Introduction
Cerebral ischemia/reperfusion (I/R) injury, one of
the most common types of stroke, often causes a variety of
cerebrovascular and cardiovascular diseases that lead to human
disability and mortality worldwide (1,2). Nerve
cell injury and apoptosis are considered the first steps in the
pathogenesis of cerebral I/R injury (3). Once cerebral I/R occurs, a large
number of oxygen free radicals and acute inflammatory reactions are
induced, resulting in apoptosis and therefore aggravating the
ischemic brain tissue injury (4).
Therefore, extensive research is needed to find effective
anti-apoptosis and anti-oxidative stress agents that can inhibit
apoptosis and oxidative stress, ameliorating cerebral I/R
injury.
Sevoflurane (Sev), a volatile anesthetic, has
potential therapeutic effects on I/R-induced injury, especially
towards the pulmonary and hepatic injuries (5,6). For
example, Xiong et al (7)
showed that Sev could attenuate ventilator-induced lung injury by
suppressing pulmonary inflammation in mice. In addition, Li et
al (8) found that Sev
preconditioning ameliorated spinal cord I/R-induced neuronal injury
by inhibiting microglial MMP-9 expression in rats. Previous
research has also demonstrated the neuroprotective effect of Sev on
severe cerebral ischemia in rats (9-11).
For example, pretreatment with Sev improved spatial learning and
memory impairment after cerebral I/R injury in rats (12). However, the underlying mechanisms
responsible for the protective effects of Sev against cerebral I/R
injury are still elusive.
The present study determined the protective effects
of Sev in a model of neuronal HT22 cell with oxygen-glucose
deprivation/reperfusion (OGD/R), and investigate the involvement of
the PI3K/AKT/GSK3β signaling pathway in molecular mechanisms. The
results of the present study suggest that Sev may be an improved
anesthetic option for patients with cerebral I/R injury.
Materials and methods
Cell culture
The mouse hippocampal neuronal HT22 cell line was
purchased from the American Type Culture Collection (ATCC) and
cultured in DMEM supplemented with 10% fetal bovine serum (FBS;
both from Sigma-Aldrich; Merck KGaA) with 4.5 g/l glucose, 100 U/ml
penicillin and 100 µg/ml streptomycin (Sigma-Aldrich; Merck KGaA)
in a humidified incubator with 5% CO2 at 37˚C. The
medium was replaced every 2 days.
Induction of OGD/R injury
The OGD/R model was built as previously described
(13). Briefly, the HT22 cells in
6-well plates (1x106 cells/well; Costar 3506, Corning
Life Sciences) were cultured under hypoxic conditions for 6 h in
glucose-free DMEM in an atmosphere of 5% CO2, 94%
N2 and 1% O2 at 37˚C.
Subsequently, the medium was discarded and DMEM containing 4.5 g/l
glucose was added, followed by culture under normoxic conditions in
a humidified 5% CO2 incubator at 37˚C for 24 h. Cells
without OGD/R treatment were used as a control.
Exposure to Sev
Cells in 6-well plates (1x106 cells/well)
were exposed to Sev in the carrier gas as previously described
(14). Briefly, different
concentrations of Sev (0, 4.1 or 8%) were delivered by a Sev
vaporizer sustained in 21% O2. The control gas was
defined as 21% O2. Cells in 6-well plates
(1x106 cells/well) were randomly placed in sealed
plastic chambers and exposed to Sev or the control gas at 4 l/min
for 6 h at 37˚C, followed by induction of OGD/R injury.
The concentration of Sev was monitored by an agent analyzer (AbbVie
Inc.)
Drug treatment
The PI3K inhibitor, wortmannin (15) was obtained from Merck KGaA and
dissolved in dimethyl sulfoxide (DMSO). HT22 cells in 6-well plates
(1x106 cells/well) were pretreated with 1 µM wortmannin
for 24 h, and then were exposed to 4.1% Sev for 6 h at 37˚C,
followed by OGD/R stimulation. The dose of wortmannin (1 µM) was
chosen as previously described (16).
Cell viability assay
At 24 h after sevo treatment, HT22 cells in 96
microplates (5x104 cells/well) were subjected to OGD/R,
and then the cell viability of HT22 cells was measured with the
Cell Counting Kit-8 assay (CCK-8; Beyotime Institute of
Biotechnology) according to the manufacturers protocols. A total of
10 µl CCK-8 reagent in DMEM was added to the cells and incubated
for another 2 h at 37˚C. Absorbances at 450 nm were measured using
a microplate reader (Bio-Rad Laboratories, Inc).
Lactate dehydrogenase (LDH) assay
At 24 h after sevo treatment, HT22 cells were
subjected to OGD/R, and LDH release in the HT22 cells was measured
using Pierce LDH Cytotoxicity Assay Kit (cat. no. 88953; Pierce;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The OD values were measured at 490 nm using a
microplate reader (Bio-Rad Laboratories, Inc).
Cell apoptosis assay
The apoptotic rate of HT22 cells was evaluated by
Annexin V-FITC and PI apoptosis detection kit I (BD Pharmingen; BD
Biosciences), following the manufacturer's protocol. At 24 h after
sevo treatment, HT22 cells were subjected to OGD/R, and then cells
were collected, washed with phosphate-buffered saline (PBS) and
resuspended in binding buffer, and double-stained with Annexin
V-FITC and PI for 10 min at room temperature in the dark. After
incubation for 15 min at room temperature in the dark, cell
apoptosis was analyzed by FACScan flow cytometer (FCM; Beckman
Coulter, Inc.) and CellQuest software version 3.3 (BD Biosciences).
The apoptosis rate was the sum of the Q2 and Q3 quadrants of the
flow cytometry images.
Caspase-3 activity
The caspase-3 activity of HT22 cells was measured
after exposure to the various treatments using the Caspase-3 Assay
kit (cat. no. C10427; Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The optical density was
measured at a wavelength of 450 nm using a microplate reader
(Bio-Rad Laboratories, Inc.).
