mmol/l
benzamidine, 0.2 mmol/l phenylmethylsulfonyl fluoride, 0.1% Triton
X-100). After centrifugation at 15.400 g for 15 min at 4˚C the
protein samples were fractionated by electrophoresis on sodium
dodecyl sulfate polyacrylamide gel. Subsequently, the separated
proteins were transferred to a polyvinylidene fluoride membrane
(#1620177, Bio-Rad Laboratories GmbH). The membranes were then
washed with PBS-Tween (0.05%) followed by the saturation with 5%
skim milk in PBS-Tween for one hour at room temperature to block
non-specific binding sites. Afterwards, the membranes were
incubated overnight at 4˚C with the primary antibody, goat anti-BiP
(#sc-1050, Santa Cruz Biotechnology, Inc.), diluted at 1:500 in 5%
skim milk. Before and after the incubation for one hour at room
temperature with the HRP-conjugated secondary antibody, donkey
anti-goat IgG (#sc-2020, Santa Cruz Biotechnology, Inc.) diluted at
1:5,000 in 5% skim milk, the membranes were washed with PBS-Tween.
Enhanced chemiluminescent substrate was added and the signal was
detected using Image Reader LAS-3000 Version 2.0 (Fuji Photo Film).
For loading control, the membranes were incubated for two hours at
room temperature with rabbit anti-glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) antibody (#sc-25778, Santa Cruz
Biotechnology, Inc.) diluted at 1:500 in 1% newborn calf serum.
Regarding the quantification of BiP expression, we
loaded two reference samples on each plot to allow a comparison of
different plots. For the analysis we used the ImageJ (Version:
1.51f; Wayne Rasband, National Institutes of Health). We selected
the lanes, plotted them and labelled the peaks. After converting
the number of pixels of each lane into percentage we divided the
percentage of each sample by the percentage of our reference
sample. These steps were performed for BiP as well as for GAPDH. We
finally divided the calculated values and received the ratio of BiP
to GAPDH expression. Using this approach, the analysis was adjusted
to inconsistent GAPDH expressions.
Statistical analyses
Statistical analysis was performed using MATLAB
R2018b (The MathWorks Inc.). Shapiro-Wilk test showed a
non-parametric distribution of the data. Therefore, the
Kruskal-Wallis test was used for the comparison of more than two
groups, together with Tukey-Kramer post-hoc correction. For the
comparison of two groups the Wilcoxon Mann-Whitney test was
applied. Data are expressed as median [minimum; maximum]. A p-value
below 0.05 was considered statistically significant. We
additionally present the Area Under the Curve (AUC) with
bootstrapped 95% confidence intervals as effect size. For this we
used the MATLAB-based MES toolbox (26). The use of AUC helps to evaluate the
strength of an effect (27).
Results
Model evaluation
Fixed pressure-controlled hemorrhagic shock was
shown to be a reliable and reproducible model (28). The mice used in this experiment did
not differ in age, body weight, or strain from previous studies
(17,18). As four animals had to be excluded,
e.g. due to death during hemorrhagic shock (n=1), malformations
(n=1), or inconsistent shock with more than three peaks above 35
mmHg (n=2), we included 21 animals in our analysis. The mean blood
volume to induce and maintain a mean arterial pressure of 30±5 mmHg
for 90 min was 0.60 [0.50; 0.65] ml (2.45 [2.10; 2.94] ml per 100 g
body weight) and did not differ significantly between the shock
groups. Furthermore, there were no significant differences between
the groups regarding the body weight at the start of the
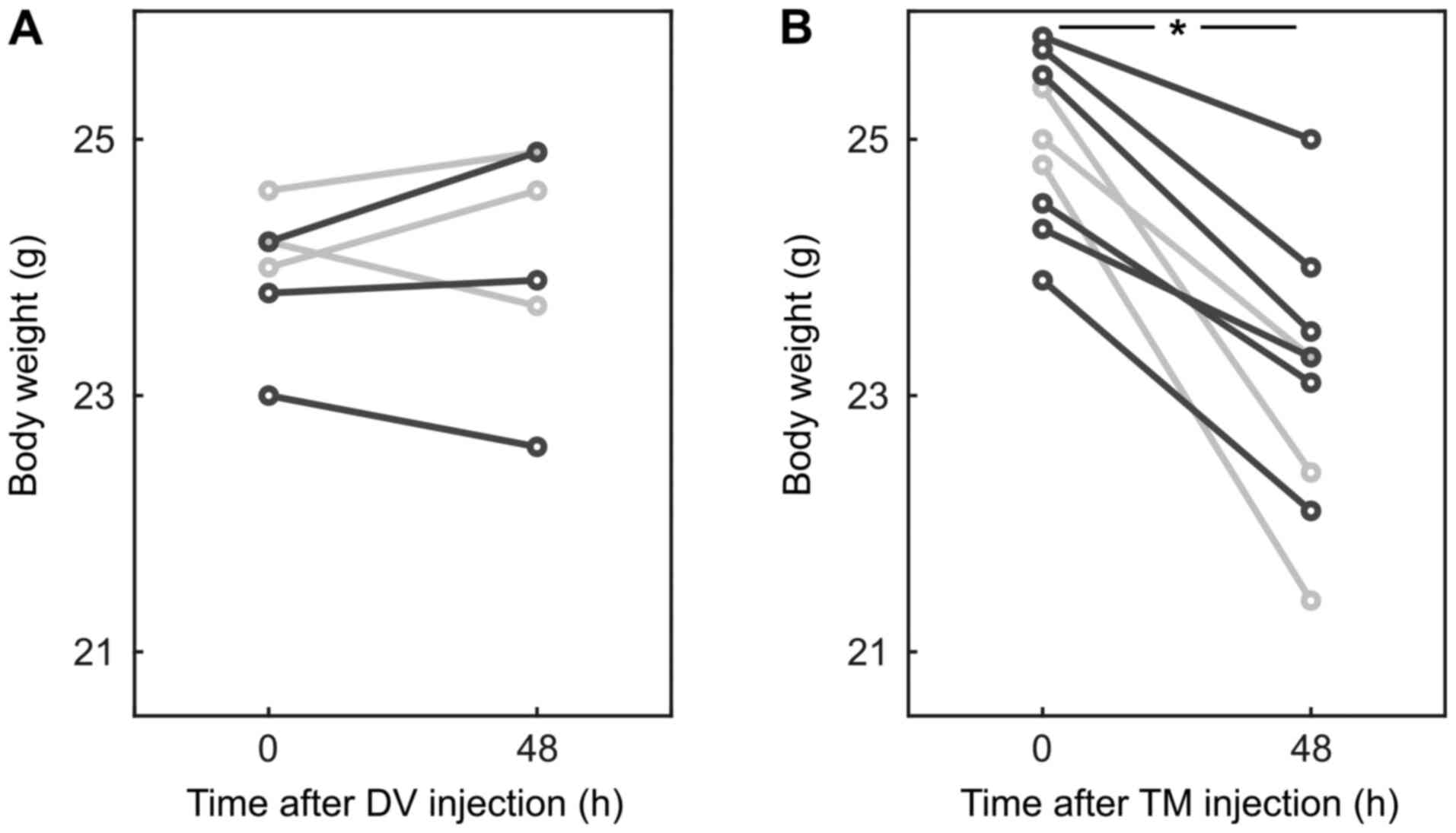
experiment. However, mice which received TM lost 6.8 [3.1; 13.7]%
of their body weight within 48 h (P=0.004; AUC 0.93 [0.78 1];
Fig. 1).
