Introduction
The neural crest (NC) is a multipotent cell
population and is one of the key features of craniofacial
development (1). Neural crest cells
(NCCs) originate from the dorsal neural tube and generate numerous
cell types, including cartilage and pigment cells (2). Defective development of NC has been
demonstrated to result in a number of syndromes, such as Treacher
Collins and DiGeorge syndrome, which are characterized by
developmental defects in the craniofacial region (3-5).
In humans, craniofacial malformations have been reported to be
associated with 75% of all congenital birth malformations (6) and are accompanied by abnormal brain
development and poor prognosis.
MicroRNAs (miRNAs/miRs) have been demonstrated to
regulate ~30% of genes in Caenorhabditis elegans, mouse and
human and serve an important role in embryonic development
(7). The Homo sapiens
(hsa)-miR-1 family includes two miRNAs, hsa-miR-1-1 and
hsa-miR-1-2, which exhibit the same mature sequence, although they
are encoded by different genes (8).
Previous studies have indicated that miR-1 participates in
regulating the development of the heart and skeletal muscle
(9,10). The outflow track of the heart, as
well as the tendons, muscles and craniofacial structures, have all
been reported to be derivatives of the NC (1). miR-1 has been revealed to be conserved
and important for embryonic development in zebrafish, especially NC
development (11). However, the
specific role of miR-1 in NCCs is currently unknown.
Zebrafish exhibit rapid embryonic development, a
short life cycle and large spawning capacity. In addition, the
embryos are transparent, so the process of embryonic skeletal
development can be observed directly (12). Therefore, zebrafish are a suitable
animal model to study the function of genes in craniofacial
development. In the present study, the miR-1-knockout zebrafish
model was a homozygous mutant, which was used to examine the role
of miR-1 in NCCs.
The current study aimed to evaluate the role of
miR-1 in the development of NC using a miR-1 deficient zebrafish
model.
Materials and methods
Animals
The wild-type (WT) zebrafish embryos were of the
Tuebingen strain. WT and homozygous miR-1 mutant zebrafish (n=60
per group) were purchased from the Institute of Model Animals of
Nanjing University. The primers used for genotyping of the miR-1
mutant zebrafish, the sequence of the mutant allele and the
identification of the genotype are presented in Tables SI and SII and Fig.
S1. The embryos were grown in Holtfreter's solution (0.05 g/l
KCL, 0.1 g/l CaCl2, 0.2 g/l NaHCO3 and 3.5
g/l Nacl) with a 14 h light/10 h dark cycle at 28.5˚C in the
zebrafish facility of the Model Animal Research Center, Nanjing
University (Nanjing, China). The zebrafish were fed three times a
day with pellet food, and their health and behavior was monitored
daily. All experiments were approved by the Ethics Committee of the
School of Stomatology of Nanjing Medical University, and all
procedures were performed according to the guidelines of the Animal
Care Committee of Nanjing Medical University.
Whole mount in situ hybridization
Whole mount in situ hybridization was
performed using standard procedures with slight modifications as
previously described (13). Probes
were synthesized using a DIG RNA labeling kit (Roche Diagnostics).
The probes of forkhead box D3 (foxd3), muscle segment
homeobox-b (msxb), snail homolog 1b (snai1b),
transcription factor AP2a (tfap2a), distal-less 3
(dlx3b), neurog1 (ngn1) (Table SIII) were designed using Primer 5.0
software (Premier Biotech, Inc.). Embryos (n=5 per group) at 24 h
post-fertilization (hpf) were collected, fixed in 4%
paraformaldehyde overnight at 4˚C and subjected to gradient
dehydration with methanol (25, 50, 75 and 100%). The embryos were
maintained in 100% methanol at -20˚C for subsequent use. Following
rehydration at gradient dilutions using methanol/PBS, the embryos
were treated with proteinase K (10 µg/ml; Gibco; Thermo Fisher
Scientific, Inc.) at room temperature for 10 min. Subsequently,
embryos were prehybridized with the hybridization solution at 65˚C
for 2 h. In situ hybridization signals were detected by
sheep anti-digoxigenin-AP Fab-fragments (Roche Diagnostics) and the
color reaction was carried out by staining with BCIP/NBT (Roche
Diagnostics) as previously described (14).