Detection of reactive oxygen species
(ROS), malonaldehyde (MDA), superoxide dismutase (SOD) and
glutathione peroxidase (GPx)
ROS production in HT22 cells was measured using
DCFH-DA (Invitrogen; Thermo Fisher Scientific, Inc.) as previously
reported (17). At 24 h after sevo
treatment, HT22 cells were subjected to OGD/R, and HT22 cells
(1x106 cells/well) were seeded in a 6-well plate for 24
h, and then stained with 20 µmol/l DCFH at 37˚C for 30 min, and
then cell images were captured using a fluorescence microscope
(Olympus Corporation) and analyzed by ImageJ software (version
1.46; National Institutes of Health).
After the designated treatment of interest, the HT22
cells were lysed using lysis buffer (Beyotime Institute of
Biotechnology), following by lysate collection for the detection of
SOD, MDA and GPx. The MDA content was measured by thiobarbituric
acid method using a Lipid Peroxidation MDA assay kit (cat. no.
S0131S), the SOD activity was detected by water-soluble tetrazolium
salt (WST-8) method using a SOD assay kit (cat. no. S0103) and the
activity of GPx was evaluated using a Cellular GPx assay kit (cat.
no. S0056), all purchased from Beyotime Institute of Biotechnology,
performed according to the manufacturer's instructions.
Western blot analysis
Western blot was performed as previously described
(18). Briefly, total protein was
isolated from HT22 cells using RIPA lysis buffer (Beyotime
Institute of Biotechnology). The concentrations of total cellular
protein were quantitated using BCA assay (Pierce; Thermo Fisher
Scientific, Inc.). The protein lysates (40 µg) were analyzed on an
8% SDS-PAGE gel and transferred onto PVDF membranes (EMD
Millipore), followed blocking with 5% skim milk at 4˚C
overnight. The membranes were probed with the following appropriate
primary antibodies: Phosphorylated (phosp)-AKT (Ser473; dilution
1:500; cat. no. 4060), AKT (dilution 1:1,000; cat. no. 4691), GSK3β
(dilution 1:1,000; cat. no. 5676), phosp-GSK3β (Ser9; dilution
1:1,000; cat. no. 9327) and β-actin (dilution 1:2,000; cat no.4970)
overnight at 4˚C, followed by incubation with
horseradish peroxidase-conjugated (HRP) antibodies (dilution
1:6,000; cat. no. 45262; Cell Signaling Technology, Inc.) for 1 h
at room temperature. All antibodies were obtained from Cell
Signaling Technology, Inc. β-actin served as an internal control.
The protein bands were visualized using ECL detection reagent
(Cytiva). The intensity of protein fragments was quantified with
Bio-Rad Laboratories Quantity One software (version 3.0; Bio-Rad
Laboratories, Inc.).
Statistical analysis
Statistical analysis was performed by SPSS software
version 14.0 (SPSS, Inc.). All data were presented as the mean ±
standard deviation. The comparisons among data were calculated by
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
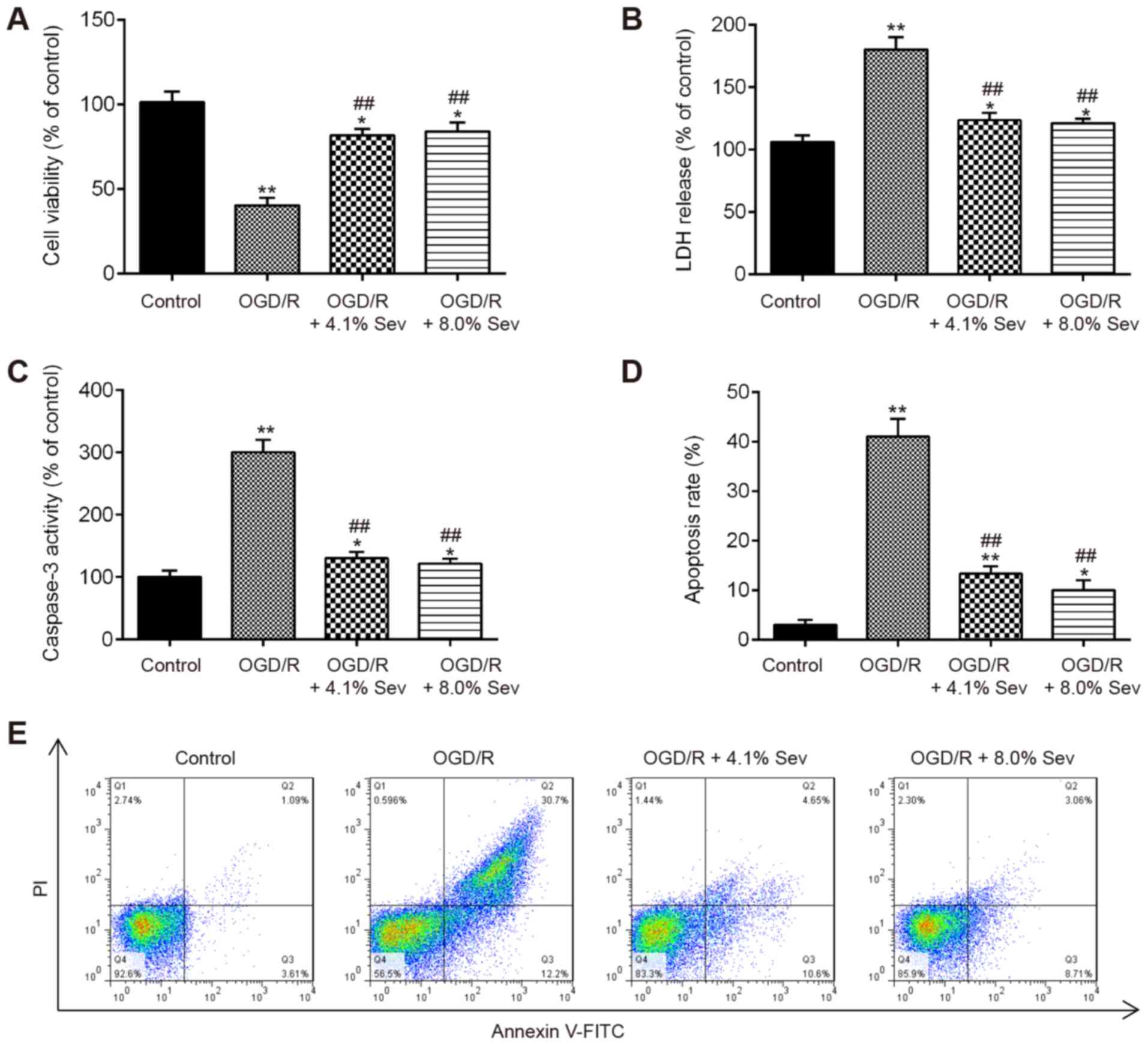
Sev ameliorates OGD/R-induced injury
in HT22 cells
To evaluate the protective effects of Sev on the
injury induced by OGD/R, HT22 cells were exposed to Sev (0, 4.1 or
8%) for 6 h, followed by induction of OGD/R injury. CCK-8 assay
showed that cell viability was significantly decreased in
OGD/R-induced cells, compared with that in the control group
(Fig. 1A). However, Sev treatment
(4.1 and 8%) significantly improved the viability of HT22 cells
receiving OGD/R stimulation (Fig.