TM preconditioning alleviated liver
damage
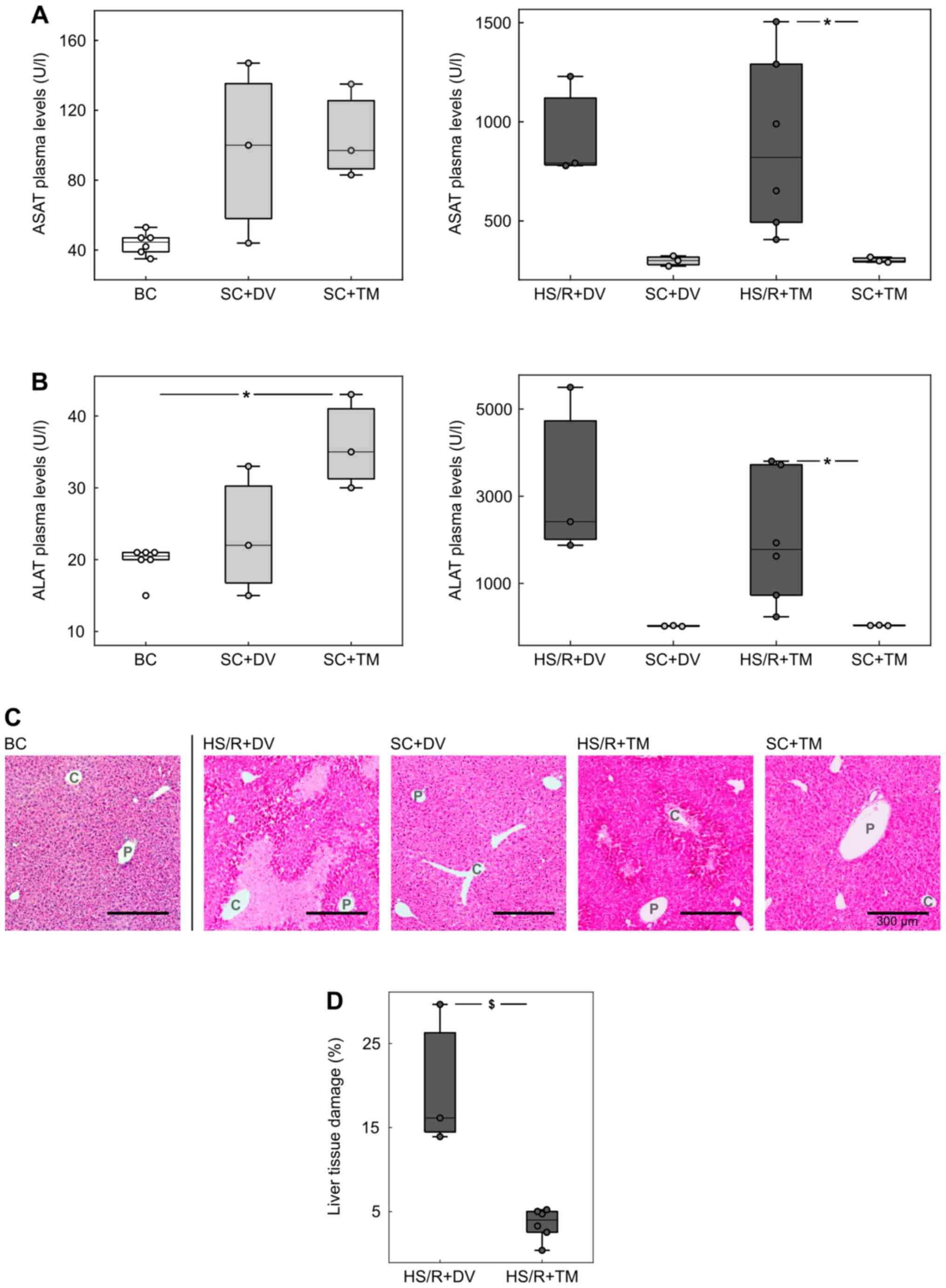
To assess the extent of liver damage, plasma
concentrations of ASAT and ALAT were measured as their plasma
levels correlate with hepatocellular injury (29) (Fig.
2A and B). The comparison of
the control groups (BC, SC+DV, SC+TM) showed no significant
differences except for the ALAT concentration in SC+TM group (35.0
[30.0; 43.0] U/l), which was significantly higher than in BC (20.5
[15.0; 21.0] U/l, P=0.047; AUC 0 [0 0]). The ASAT/ALAT levels in
the shock groups were similar: 1141.5 [312.0; 2510.0] U/l and
1778.0 [235.0; 3805.0] U/l in HS/R+TM group vs. 1084.0 [1058.0;
1958.0] U/l and 2417 [1876.0; 5499.0] U/l in the HS/R+DV group. The
comparison of the shock groups with their corresponding sham groups
demonstrated more than 10-fold higher transaminases concentrations
in the HS/R groups. This difference was significant for TM groups
(P=0.024; AUC 0 [0 0]) and non-significant for the DV groups
(P=0.1).
Additionally, H&E-staining of liver tissue
sections was performed to confirm the results of the plasma
measurements (Fig. 2C and D). The evaluation of baseline and sham
controls showed no obvious signs of hepatocellular damage. In
contrast to this finding, there were cell death areas spreading
centrifugally from the central vein in both shock groups. The
quantification of these areas revealed that the percentage of
damaged liver tissue was significantly lower in the HS/R+TM group
(4.0 [0.4; 13.9]%) compared to the HS/R+DV group (16.1 [5.2;
30.0]%; P=0.024). This finding is underlined by an AUC of 0 [0
0].