Alcian blue staining
Zebrafish embryos at 96 hpf (n=5 per group) were
fixed in 4% paraformaldehyde overnight at 4˚C, dehydrated with
ethanol (50 and 100%) and stained with Alcian blue (0.15 mg/ml;
Sigma-Aldrich; Merck KGaA) overnight at room temperature (15). After the embryonic cartilage was
stained, the embryos were incubated in saturated boron overnight at
room temperature. Subsequently, bleaching was performed in a
mixture of 3% H2O2 and 1% KOH at a ratio of
15:85 v/v, and the specimens were digested in 1% trypsin at 37˚C
until all embryonic tissues were degraded. The digestion mixture
was replaced with 1% KOH solution, and subsequently the specimens
were incubated in a glycerol gradient (25, 50, 75 and 100%). The
embryos were observed using a stereomicroscope (magnification,
x20).
Histology and immunostaining
For whole mount immunostaining, the embryos (n=5 per
group) were fixed in 4% paraformaldehyde overnight at 4˚C
pretreated with proteinase K (10 µg/ml) at 37˚C for 30 min and
blocked in 2% BSA solution (BioFroxx) at room temperature for 1 h.
Subsequently, the embryos were stained with an anti-phospho-histone
H3 (PHH3; 1:100; cat. no. 3377; Cell Signaling Technology, Inc.)
antibody overnight at 4˚C and then with an anti-rabbit Alexa
Fluor@594 (1:400; Jackson ImmunoResearch Laboratories, Inc.) at 4˚C
for 2 h. A TUNEL assay was performed using an In Situ Cell
Death Detection kit (Roche Diagnostics) as previously described
(16). The staining was
subsequently observed using laser scanning confocal microscopy
(magnification, x20) (17).
Culture of NCCs
Mouse embryos were harvested at gestational day E9.5
according to a previously published protocol (18). The pregnant adult mice (n=3; Model
Animal Research Center, Yangzhou University) were sacrificed by
cervical dislocation following anesthesia with sodium pentobarbital
(50 mg/kg). The first branchial arch of the embryos was separated,
washed in PBS and dissociated in 0.25% trypsin-EDTA (Gibco; Thermo
Fisher Scientific, Inc.) for 30 min at 37˚C. The separated cells
were plated into a dish in Nutrient Mixture F-12 (1:1)media (Gibco;
Thermo Fisher Scientific, Inc.) with 15% FBS (ScienCell Research
Laboratories, Inc.), supplemented with 100 U/ml
penicillin-streptomycin (PS), 100 mg/ml L-glutamate, 0.1 mM minimum
essential medium non-essential amino acids (Gibco; Thermo Fisher
Scientific, Inc.) and 1,000 U/ml leukemia inhibitory factor
(PeproTech, Inc.). The inhibitor (5'-AUA CAUACUUCUUUACAUUCCA-3';
100 nM) and mimics (forward, 5'-UGGAAUGUAAAGAAGUAUGUAU-3'; and
reverse, 5'-AUACAUACUUCUUUACAUUCCA-3'; 50 nM) of miR-1 and their
negative controls (NCs; inhibitor NC, 5'-CAG
UACUUUUGUGUAGUACAAA-3'; mimics NC, 5'-UUU GUACUACACAAAAGUACUG-3';
5'-CAGUACUUUUG UGUAGUACAAA-3'; all Guangzhou RiboBio Co., Ltd.)
were transfected into NCCs when cells grew to 50% density using a
riboFECT™ CP Transfection kit (Guangzhou RiboBio Co., Ltd.)
according to the manufacturer's protocol.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 assay (Dojindo Molecular Technologies, Inc.)
was used to evaluate cell proliferation according to the
manufacturer's protocol. NCCs were seeded at 2x103
cells/well in 96-well plates. The cells were incubated at 37˚C for
12 h (day 0) and transfected with the miR-1 inhibitor. CCK-8
reagent in Nutrient Mixture F-12 media was added to each well at
the specified time points (days 0, 1, 2, 3 and 4). Following 1-h
incubation at 37˚C, the optical density was measured at 450 nm
using a microplate reader with three blank controls.