1A). The LDH assay showed that Sev treatment (4.1 and 8%)
markedly decreased LDH leakage in OGD/R-induced HT22 cells,
compared with the OGD/R group (Fig.
1B). Given that apoptosis is a key event in the pathogenesis of
cerebral I/R injury (19), Sev
treatment was investigated for its potential to affect the
apoptosis in OGD/R-induced HT22 cells. As shown in Fig. 1C, OGD/R significantly increased the
caspase-3 activity, compared with the control group, whereas Sev
treatment attenuated the promoting effect of OGD/R on the caspase-3
activity. Moreover, it was observed that OGD/R significantly
promoted the apoptosis portion of HT22 cells, whereas Sev treatment
decreased the apoptosis portion of HT22 cells (Fig. 1D and E). These data indicated that Sev treatment
ameliorates OGD/R-induced cell injury and apoptosis in HT22
cells.
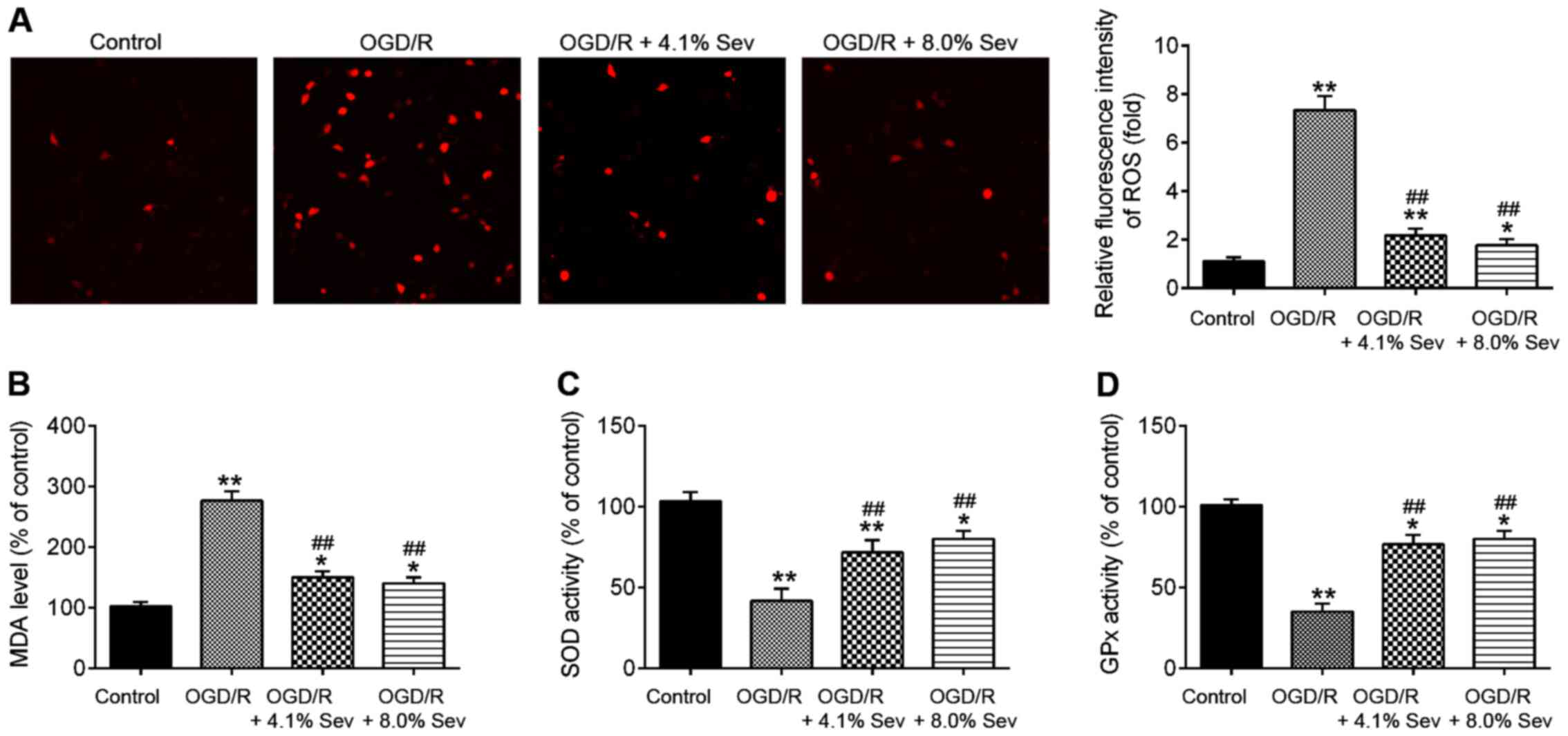
Sev inhibits OGD/R-induced oxidative
stress in HT22 cells
Oxidative stress is another key factor in brain
damage after cerebral I/R (20).
Previous studies suggested that cerebral I/R could cause oxidative
stress and then lead to over-production of ROS. Excessive ROS
promote cell death and result in brain damage. Malondialdehyde
(MDA) is the parameter of oxidative stress and is the lipid
peroxidation product that is used to determine the level of damage
of cell membrane lipid by ROS (21). Superoxide dismutase (SOD) and
glutathione peroxidase (GSH-Px) are antioxidant enzymes to protect
cells from oxidative damage (22).