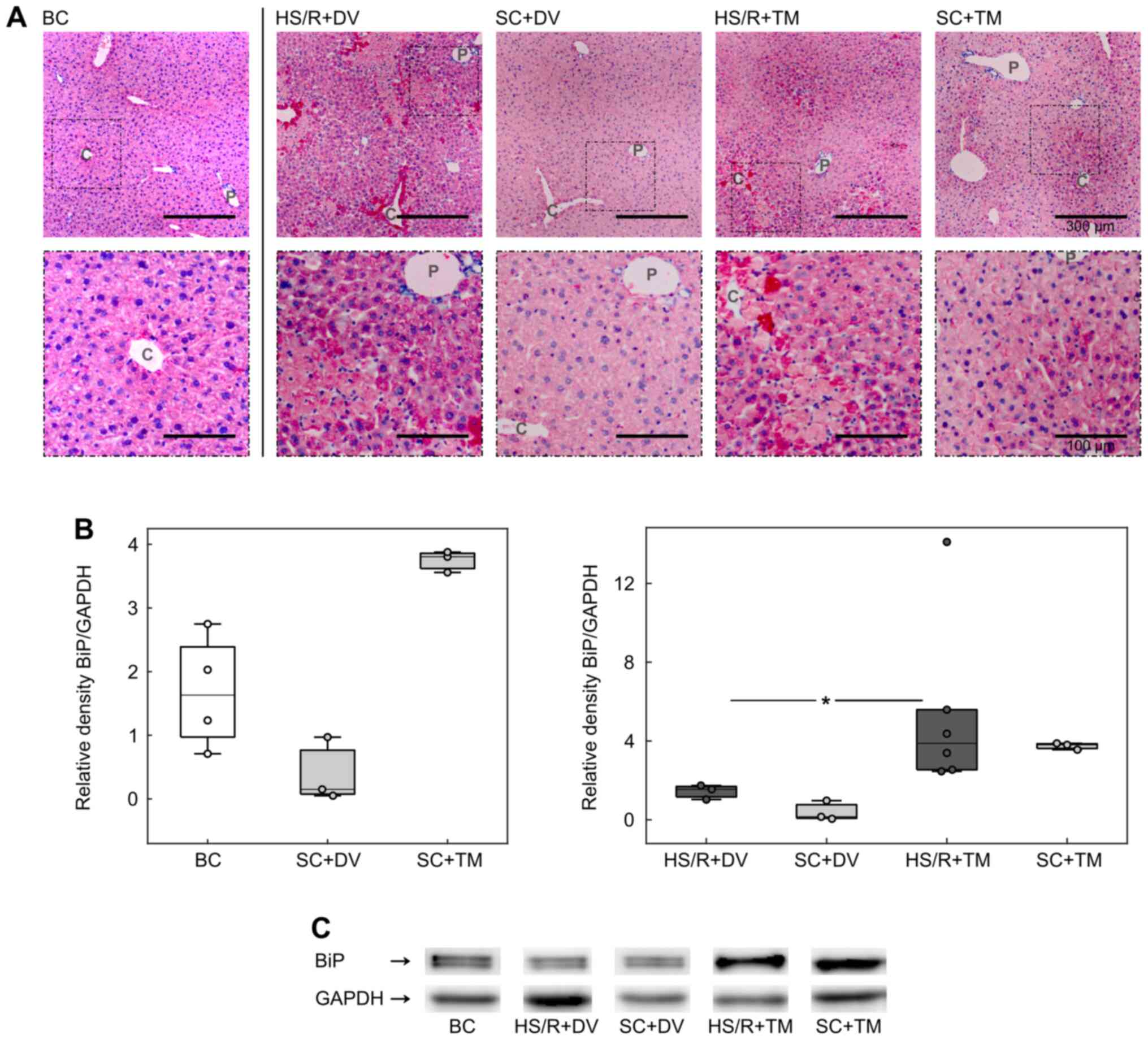
BiP expression was upregulated by TM
preconditioning
To investigate the influence of TM preconditioning
on the ER, we analyzed the expression of BiP, a master regulator of
ER function and a known ER stress marker (8,30).
Immunohistochemistry displayed a homogeneous staining of the liver
sections in BC and SC+DV groups (Fig.
3A). In the HS/R+DV group the vital parenchyma was also
homogenously stained but the staining intensity appeared to be
higher compared to its corresponding sham group. Furthermore, there
were single, intensely stained cells adjacent to the cell death
areas. These cells were also seen in the HS/R+TM group. However,
the BiP baseline expression pattern in HS/R+TM and SC+TM varied
from all other groups. Liver tissue sections of mice, which
received TM, displayed an increasing gradient of BiP expression
from the periportal field to the central vein.
For the evaluation of BiP expression in whole liver
homogenates western blotting was performed (Fig. 3B and 3C). The analysis demonstrated an increased
BiP expression in both TM groups. Compared to the HS/R+DV (1.55
[1.03; 1.74]), the HS/R+TM group (3.88 [2.46; 14.11]) showed a
significantly higher BiP/GAPDH ratio depicted by P=0.024 and an AUC
of 1 [1 1].
Topographical changes in the UPR after
TM preconditioning
Since PERK undergoes autophosphorylation upon ER
stress, phosphorylated PERK (pPERK) indicates its activation
(9). The pPERK staining was
predominated by an increased staining intensity around the
periportal field (Fig. 4A). This
pattern was observed in the BC, SC+DV, SC+TM, and HS/R+DV group.
However, the difference in the periportal and pericentral staining
intensity appeared to be smaller in the SC+TM group compared to the
SC+DV group. Liver tissue sections of HS/R+TM group showed a
similar pericentral as well as periportal expression level
displayed by a homogeneous, intense staining.
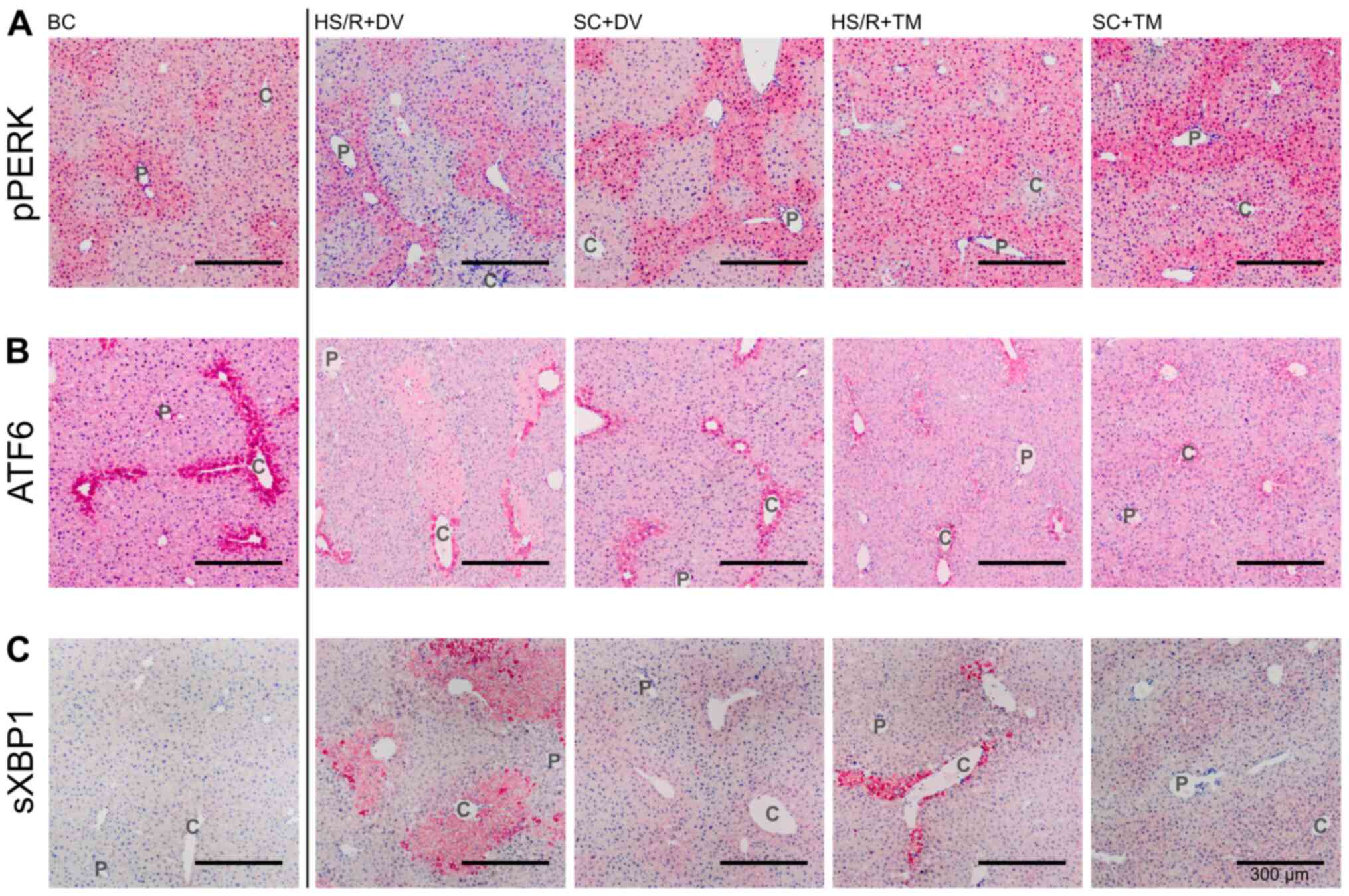 | Figure 4Topographical changes of UPR
signaling. Representative stains of the immunohistochemical proof
of pPERK, ATF6, and sXBP1. Vessels of the periportal field (P) and
central veins (C) are exemplified. Immunohistochemical staining for
detection of (A) pPERK, (B) ATF6 and (C) sXBP1 in the different
groups. Scale bar, 300 µm. pPERK, phosphorylated protein kinase
RNA-like endoplasmic reticulum kinase; ATF6, activating
transcription factor 6; sXBP1, spliced Version of X-box binding
protein 1; SC, sham control; HS/R, hemorrhagic shock and