Wound healing assay
NCCs were seeded at 1.5x105 cells/well in
a 6-well plate. Following transfection with the miR-1 inhibitor, a
wound was created with a 10 µl pipette tip, and the cells were
incubated in serum-free medium at 37˚C for 24 h. Cell migration was
observed at 0, 12 and 24 h using a Leica DMIRE2 microscope
(magnification, x100; Leica Microsystems GmbH) in phase contrast
mode (19).
Cell cycle and apoptosis analysis
NCCs transfected with the miR-1 inhibitor were
cultured for 24 h without PS. Following an additional 48-h culture,
cell cycle and apoptosis analyses were performed as previously
described (20) using flow
cytometry (FACSCalibur; BD Biosciences). For cell cycle analysis,
the cells (~1x104) were fixed overnight with 75% ethanol
at 4˚C in the dark, and then stained with propidium iodide (PI;
MultiSciences Biotech Co., Ltd.) containing RNase. Apoptosis was
detected by harvesting cells using a centrifuge at room temperature
(5 min, 210 x g) and staining the cells (~1x104) with an
Annexin V-FITC Apoptosis Detection kit (BD Pharmingen; BD
Biosciences).
PI-/Annexin V-FITC+ cells
(early apoptotic population) and PI+/Annexin
V-FITC+ cells (late apoptotic population) were sorted.
Analysis was performed using FACScan cytometer (BD Biosciences) and
FlowJo V7 software (Tree Star).
Apoptotic cell staining
NCCs were seeded at 6x104 cells/well in a
24-well plate, transfected with the miR-1 inhibitor and then
cultured for 3 days at 37˚C. Acridine orange/ethidium bromide
(AO/EB) staining was performed using an AO/EB Detection kit
(Biotechgate) according to the manufacturer's instructions. After
fixing in 4% paraformaldehyde at 4˚C for 15 min, TUNEL staining of
NCCs was performed using an In Situ Cell Death Detection kit
(Roche Diagnostics) according to the manufacturer's instructions.
The cells were observed using fluorescence microscopy
(magnification, x400; Leica Microsystems, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total NCCs RNA was isolated using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), as previously
described (21). The miRNA
expression levels of miR-1 relative to the internal control U6 were
evaluated after reverse transcription (37˚C for 60 min and 85˚C for
5 min) using an All-in-One™ miRNA RT-qPCR Detection kit
(GeneCopoeia, Inc.), the thermocycling conditions were set
according to the manufacturer's instructions. The miR-1
(5'-CGCGTGGAATGTAAAGAA GTATGTA-3') and U6 (5'-AAAACAGCAATATGGAGC
GC-3') forward primers and universal reverse primers (cat. no.
QP015) were purchased from GeneCopoeia, Inc. Relative expression
levels were calculated using the 2-ΔΔCq method (22).
Western blot analysis
Proteins of cells were extracted using RIPA buffer
(Beyotime Institute of Biotechnology), boiled for 5 min and
analyzed its protein concentrations using a Bicinchoninic Acid
Protein Assay Kit (Beyotime Institute of Biotechnology). The
proteins were separated using 10% or 12% SDS-PAGE as previously
described (19), transferred onto
PVDF membranes and then blocked with 5% BSA at room temperature for
2 h. Primary antibodies against BCL-2, poly ADP-ribose polymerase
(PARP), pro- and cleaved-caspase-9 (1:500; cat. nos. 12789, 13371
and 10380, respectively; Wuhan Sanying Biotechnology), pro- and
cleaved-caspase-3 (1:1,000; cat. no. 9661S; Cell Signaling
Technology, Inc.) and β-actin (1:1,000; cat. no. sc-47778; Santa
Cruz Biotechnology, Inc.) were used overnight at 4˚C. Next, the
membranes were incubated with HRP-labeled Goat Anti-Rabbit or
HRP-labeled Goat Anti-mouse secondary antibody (1:8,000, cat. nos.