Therefore, the detection of MDA, GSH-Px levels, and SOD activity
reflect the degree of oxidative damage in cells and tissues. To
examine whether Sev ameliorates OGD/R-induced cell injury by
regulating oxidative stress, the markers of oxidative stress,
namely ROS, MDA, SOD and GPx, were detected after OGD/R injury and
Sev treatment. As shown in Fig. 2A
and B, ROS and MDA production
levels were significantly increased in OGD/R-treated HT22 cells
compared with those in the control group. However, the increased
production of ROS and MDA in OGD/R-induced HT22 cells was decreased
by Sev treatment (Fig. 2A and
B). In addition, it was observed
that OGD/R-induced a significant decrease in the activity of SOD
and GPx in HT22 cells, whereas Sev attenuated the inhibitory
effects of OGD/R on the activity of SOD and GPx (Fig. 2C and D). All data suggest that Sev treatment may
improve OGD/R-induced injury by suppressing oxidative stress in
HT22 cells.
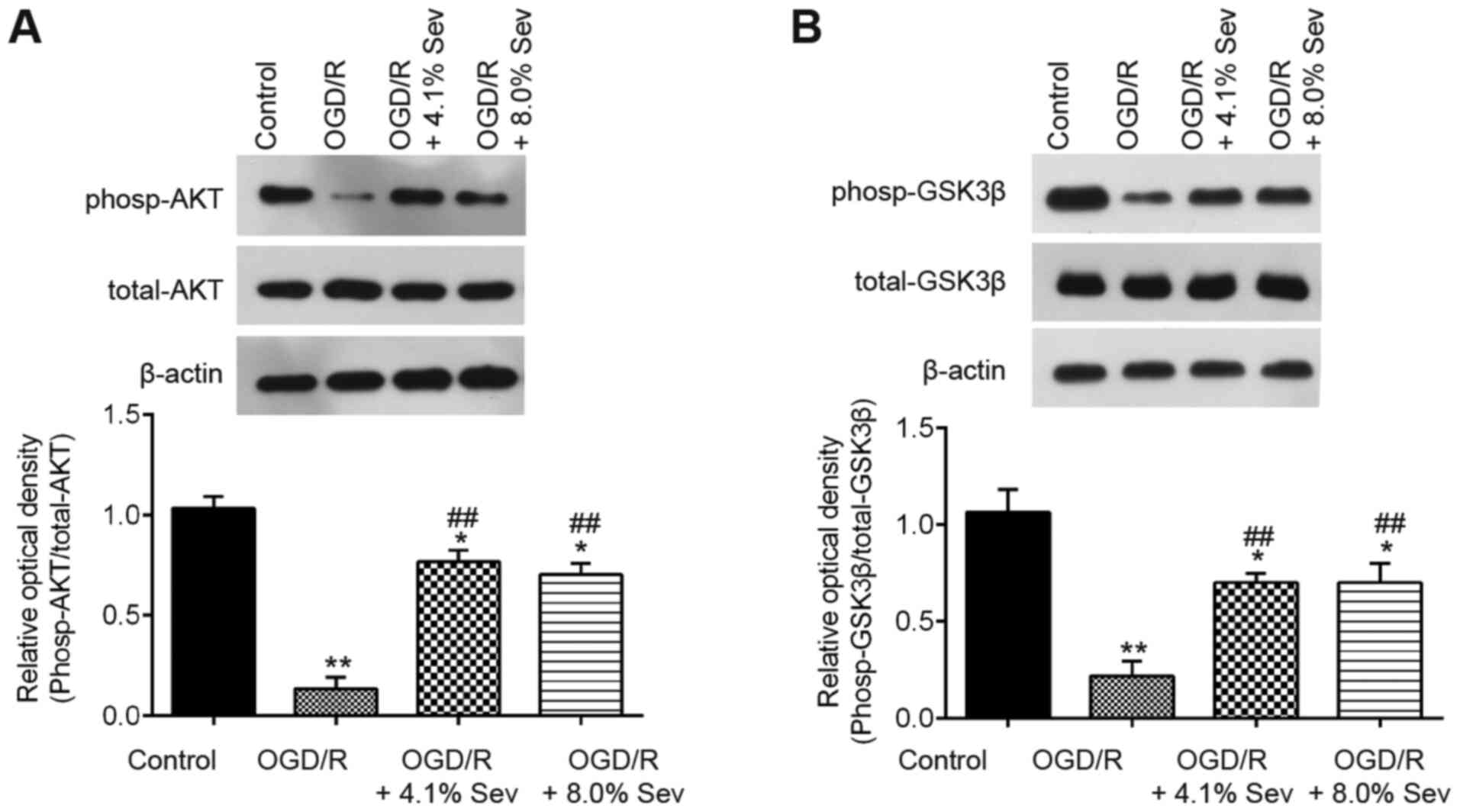
Sev induces phosphorylation of GSK3β
via the PI3K/AKT pathway
It has been previously reported that Sev provides
neuroprotection in adult animals via the PI3K/Akt/GSK3β signaling
pathway, which has a key role in cerebral I/R injury (23,24).