reperfusion; DV, drug vehicle; TM, tunicamycin; BC, baseline
control; UPR, unfolded protein response. |
ATF6 is constitutively expressed (31). Upon ER stress, its activation is
initiated by the dissociation from BiP, followed by proteolytic
cleavage in the Golgi compartment (32). The ATF6 expression pattern was
characterized by a homogeneous staining of the vital liver
parenchyma and one row of intensely stained cells around the
central vein (Fig. 4B). In the
SC+TM group the pericentral cells were not as intensely stained as
in the other groups. In this group, a smooth transition from the
intensely stained pericentral area to the remaining liver
parenchyma was detected.
Since activated IRE1 splices XBP1 mRNA, measuring
spliced XBP1 (sXBP1) is a reliable, indirect method of assessing
IRE1 activation (30). In the BC
group only a slight, homogeneous expression could be detected
(Fig. 4C). The staining pattern in
SC+DV was characterized by an increased pericentral intensity that
faded out centrifugally. Whereas in HS/R+DV a sharp transition
between intensely stained cell death areas and slightly stained
vital parenchyma was found the staining pattern in SC+TM and
HS/R+TM was similar to SC+DV.
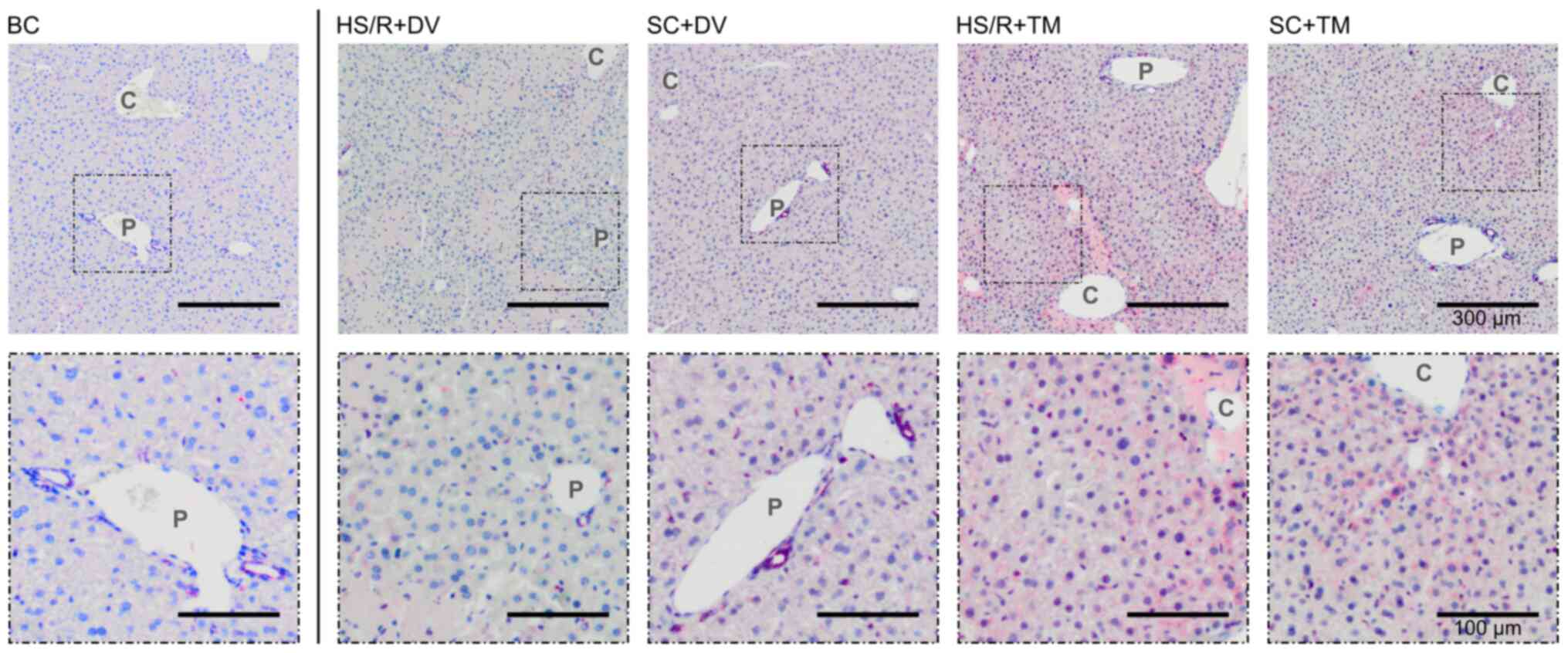
TM preconditioning induced pericentral
autophagy
Beclin1 is a known marker of autophagy, which is
naturally expressed in biliary epithelium (33). In the BC, SC+DV, and HS/R+DV group
Beclin1 was scarcely expressed indicated by a weak and partly
missing staining of the vital parenchyma (Fig. 5). In contrast to this finding, both
TM groups showed an increased Beclin1 expression pericentrally.
Similar to the BiP staining pattern an increasing staining
intensity from the periportal field to the central vein was
observed.
Discussion
As previously shown by Jian et al (16), ER stress plays an important role in
liver injury following HS/R. We confirmed this finding in our
preceding study: The injection of the ER stress inhibitor
tauroursodeoxycholic acid (TUDCA) during reperfusion mitigated
hepatocellular damage, whereas the administration of the ER stress
inductor TM during reperfusion increased hepatocellular damage
(18). In addition, we conducted a
detailed timeline investigation of the temporal dynamics of the
expression of UPR signaling proteins and liver injury (17). Our analysis revealed a maximum of
hepatocellular damage 14 h after shock induction. Since the focus
of the present study was on liver damage, we chose to sacrifice
mice 14 h after hemorrhagic shock induction.