474-1506 and 474-1806, respectively; SeraCare KPL) at room
temperature for 1 h and visualized by enhanced chemiluminescence
(Thermo Fisher Scientific, Inc.). Semi-quantitative analysis was
performed using ImageJ 1.52 software (National Institutes of
Health).
Statistical analysis
All data are presented as the mean ± standard
deviation. Differences between two groups were analyzed using
GraphPad Prism v6 statistical software (GraphPad Software Inc.).
Unpaired Student's t-test was used for the statistical analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Loss of miR-1 in zebrafish results in
craniofacial, pigment cell and cardiac defects
In the present study, a miR-1 mutant zebrafish model
was generated to evaluate the role of miR-1 in the development of
NC. The expression of miR-1 was primarily localized in the head,
pharyngeal arch and pectoral fin of zebrafish (Fig. 1A). In the miR-1 mutant zebrafish,
the expression of miR-1 was suppressed in the aforementioned areas
(Fig. 1B).
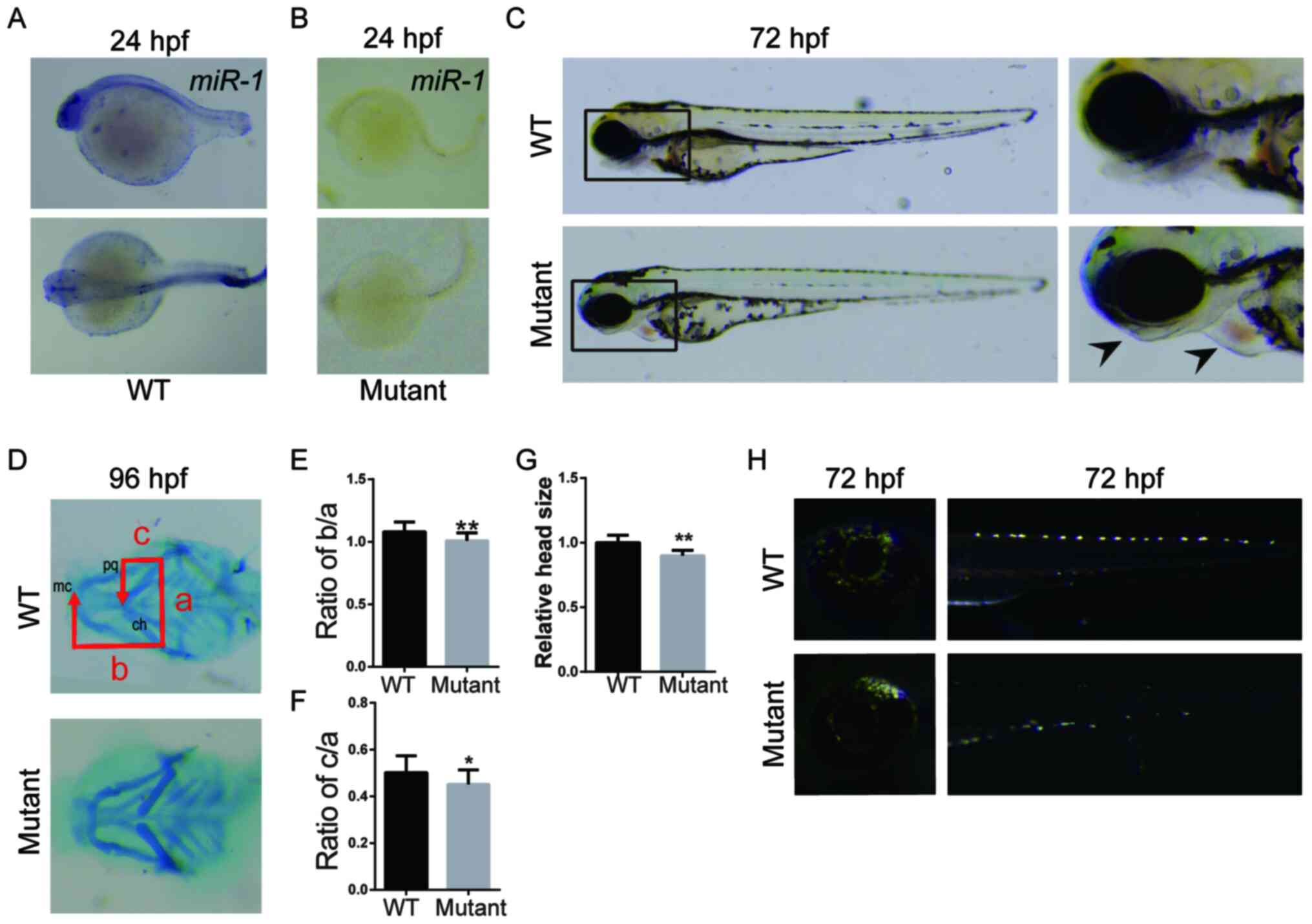 | Figure 1Loss of miR-1 in zebrafish results in
craniofacial, pigment cell and cardiac defects. In situ
hybridization of miR-1 in (A) wild-type and (B) mutant zebrafish at
24 hpf. (C) Zebrafish larvae at 72 hpf with mutants exhibiting
mandibular retrognathia (left arrow) and edema around the heart
(right arrow). (D) Alcian blue staining at 96 hpf. (E) Quantitative
analysis of b/a. (F) Quantitative analysis of c/a. a, width between
ch; b, distance between mc and ch; and c, the distance between pq
and ch. (G) Quantitative analysis of head size. (H) Distribution
and number of iridophores at 72 hpf. Data are presented as the mean
± standard deviation (n=5). *P<0.05 and
**P<0.01 vs. WT. WT, wild-type; hpf, hours
post-fertilization; miR-1, microRNA-1; mc, Meckel's cartilage; pq,
palatoquadrate; ch, ceratohyal cartilage. |
A number of abnormal phenotypes were observed in the
mutant compared with the WT zebrafish, including a smaller head, a