To explore the potential molecular mechanisms of Sev against
OGD/R-induced cell injury in HT22 cells, western blotting analysis
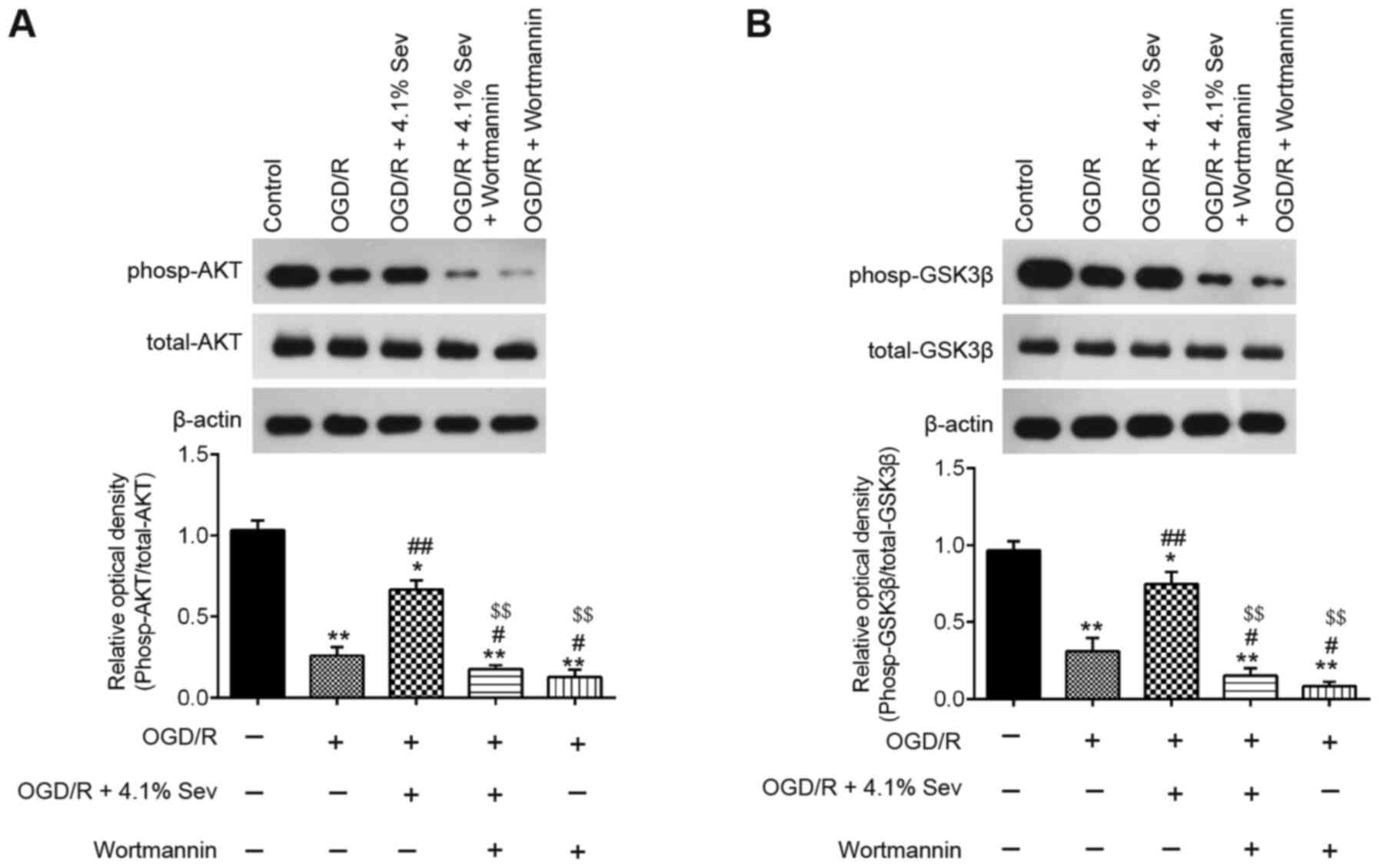
was performed to detect the phosp-AKT, AKT, phosp-GSK3β and GSK3β
levels in OGD/R-induced cells. As shown in Fig. 3A, phosp-AKT expression was markedly
downregulated in OGD/R-induced cells compared with that in control
cells, whereas Sev treatment attenuated the inhibitory effect of
OGD/R on the expression of phosp-AKT. Furthermore, Sev treatment
ameliorated the decrease in phosp-GSK3β expression induced by OGD/R
in HT22 cells (Fig. 3B). These
results indicated that Sev could reactivate the PI3K/AKT/GSK3β
pathway in HT22 cells after OGD/R induction.
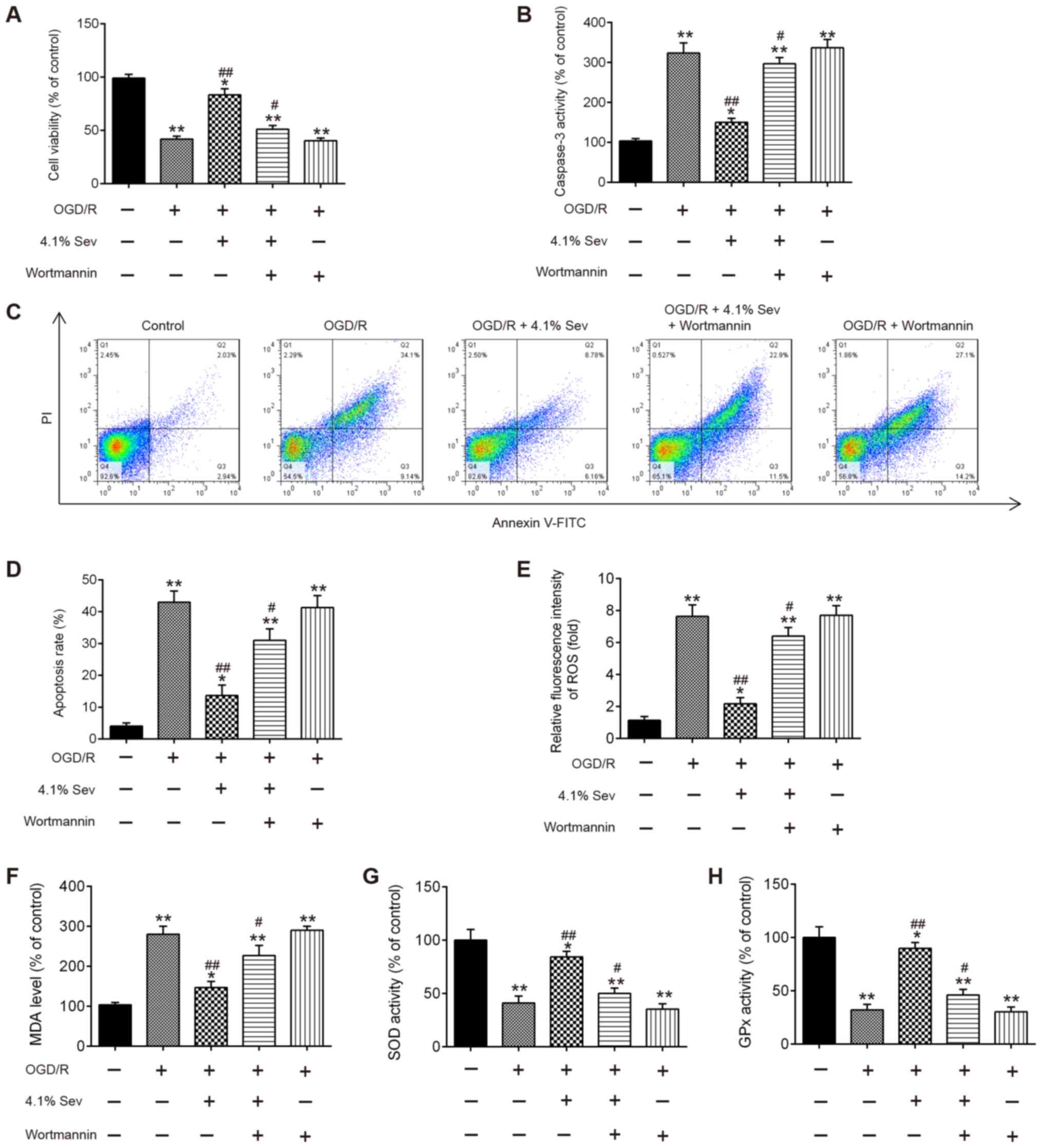
PI3K kinase inhibitor wortmannin
inhibits the protective effect of Sev on OGD/R-induced cell
injury
To investigate the effects of the PI3K/AKT/GSK3β
signaling pathway on the neuroprotective effect exerted by Sev,
wortmannin, a selective PI3K inhibitor, was added to HT22 cells in
combination with Sev for 30 min before subjecting them to OGD/R at
a concentration of 10 µM. As shown in Fig. 4A, Sev treatment (4.1%) markedly
promoted the cell viability in OGD/R-induced HT22 cells. However,
the protection of Sev was significantly alleviated by the presence
of 10 µM wortmannin, whereas wortmannin alone did not influence the
cell viability of HT22 cells compared with OGD/R-induced cells
alone (Fig. 4A). Additionally, the
results showed that pretreatment with wortmannin partially reversed
the inhibitory effect of Sev on caspase-3 activity and apoptosis in
OGD/R-induced cells (Fig. 4B-D).
Moreover, blockade of PI3K by wortmannin also markedly reversed the
protective effect of Sev on oxidative stress induced by OGD/R
(Fig. 4E-H). All these data
suggested that Sev may suppress OGD/R-induced apoptosis and
oxidative stress through the PI3K/AKT/GSK3β signaling pathway.
Sev-reactivated PI3K/AKT/GSK3β
signaling pathway is inhibited by wortmannin in OGD/R-induced
cells
To confirm whether Sev regulated the PI3K/AKT/GSK3β
signaling pathway in OGD/R-induced cell injury, phosp-AKT, AKT,
phosp-GSK3β and GSK3β expression levels were detected in
OGD/R-induced HT22 cells treated with Sev and pretreated with
wortmannin. As shown in Fig. 5A and
B, Sev treatment significantly
increased the levels of phosp-AKT and phosp-GSK3β compared with
OGD/R-induced cells alone, but these effects were inhibited by
wortmannin treatment. These data indicate that Sev alleviates
OGD/R-induced cell injury through reactivating the PI3K/AKT/GSK3β
signaling pathway.
Discussion
In the present study, the results demonstrated that
Sev improves OGD/R-induced neuronal injury by suppressing cell
apoptosis and oxidative stress. Moreover, data revealed that the