TM is a pharmaceutical ER stress inducer and acts
via an inhibition of N-glycosylation, causing an accumulation of
unfolded glycoproteins in the ER (30,34).
In accordance with previous studies, the results of the western
blot analysis of whole-organ homogenates demonstrated an increased
BiP expression following TM pretreatment (35). As prior publications on IRI and HS/R
have already demonstrated that an increased expression of BiP is
accompanied by an upregulation of pPERK, IRE1, and ATF6 (16,36,37)
and as the focus of the present study was also on the topographical
distribution of ER stress, we decided to only analyze BiP in whole
liver homogenates. We chose BiP since it is a master regulator of
ER function. As a member of the heat shock protein 70 family, BiP
is a highly conserved molecular chaperone (8). However, it not only facilitates
protein folding, but is also an essential component of quality
control mechanisms of the secretory pathway and regulates
endoluminal calcium concentration (38,39).
Additionally, and of the utmost importantance for the present
study, BiP is a well-established marker of ER stress as its
expression is induced by mal-/unfolded proteins (40,41).
Interestingly, immunohistochemistry showed that BiP
was not homogeneously increased but rather focused in the
pericentral area. This topographic distribution pattern might be
explained by the unique blood supply of the liver, which leads to a
decreasing oxygen gradient from the periportal region towards the
pericentral area (42). In several
studies, Paxian et al (43,44)
demonstrated the resulting susceptibility to external stressors of
pericentral hepatocytes, especially during hemorrhagic shock and
the oxidant stress upon reperfusion. Consequently, these cells
respond more sensitively to TM than periportal cells, indicated in
the present study by the increased pericentral expression of ER
stress marker BiP in both TM groups (44). Our finding of a topographic
correlation of an upregulated BiP induction with the diminution of
hepatocellular damage suggests that BiP has beneficial effects.
This assumption is also supported by a recent publication from Bi
et al (45), which
demonstrated that an overexpression of BiP mitigated myocardial
IRI. In line with the results of Paxian et al (43), the protective effect was mediated by
inhibiting an accumulation of reactive oxygen species. Taking our
observations and the current literature into account, we conclude
that a pre-hemorrhagic BiP induction by TM administration mitigates
post-hemorrhagic hepatocellular injury.
Whereas TM injection significantly altered BiP
expression, its influence on the topographic patterns of ATF6 and
IRE1 was limited. This difference might be based on the degradation
of the proteins: The half-life of BiP is approximately 46 h, while
ATF6 and IRE1 are degraded with a half-life of about 2 and 3 h,
respectively (46-48).
Consequently, the effect of TM on their topographic patterns might
already have faded away 62 h after the injection. In contrast, PERK
signaling, represented by pPERK, was markedly influenced by TM
pretreatment. The homogeneous staining intensity in HS/R+TM group
suggests an upregulation of the PERK pathway in the intermediary
and pericentral zone. Even though PERK can contribute to cell
death, we theorize that PERK signaling in the context of IRI and
HS/R is primarily protective (49).
The pro-survival effect might be mediated by the activation of the
antioxidant response element via ATF4 and nuclear factor erythroid
2-related factor 2 (Nrf2), resulting in an upregulation of
protective enzymes (50). Since
PERK activation thereby promotes beneficial effects, the increased
pericentral expression of pPERK may explain the reduction of
centrilobular cell death areas in the HS/R+TM group. Leung et
al (51) recently demonstrated
in a murine HS/R model that Nrf2 plays a crucial role in the
generation of protective factors induced by stress preconditioning
and thus confirmed the importance of the PERK/ATF4/Nrf2 signaling
branch.
In addition to its function as an activator of Nrf2,
ATF4 is a key signal for ER stress induced autophagy (52). Although autophagy can play dual
roles and may promote cell death, multiple studies attribute
beneficial effects to the autophagic process during hypoperfusion
or ischemia (53-55).
Chandrika et al (53)
demonstrated in the context of renal IRI that ER-stress induced
autophagy provides cytoprotection. Yan et al (56) induced a subarachnoid hemorrhage in
rats and described an autophagy dependent mitigation of early brain
injury. Moreover, autophagy was shown to be hepatoprotective during
a low-flow state, e.g. caused by septic shock (55). Previous studies reported that for
correct autophagosome formation BiP is an obligatory component
(57). Our immunohistochemical
analysis supports this finding as the expression patterns of BiP
and the autophagy marker Beclin1 were similar in the TM groups
(33). Furthermore, our results
underpin the hypothesis of Zhang et al (19), attributing a protective role to BiP
dependent autophagy induction in the context of IRI. In the HS/R+TM
group, Beclin1 expression was upregulated around the central vein,
which indicates an increase in autophagic activity in the
pericentral zone. With regard to the diminished liver damage in the
HS/R+TM group, the topographic distribution of the cell death areas
and Beclin1, the present study underlines the protective role of
autophagy and identifies its activation as a beneficial
mechanism.
Hepatic injury was analyzed by evaluating
H&E-stained liver tissue sections and measuring serum
transaminases. We chose to use the umbrella term ‘liver damage’
because of the vague understanding of cell death mechanisms during
HS/R. Although necrosis has been postulated as the predominant cell
death mechanism, the occurrence of apoptosis and autophagy-related
cell death has similarly been reported (58,59).
In the present work, we focused on ER stress as an underlying
mechanism of the IRI as well as on the impact of ER stress
preconditioning on organ damage. We did not investigate the exact
modalities of ER stress associated cell death. In contrast to the
long-standing assumption that prolonged ER stress only triggers
apoptotic cell death, recent studies have demonstrated an ER stress
induced caspase-independent cell death mechanism (60-62).
Therefore, we suggest taking into account histomorphologic signs of
both cell death modalities, when investigating post-hemorrhagic
hepatocellular damage.
Interestingly, the results of the methods employed
were not totally consistent. The discrepancy in the results of the
two methods might be explained by the lengthy half-life of the
liver transaminases on one hand and the liver's ability to
regenerate on the other hand (63).
Li et al (64) and Pajaud
et al (65) determined the
enormous regenerative potential of the murine liver as their data
shows a completion of hepatocyte proliferation 72 h after
two-thirds partial hepatectomy. The upregulation of early liver
regeneration was particularly remarkable considering the
debilitating, pre-hemorrhagic body weight loss of mice receiving
TM. This body weight loss might be explained by the interplay of
ER-stress and inflammation since it is known that ATF4 induces
interleukin-6, which promotes adipose tissue lipolysis (66,67).
Therefore, ER-stress induction by TM may initiate an inflammatory
response which, in turn, decreased body weight.
Since we focused on a fixed point in time, we can
only speculate whether TM preconditioning lowered organ damage or
if it merely accelerated liver regeneration. Previous studies
focusing on IRI or partial hepatectomy (PH) demonstrated the
enormous potential of the liver to regenerate (64,65,68).