PI3K/AKT/GSK3β axis is involved in the protective effects of Sev.
The findings of the current study suggested that Sev has the
potential to protect neurons from cerebral ischemic injury.
Sev, a volatile anesthetic with minimal pungency,
low solubility and low toxicity, is used widely in anesthetic
practice (25), and has been
documented to play a neuroprotective role following severe cerebral
ischemia. For example, Canas et al (10) showed that Sev has a neuroprotective
effect in an in vitro model of ischemia-reoxygenation.
Wise-Faberowski et al (26)
found that Sev can attenuate OGD-induced neuronal cell death. Ye
et al (19) reported that
delayed application of Sev after reperfusion provides
neuroprotection by activating the PI3K/Akt pathway, and Wen et
al (27) demonstrated the
neuroprotection of Sev against I/R-induced brain injury through the
inhibition of JNK3/caspase-3 by enhancing the Akt signaling
pathway. Together, the results of these previous studies make Sev
an attractive agent for the preservation of neuronal function. In
the present study, using an OGD/R model, Sev pretreatment was found
to markedly increase cell viability and attenuate LDH leakage in
HT22 cells following OGD/R. In addition, Sev inhibited
OGD/R-induced apoptosis, as evidenced by the decrease in caspase-3
activity and cleaved-caspase-3 protein expression in the HT22
cells. These data suggested that Sev can efficiently attenuate
OGD/R-induced neuronal cell injury and apoptosis.
It is well known that oxidative stress is an
important factor in the pathogenesis of cerebral I/R injury, which
is caused by increased production of ROS and decreased activity
levels of scavenger enzymes, such as SOD and GPx (28). ROS can interfere with the regulation
of gene expression, thus altering the relative expression levels of
signaling proteins, influencing intracellular signaling cascades
(29). Under pathological
conditions, such as cerebral ischemia, the aberrant production of
ROS can result in the oxidation of cellular components, leading to
cell damage (30). Hence,
investigating whether Sev plays a role in ROS production after
cerebral I/R injury was a focus of the present study. Sev was found
to decrease the production of ROS and MDA, and to elevate the SOD
and GPx activity induced by OGD/R in HT22 cells, suggesting that
Sev protects neuronal cells from OGD/R injury by inhibiting
oxidative stress.
PI3K/AKT, an anti-apoptotic and pro-survival kinase
signaling cascade, plays an important role in I/R induced brain
injury (31-33).
Activation of AKT induces the phosphorylation of GSK3β at serine 9
(34,35). In fact, the PI3K/AKT/GSK3β signaling
pathway plays a key role in modulating cellular survival/death
after hypoxia and I/R (36-38).
Recently, Gerace et al (39)
indicated that inhibition of AKT/GSK3β signaling pathway by the
PI3K inhibitor LY294002 or two GSK3β inhibitors exerts a
neuroprotective effect against cerebral I/R injury induced by OGD/R
(39). In addition, suppression of
PI3K/AKT/GSK3β signaling pathway by the PI3K inhibitor LY294002 and
GSK3β inhibitor LiCl exhibited neuroprotective properties against
cerebral ischaemia-induced apoptosis (40). Therefore, we hypothesize that the
neuroprotective effect of Sev on cerebral I/R injury occurs through
the modulation of the PI3K/AKT/GSK3β signaling pathway. Consistent
with the aforementioned studies, the activation of the PI3K/Akt
pathway was also demonstrated to mediate the protective effect of
Sev on OGD/R-induced cell injury in the present study. However, it
is worthy of note that wortmannin, a selective PI3K inhibitor,
abolished the neuroprotective effect of Sev by suppressing the
phosphorylation of AKT and GSK3β proteins. Collectively, these
results suggested that Sev improves OGD/R-induced neuronal injury
by activating the PI3K/AKT/GSK3β signaling pathway.
In summary, the results of the present study
demonstrated that Sev alleviates OGD/R-induced neuronal injury
through regulating the PI3K/AKT/GSK3β signaling pathway. These
findings suggest that the application of Sev may help to protect
neurons against cerebral I/R injury.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by Shanghai Municipal
Commission of Health and Family Planning (201740008).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QY, HD, YJ and YZ performed the experiments,
analyzed the data and wrote the study. JZ conceived and designed
the study and contributed reagents to the study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Feigin VL: Stroke epidemiology in the
developing world. Lancet. 365:2160–2161. 2005.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Surinkaew P, Sawaddiruk P, Apaijai N,
Chattipakorn N and Chattipakorn SC: Role of microglia under cardiac
and cerebral ischemia/reperfusion (I/R) injury. Metab Brain Dis.
33:1019–1030. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Schrepfer E and Scorrano L: Mitofusins,
from mitochondria to metabolism. Mol Cell. 61:683–694.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu H, Tang C, Tai LW, Yao W, Guo P, Hong
J, Yang X, Li X, Jin Z, Ke J and Wang Y: Flurbiprofen axetil
attenuates cerebral ischemia/reperfusion injury by reducing
inflammation in a rat model of transient global cerebral
ischemia/reperfusion. Biosci Rep. 38(BSR20171562)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang Y, Li D, Luo J, Chen S, Dou X, Han M
and Zhang H: Pharmacological postconditioning with sevoflurane
activates PI3K/AKT signaling and attenuates cardiopulmonary
bypass-induced lung injury in dog. Life Sci. 173:68–72.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lu CC, Tsai CS, Ho ST, Chen WY, Wong CS,
Wang JJ, Hu OYP and Lin CY: Pharmacokinetics of sevoflurane uptake
into the brain and body. Anaesthesia. 58:951–956. 2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xiong XQ, Lin LN, Wang LR and Jin LD:
Sevoflurane attenuates pulmonary inflammation and
ventilator-induced lung injury by upregulation of HO-1 mRNA
expression in mice. Int J Nanomedicine. 6:1075–1081.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li XQ, Cao XZ, Wang J, Fang B, Tan WF and
Ma H: Sevoflurane preconditioning ameliorates neuronal deficits by
inhibiting microglial MMP-9 expression after spinal cord
ischemia/reperfusion in rats. Mol Brain. 7(69)2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Park HP, Jeong EJ, Kim MH, Hwang JW, Lim
YJ, Min SW, Kim CS and Jeon YT: Effects of sevoflurane on neuronal
cell damage after severe cerebral ischemia in rats. Korean J
Anesthesiol. 61:327–331. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Canas PT, Velly LJ, Labrande CN, Guillet
BA, Sautou-Miranda V, Masmejean FM, Nieoullon AL, Gouin FM, Bruder
NJ and Pisano PS: Sevoflurane protects rat mixed cerebrocortical
neuronal-glial cell cultures against transient oxygen-glucose
deprivation: Involvement of glutamate uptake and reactive oxygen
species. Anesthesiology. 105:990–998. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang Z, Ye Z, Huang G, Wang N, Wang E and
Guo Q: Sevoflurane post-conditioning enhanced hippocampal neuron
resistance to global cerebral ischemia induced by cardiac arrest in
rats through PI3K/Akt survival pathway. Front Cell Neurosci.
10(271)2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hu X, Zhang Y, Li W, Liu J and Li Y:
Preconditioning with sevoflurane ameliorates spatial learning and
memory deficit after focal cerebral ischemia-reperfusion in rats.
Int J Dev Neurosci. 31:328–333. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang G, Wang T, Zhang Y, Li F, Yu B and
Kou J: Schizandrin protects against OGD/R-induced neuronal injury
by suppressing autophagy: Involvement of the AMPK/mTOR pathway.
Molecules. 24(3624)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liang H, Gu M, Yang C, Wang H, Wen X and
Zhou Q: Sevoflurane inhibits invasion and migration of lung cancer
cells by inactivating the p38 MAPK signaling pathway. J Anesth.
26:381–392. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Pyo H, Yang MS, Jou I and Joe EH:
Wortmannin enhances lipopolysaccharide-induced inducible nitric
oxide synthase expression in microglia in the presence of
astrocytes in rats. Neurosci Lett. 346:141–144. 2003.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rössler OG, Giehl KM and Thiel G:
Neuroprotection of immortalized hippocampal neurones by
brain-derived neurotrophic factor and Raf-1 protein kinase: Role of
extracellular signal-regulated protein kinase and
phosphatidylinositol 3-kinase. J Neurochem. 88:1240–1252.
2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tang Y, Vater C, Jacobi A, Liebers C, Zou
X and Stiehler M: Salidroside exerts angiogenic and cytoprotective
effects on human bone marrow-derived endothelial progenitor cells
via Akt/mTOR/p70S6K and MAPK signalling pathways. Br J Pharmacol.