Furthermore, it has been shown that liver regeneration can be
promoted through heat-shock proteins (HSPs) (69). As HSPs are upregulated through ER
stress, we assume that TM preconditioning mediates its beneficial
effects by accelerating liver regeneration via induction of HSPs,
e.g. BiP (8,70,71).
However, to fully elucidate the temporal dynamics, a detailed time
trial is needed. This trial should also include the analysis of
transaminase and protein levels 48 h after TM injection. An
upregulation of HSP expression just prior to hemorrhagic shock
induction could support our above-mentioned assumption.
Since TM is not soluble in aqueous solution at pH
7.4, the manufacturer recommends using DMSO as a solvent. Since
DMSO increases serum transaminases, its influence should also be
considered when evaluating the results of the histological and
laboratory analysis as it could be another explanation for the
aforementioned difference (72).
However, the dosage applied in the present study was more than
100-fold below its median lethal dose, and in our previous studies
we did not detect considerable differences comparing DV groups with
mice undergoing sham or HS/R procedure without any drug injection
(18,73). Consequently, we assigned only three
animals to each control group as we did not expect to augment
scientific knowledge by including more mice and might thereby avoid
raising ethical issues.
Furthermore, Beclin1 detection is no absolute
criteria for determining autophagic status even though it is an
established marker of autophagy onset (33,74)
Complementary to our analyses, it would be worth evaluating the
expression of e.g. p62 or microtubule associated protein 1
light-chain 3 as these proteins are required for the formation of
ubiquitinated protein aggregates and their delivery to the
autophagy system (74). Detecting
these proteins could confirm our conclusion and enable a deeper
analysis of the pericentral autophagic processes. Nevertheless,
looking at our data, there are further indicators of the induction
of autophagy by TM preconditioning in addition to the increased
Beclin1 expression. Furthermore, PERK activation was enhanced
around the central vein in HS/R+TM group compared to all other
groups. One target of the PERK pathway is ATF4, a key signal for
autophagy induced by ER-stress (52). Regarding the concomitant
upregulation of BiP, which is an obligatory component of autophagy,
these immunohistochemical findings support the assumption of a
pericentral autophagy induction by TM preconditioning (57). Furthermore, we performed an
immunohistochemical proof of CCAAT/Enhancer Binding Protein
Homologous Protein (CHOP; data not shown), which is a target gene
of ATF4 and has been shown to promote the transcription of several
autophagy genes (49,75). In addition, an increased CHOP
expression downregulates protein B-Cell Lymphoma 2 (Bcl-2)
(76). As Bcl-2 inhibits
Beclin1-dependent autophagy, its downregulation facilitates
autophagy induction (77). In the
present study, the immunohistochemical analysis of liver tissue
sections demonstrated an increased CHOP expression around the
central vein in TM groups. As this finding suggests an upregulation
of autophagy genes and a suppression of autophagy inhibition, it
confirmed the above-mentioned results and supports the hypothesis
of a pericentral autophagy induction by TM preconditioning. To
further elucidate the importance of this finding, a selective
post-hemorrhagic autophagy induction would be useful. Since TM
influences BiP expression as well autophagy, this approach would
help to differentiate the impact of these two mechanisms and
highlight their clinical significance (41,78).
A comparison of BiP western blot and
immunohistochemistry reveals the strengths and weaknesses of both
methods. Since the semi quantitative analysis of whole liver
homogenates did not contain any information about the topographic
distribution of BiP, we also performed an immunohistochemical proof
of BiP. Unexpectedly, the results of the two methods did not
completely overlap. Our evaluation of the immunohistochemical
staining suggested the highest BiP expression was in the HS/R+DV
group. In contrast to this finding, the western blot analysis
showed the highest BiP expression in the TM groups. The strong
pericentral upregulation of BiP in TM groups may have outweighed
the lower expression in the remaining zones, whereas in the HS/R+DV
group the BiP expression in the vital liver parenchyma was not
enough to compensate for the cell death areas. To backup this
assumption, for the future we propose microdissecting the
individual liver zones followed by western blot or PCR analyses.
This approach makes possible the detection of topographical changes
and their simultaneous quantification.
In the present study, we confirmed previous results
reporting a significant role for the UPR in IRI. In addition, we
demonstrated that the injection of the ER stress inducer
tunicamycin mitigates post-hemorrhagic hepatocellular injury. By
analyzing topographic expression patterns, we identified an
upregulated BiP expression and a concomitant autophagy induction as
potential beneficial mechanisms. In conclusion, ER stress
preconditioning alleviates post-hemorrhagic liver damage and may
lead to novel therapeutic targets.
Acknowledgements
The authors would like to thank Prof. Dr. Gerhard
Schmidmaier (Department of Trauma Surgery, University of
Heidelberg, Heidelberg) for his valuable support and are grateful
for the skillful technical assistance of Ms Birgit Frey (Department
of Trauma Surgery, University of Heidelberg, Germany).
Funding
This research was funded by the University of
Heidelberg, Germany.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
The conception and design of the study was performed
by DPO and SK, the acquisition of data was carried out by DPO and
AKW. DPO, AKW, NLG and SK analyzed and interpreted the data. DPO
drafted the article and all authors revised it critically for
important intellectual content. All authors approved the final
version to be submitted.
Ethics approval and consent to
participate
All study protocols were reviewed and approved by
the section for agriculture and veterinary services of the Regional
Council, Karlsruhe, Germany (35-9185.81/G-65/13).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heron M: Deaths: Leading Causes for 2012.
Natl Vital Stat Rep. 64:1–94. 2015.PubMed/NCBI
|
|
2
|
GBD 2016 Causes of Death Collaborators.
Global, regional, and national age-sex specific mortality for 264
causes of death, 1980-2016: A systematic analysis for the global
burden of disease study 2016. Lancet. 390:1151–1210.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rhee P, Joseph B, Pandit V, Aziz H,
Vercruysse G, Kulvatunyou N and Friese RS: Increasing trauma deaths
in the United States. Ann Surg. 260:13–21. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Eastridge BJ, Holcomb JB and Shackelford
S: Outcomes of traumatic hemorrhagic shock and the epidemiology of
preventable death from injury. Transfusion. 59:1423–1428.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kauvar DS, Lefering R and Wade CE: Impact
of hemorrhage on trauma outcome: An overview of epidemiology,
clinical presentations, and therapeutic considerations. J Trauma.
60 (Suppl 6):S3–S11. 2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sciandra JJ, Subjeck JR and Hughes CS:
Induction of glucose-regulated proteins during anaerobic exposure
and of heat-shock proteins after reoxygenation. Proc Natl Acad Sci
USA. 81:4843–4847. 1984.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hetz C, Chevet E and Oakes SA:
Proteostasis control by the unfolded protein response. Nat Cell
Biol. 17:829–838. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Wang J, Lee J, Liem D and Ping P: HSPA5
gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum.
Gene. 618:14–23. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Ye J, Rawson RB, Komuro R, Chen X, Davé
UP, Prywes R, Brown MS and Goldstein JL: ER stress induces cleavage
of membrane-bound ATF6 by the same proteases that process SREBPs.
Mol Cell. 6:1355–1364. 2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: ATF6 activated by proteolysis binds in the
presence of NF-Y (CBF) directly to the cis-acting element
responsible for the mammalian unfolded protein response. Mol Cell
Biol. 20:6755–6767. 2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yoshida H, Matsui T, Hosokawa N, Kaufman
RJ, Nagata K and Mori K: A time-dependent phase shift in the
mammalian unfolded protein response. Dev Cell. 4:265–271.
2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Harding HP, Zhang Y and Ron D: Protein
translation and folding are coupled by an
endoplasmic-reticulum-resident kinase. Nature. 397:271–274.
1999.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995.
1998.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jian B, Hsieh CH, Chen J, Choudhry M,
Bland K, Chaudry I and Raju R: Activation of endoplasmic reticulum
stress response following trauma-hemorrhage. Biochim Biophys Acta.
1782:621–626. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wolpert A, Obert D, Frey B, Lee YS and
Korff S: Hepatic topographical changes of endoplasmic reticulum
stress and unfolded protein response signaling after hemorrhagic
shock and reperfusion. J Surg Res. 231:278–289. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Obert DP, Wolpert AK and Korff S:
Modulation of endoplasmic reticulum stress influences
ischemia-reperfusion injury after hemorrhagic shock. Shock.
52:e76–e84. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang XY, Zhang TT, Song DD, Zhou JH, Han
R, Qin ZH and Sheng R: Endoplasmic reticulum chaperone GRP78 is
involved in autophagy activation induced by ischemic
preconditioning in neural cells. Mol Brain. 8(20)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li J, Lai X, Chen Y, Niu B and Gong J:
Endotoxin tolerance attenuates liver ischemia/reperfusion injury by
down-regulation of interleukin-1 receptor-associated kinase 4 in
kupffer cells. Transplant Proc. 43:2531–2535. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Thielmann M, Kottenberg E, Kleinbongard P,
Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters
J, et al: Cardioprotective and prognostic effects of remote
ischaemic preconditioning in patients undergoing coronary artery
bypass surgery: A single-centre randomised, double-blind,
controlled trial. Lancet. 382:597–604. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Birnbaum Y, Hale SL and Kloner RA:
Ischemic preconditioning at a distance: Reduction of myocardial
infarct size by partial reduction of blood supply combined with
rapid stimulation of the gastrocnemius muscle in the rabbit.
Circulation. 96:1641–1646. 1997.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hildebrand F, Hubbard WJ, Choudhry MA,
Frink M, Pape HC, Kunkel SL and Chaudry IH: Kupffer cells and their
mediators: The culprits in producing distant organ damage after
trauma-hemorrhage. Am J Pathol. 169:784–794. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wallig MA, Haschek WM, Rousseaux CG, Bolon
B and Mahler BW: Fundamentals of toxicologic pathology.
Elsevier/Academic Press, London, 2018.
|
|
25
|
Matsuo A, Watanabe A, Takahashi T,
Futamura M, Mori S, Sugiyama Y, Takahashi Y and Saji S: A simple
method for classification of cell death by use of thin layer
collagen gel for the detection of apoptosis and/or necrosis after
cancer chemotherapy. Jpn J Cancer Res. 92:813–819. 2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hentschke H and Stuttgen MC: Computation
of measures of effect size for neuroscience data sets. Eur J
Neurosci. 34:1887–1894. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Amrhein V, Greenland S and McShane B:
Scientists rise up against statistical significance. Nature.
567:305–307. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pfeifer R, Lichte P, Schreiber H, Sellei
RM, Dienstknecht T, Sadeghi C, Pape HC and Kobbe P: Models of
hemorrhagic shock: Differences in the physiological and
inflammatory response. Cytokine. 61:585–590. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
James O, Lesna M, Roberts SH, Pulman L,
Douglas AP, Smith PA and Watson AJ: Liver damage after paracetamol
overdose. Comparison of liver-function tests, fasting serum bile
acids, and liver histology. Lancet. 2:579–581. 1975.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Oslowski CM and Urano F: Measuring ER
stress and the unfolded protein response using mammalian tissue
culture system. Methods Enzymol. 490:71–92. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Haze K, Yoshida H, Yanagi H, Yura T and
Mori K: Mammalian transcription factor ATF6 is synthesized as a
transmembrane protein and activated by proteolysis in response to
endoplasmic reticulum stress. Mol Biol Cell. 10:3787–3799.
1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676.
1999.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Tkacz JS and Lampen O: Tunicamycin
inhibition of polyisoprenyl N-acetylglucosaminyl pyrophosphate
formation in calf-liver microsomes. Biochem Biophys Res Commun.
65:248–257. 1975.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Prachasilchai W, Sonoda H, Yokota-Ikeda N,
Oshikawa S, Aikawa C, Uchida K, Ito K, Kudo T, Imaizumi K and Ikeda
M: A protective role of unfolded protein response in mouse ischemic
acute kidney injury. Eur J Pharmacol. 592:138–145. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Thacker SA, Robinson P, Abel A and Tweardy
DJ: Modulation of the unfolded protein response during hepatocyte
and cardiomyocyte apoptosis in trauma/hemorrhagic shock. Sci Rep.
3(1187)2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Duvigneau JC, Kozlov AV, Zifko C, Postl A,
Hartl RT, Miller I, Gille L, Staniek K, Moldzio R, Gregor W, et al:
Reperfusion does not induce oxidative stress but sustained
endoplasmic reticulum stress in livers of rats subjected to
traumatic-hemorrhagic shock. Shock. 33:289–298. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ellgaard L and Helenius A: Quality control
in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 4:181–191.
2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Schauble N, Lang S, Jung M, Cappel S,
Schorr S, Ulucan O, Linxweiler J, Dudek J, Blum R, Helms V, et al:
BiP-mediated closing of the Sec61 channel limits Ca2+
leakage from the ER. EMBO J. 31:3282–3296. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kozutsumi Y, Segal M, Normington K,
Gething MJ and Sambrook J: The presence of malfolded proteins in
the endoplasmic reticulum signals the induction of
glucose-regulated proteins. Nature. 332:462–464. 1988.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Paxian M, Keller SA, Cross B, Huynh TT and
Clemens MG: High-resolution visualization of oxygen distribution in
the liver in vivo. Am J Physiol Gastrointest Liver Physiol.
286:G37–G44. 2004.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Paxian M, Bauer I, Kaplan D, Bauer M and
Rensing H: Hepatic redox regulation of transcription factors
activator protein-1 and nuclear factor-kappaB after hemorrhagic
shock in vivo. Antioxid Redox Signal. 4:711–720. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Paxian M, Bauer I, Rensing H, Jaeschke H,
Mautes AEM, Kolb SA, Wolf B, Stockhausen A, Jeblick S and Bauer M:
Recovery of hepatocellular ATP and ‘pericentral apoptosis’ after
hemorrhage and resuscitation. FASEB J. 17:993–1002. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bi X, Zhang G, Wang X, Nguyen C, May HI,
Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, et al:
Endoplasmic reticulum chaperone GRP78 protects heart from
ischemia/reperfusion injury through akt activation. Circ Res.
122:1545–1554. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Rutkowski DT, Arnold SM, Miller CN, Wu J,
Li J, Gunnison KM, Mori K, Akha AAS, Raden D and Kaufman RJ:
Adaptation to ER stress is mediated by differential stabilities of
pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol.