171:2440–2456. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Landucci E, Lattanzi R, Gerace E,
Scartabelli T, Balboni G, Negri L and Pellegrini-Giampietro DE:
Prokineticins are neuroprotective in models of cerebral ischemia
and ischemic tolerance in vitro. Neuropharmacology. 108:39–48.
2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ye Z, Xia P, Cheng ZG and Guo Q:
Neuroprotection induced by sevoflurane-delayed post-conditioning is
attributable to increased phosphorylation of mitochondrial GSK-3β
through the PI3K/Akt survival pathway. J Neurol Sci. 348:216–225.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen H, Yoshioka H, Kim GS, Jung JE, Okami
N, Sakata H, Maier CM, Narasimhan P, Goeders CE and Chan PH:
Oxidative stress in ischemic brain damage: Mechanisms of cell death
and potential molecular targets for neuroprotection. Antioxid Redox
Signal. 14:1505–1517. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xu HX, Pan W, Qian JF, Liu F, Dong HQ and
Liu QJ: MicroRNA-21 contributes to the puerarin-induced
cardioprotection via suppression of apoptosis and oxidative stress
in a cell model of ischemia/reperfusion injury. Mol Med Rep.
20:719–727. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Siti HN, Kamisah Y and Kamsiah J: The role
of oxidative stress, antioxidants and vascular inflammation in
cardiovascular disease (Review). Vascul Pharmacol. 71:40–56.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lai Z, Zhang L, Su J, Cai D and Xu Q:
Sevoflurane postconditioning improves long-term learning and memory
of neonatal hypoxia-ischemia brain damage rats via the
PI3K/Akt-mPTP pathway. Brain Res. 1630:25–37. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li P, Zhang Y and Liu H: The role of
Wnt/β-catenin pathway in the protection process by dexmedetomidine
against cerebral ischemia/reperfusion injury in rats. Life Sci.
236(116921)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Patel SS and Goa KL: Sevoflurane. A review
of its pharmacodynamic and pharmacokinetic properties and its
clinical use in general anaesthesia. Drugs. 51:658–700.
1996.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wise-Faberowski L, Raizada MK and Sumners
C: Desflurane and sevoflurane attenuate oxygen and glucose
deprivation-induced neuronal cell death. J Neurosurg Anesthesiol.
15:193–199. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wen XR, Fu YY, Liu HZ, Wu J, Shao XP,
Zhang XB, Tang M, Shi Y, Ma K, Zhang F, et al: Neuroprotection of
sevoflurane against ischemia/reperfusion-induced brain injury
through inhibiting JNK3/caspase-3 by enhancing akt signaling
pathway. Mol Neurobiol. 53:1661–1671. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Coyle JT and Puttfarcken P: Oxidative
stress, glutamate, and neurodegenerative disorders. Science.
262:689–695. 1993.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shen MH, Zhang CB, Zhang JH and Li PF:
Electroacupuncture attenuates cerebral ischemia and reperfusion
injury in middle cerebral artery occlusion of rat via modulation of
apoptosis, inflammation, oxidative stress, and excitotoxicity. Evid
Based Complement Alternat Med. 2016(9438650)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sosunov SA, Ameer X, Niatsetskaya ZV,
Utkina-Sosunova I, Ratner VI and Ten VS: Isoflurane anesthesia
initiated at the onset of reperfusion attenuates oxidative and
hypoxic-ischemic brain injury. PLoS One.
10(e0120456)2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chen L, Wei X, Hou Y, Liu X, Li S, Sun B,
Liu X and Liu H: Tetramethylpyrazine analogue CXC195 protects
against cerebral ischemia/reperfusion-induced apoptosis through
PI3K/Akt/GSK3β pathway in rats. Neurochem Int. 66:27–32.
2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu H, Liu X, Wei X, Chen L, Xiang Y, Yi F
and Zhang X: Losartan, an angiotensin II type 1 receptor blocker,
ameliorates cerebral ischemia-reperfusion injury via
PI3K/Akt-mediated eNOS phosphorylation. Brain Res Bull. 89:65–70.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang J, Deng Z, Liao J, Song C, Liang C,
Xue H, Wang L, Zhang K and Yan G: Leptin attenuates cerebral
ischemia injury through the promotion of energy metabolism via the
PI3K/Akt pathway. J Cereb Blood Flow Metab. 33:567–574.
2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Martin M, Rehani K, Jope RS and Michalek
SM: Toll-like receptor-mediated cytokine production is
differentially regulated by glycogen synthase kinase 3. Nat
Immunol. 6:777–784. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Jope RS and Johnson GVW: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Arslan F, Lai RC, Smeets MB, Akeroyd L,
Choo A, Aguor ENE, Timmers L, van Rijen HV, Doevendans PA,
Pasterkamp G, et al: Mesenchymal stem cell-derived exosomes
increase ATP levels, decrease oxidative stress and activate
PI3K/Akt pathway to enhance myocardial viability and prevent
adverse remodeling after myocardial ischemia/reperfusion injury.
Stem Cell Res. 10:301–312. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li Y, Zhu W, Tao J, Xin P, Liu M, Li J and
Wei M: Fasudil protects the heart against ischemia-reperfusion
injury by attenuating endoplasmic reticulum stress and modulating
SERCA activity: The differential role for PI3K/Akt and JAK2/STAT3
signaling pathways. PLoS One. 7(e48115)2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang JF, Zhang L, Shi LL, Zhao ZH, Xu H,
Liang F, Li HB, Zhao Y, Xu X, Yang K and Tian YF: Parthenolide
attenuates cerebral ischemia/reperfusion injury via Akt/GSK-3β
pathway in PC12 cells. Biomed Pharmacother. 89:1159–1165.
2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Gerace E, Scartabelli T,
Pellegrini-Giampietro DE and Landucci E: Tolerance induced by
(S)-3,5-dihydroxyphenylglycine postconditioning is mediated by the
PI3K/Akt/GSK3β signalling pathway in an in vitro model of cerebral
ischemia. Neuroscience. 433:221–229. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lee DH, Lee YJ and Kwon KH:
Neuroprotective effects of astaxanthin in oxygen-glucose
deprivation in SH-SY5Y cells and global cerebral ischemia in rat. J
Clin Biochem Nutr. 47:121–129. 2010.PubMed/NCBI View Article : Google Scholar
|