4(e374)2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gao B, Lee SM, Chen A, Zhang J, Zhang DD,
Kannan K, Ortmann RA and Fang D: Synoviolin promotes IRE1
ubiquitination and degradation in synovial fibroblasts from mice
with collagen-induced arthritis. EMBO Rep. 9:480–485.
2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sato Y, Nadanaka S, Okada T, Okawa K and
Mori K: Luminal domain of ATF6 alone is sufficient for sensing
endoplasmic reticulum stress and subsequent transport to the Golgi
apparatus. Cell Struct Funct. 36:35–47. 2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Fawcett TW, Martindale JL, Guyton KZ, Hai
T and Holbrook NJ: Complexes containing activating transcription
factor (ATF)/cAMP-responsive-element-binding protein (CREB)
interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF
composite site to regulate Gadd153 expression during the stress
response. Biochem J. 339:135–141. 1999.PubMed/NCBI
|
|
50
|
He CH, Gong P, Hu B, Stewart D, Choi ME,
Choi AM and Alam J: Identification of activating transcription
factor 4 (ATF4) as an Nrf2-interacting protein. Implication for
heme oxygenase-1 gene regulation. J Biol Chem. 276:20858–20865.
2001.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Leung CH, Caldarone CA, Guan R, Wen XY,
Ailenberg M, Kapus A, Szaszi K and Rotstein OD: Nrf2 regulates the
hepatoprotective effects of remote ischemic conditioning in
hemorrhagic shock. Antioxid Redox Signal. 30:1760–1773.
2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Matsumoto H, Miyazaki S, Matsuyama S,
Takeda M, Kawano M, Nakagawa H, Nishimura K and Matsuo S: Selection
of autophagy or apoptosis in cells exposed to ER-stress depends on
ATF4 expression pattern with or without CHOP expression. Biol Open.
2:1084–1090. 2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chandrika BB, Yang C, Ou Y, Feng X, Muhoza
D, Holmes AF, Theus S, Deshmukh S, Haun RS and Kaushal GP:
Endoplasmic reticulum stress-induced autophagy provides
cytoprotection from chemical hypoxia and oxidant injury and
ameliorates renal ischemia-reperfusion injury. PLoS One.
10(e0140025)2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hsieh YC, Athar M and Chaudry IH: When
apoptosis meets autophagy: Deciding cell fate after trauma and
sepsis. Trends Mol Med. 15:129–138. 2009.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Oami T, Watanabe E, Hatano M, Teratake Y,
Fujimura L, Sakamoto A, Ito C, Toshimori K, Swanson PE and Oda S:
Blocking liver autophagy accelerates apoptosis and mitochondrial
injury in hepatocytes and reduces time to mortality in a murine
sepsis model. Shock. 50:427–434. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yan F, Li J, Chen J, Hu Q, Gu C, Lin W and
Chen G: Endoplasmic reticulum stress is associated with
neuroprotection against apoptosis via autophagy activation in a rat
model of subarachnoid hemorrhage. Neurosci Lett. 563:160–165.
2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Cursio R, Colosetti P and Gugenheim J:
Autophagy and liver ischemia-reperfusion injury. Biomed Res Int.
2015(417590)2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Cursio R, Colosetti P, Saint-Paul MC,
Pagnotta S, Gounon P, Iannelli A, Auberger P and Gugenheim J:
Induction of different types of cell death after normothermic liver
ischemia-reperfusion. Transplant Proc. 42:3977–3980.
2010.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Sovolyova N, Healy S, Samali A and Logue
SE: Stressed to death-mechanisms of ER stress-induced cell death.
Biol Chem. 395:1–13. 2014.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Saveljeva S, Mc Laughlin SL, Vandenabeele
P, Samali A and Bertrand MJ: Endoplasmic reticulum stress induces
ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell
Death Dis. 6(e1587)2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Santofimia-Castano P, Lan W, Bintz J,
Gayet O, Carrier A, Lomberk G, Neira JL, González A, Urrutia R,
Soubeyran P and Iovanna J: Inactivation of NUPR1 promotes cell
death by coupling ER-stress responses with necrosis. Sci Rep.
8(16999)2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kim WR, Flamm SL, Di Bisceglie AM and
Bodenheimer HC: Public Policy Committee of the American Association
for the Study of Liver Disease. Serum activity of alanine
aminotransferase (ALT) as an indicator of health and disease.
Hepatology. 47:1363–1370. 2008.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Li D, Li J, Wang G, Qin Y, Niu Z, Li Z and
Xu C: Delayed liver regeneration after partial hepatectomy in aged
Nos2 knockout mice. Cell J. 19:218–230. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Pajaud J, Ribault C, Ben Mosbah I, Rauch
C, Henderson C, Bellaud P, Aninat C, Loyer P, Morel F and Corlu A:
Glutathione transferases P1/P2 regulate the timing of signaling
pathway activations and cell cycle progression during mouse liver
regeneration. Cell Death Dis. 6(e1598)2015.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Han J, Meng Q, Shen L and Wu G:
Interleukin-6 induces fat loss in cancer cachexia by promoting
white adipose tissue lipolysis and browning. Lipids Health Dis.
17(14)2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Iwasaki Y, Suganami T, Hachiya R,
Shirakawa I, Kim-Saijo M, Tanaka M, Hamaguchi M, Takai-Igarashi T,
Nakai M, Miyamoto Y and Ogawa Y: Activating transcription factor 4
links metabolic stress to interleukin-6 expression in macrophages.
Diabetes. 63:152–161. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Karatzas T, Neri AA, Baibaki ME and Dontas
IA: Rodent models of hepatic ischemia-reperfusion injury: Time and
percentage-related pathophysiological mechanisms. J Surg Res.
191:399–412. 2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Oka Y, Akagi Y, Kinugasa T, Ishibashi N,
Iwakuma N, Shiratsuchi I and Shirouzu K: Heat-shock pre-treatment
reduces liver injury and aids liver recovery after partial
hepatectomy in mice. Anticancer Res. 33:2887–2894. 2013.PubMed/NCBI
|
|
70
|
Chien CY, Chien CT and Wang SS:
Progressive thermopreconditioning attenuates rat cardiac
ischemia/reperfusion injury by mitochondria-mediated antioxidant
and antiapoptotic mechanisms. J Thorac Cardiovasc Surg.
148:705–713. 2014.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Liu Y and Chang A: Heat shock response
relieves ER stress. EMBO J. 27:1049–1059. 2008.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Pestel S, Martin HJ, Maier GM and Guth B:
Effect of commonly used vehicles on gastrointestinal, renal, and
liver function in rats. J Pharmacol Toxicol Methods. 54:200–214.
2006.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Willson JE, Brown DE and Timmens EK: A
toxicologic study of dimethyl sulfoxide. Toxicol Appl Pharmacol.
7:104–112. 1965.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
B'Chir W, Chaveroux C, Carraro V, Averous
J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P and
Bruhat A: Dual role for CHOP in the crosstalk between autophagy and
apoptosis to determine cell fate in response to amino acid
deprivation. Cell Signal. 26:1385–1391. 2014.PubMed/NCBI View Article : Google Scholar
|
|
76
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006.PubMed/NCBI View Article : Google Scholar
|