decreased mandibular arch length and mandibular retrognathia
(Fig. 1C-G). In the mutant
zebrafish, the number of melanocytes and iridophores (NCC-derived
pigment cells) was decreased (Fig.
1C and H). In addition, the
mutant zebrafish exhibited abnormal cardiac development with
enlarged hearts compared with those of the WT zebrafish (Fig. 1C). A quantitative analysis of the
association among the palatal square joint, McGuire cartilage and
hyoid small horn cartilage has been previously performed (23). In the present study, the head size
and mandibular arch length were decreased in the mutant group
compared with those in the WT group (Fig. 1D-G).
These results suggested that the lack of miR-1
affected the development of NCCs in zebrafish, which resulted in
severe phenotypic defects.
miR-1 affects NCC apoptosis during the
migration and differentiation periods in embryonic development
To determine which stage of NC development was
affected by miR-1, the expression of marker genes of specific
stages (foxd3, msxb, snai1b, tfap2a, dlx3b and ngn1)
was detected via in situ hybridization. These genes have
been demonstrated to be associated with the induction,
specialization, migration and differentiation of NCCs (24,25).
Foxd3 has been indicated to serve an
important role in NC derived tissues, such as the pharyngeal arch
(24). In the present study, two
regions of NCCs were labelled with the foxd3 probe in WT and
mutant zebrafish; however, the mutant group appeared to exhibit
lower expression levels of foxd3 compared with that in the
WT group (Fig. 2A). This indicated
that the migration of NCCs may be suppressed in the miR-1 mutant
group. Msxb in zebrafish has also been reported to be
required for NCC survival (25).
However, msxb expression exhibited no difference between the
two groups (Fig. 2B). Therefore, it
was speculated that the lack of miR-1 affected the migration, but
not the survival of NCCs.
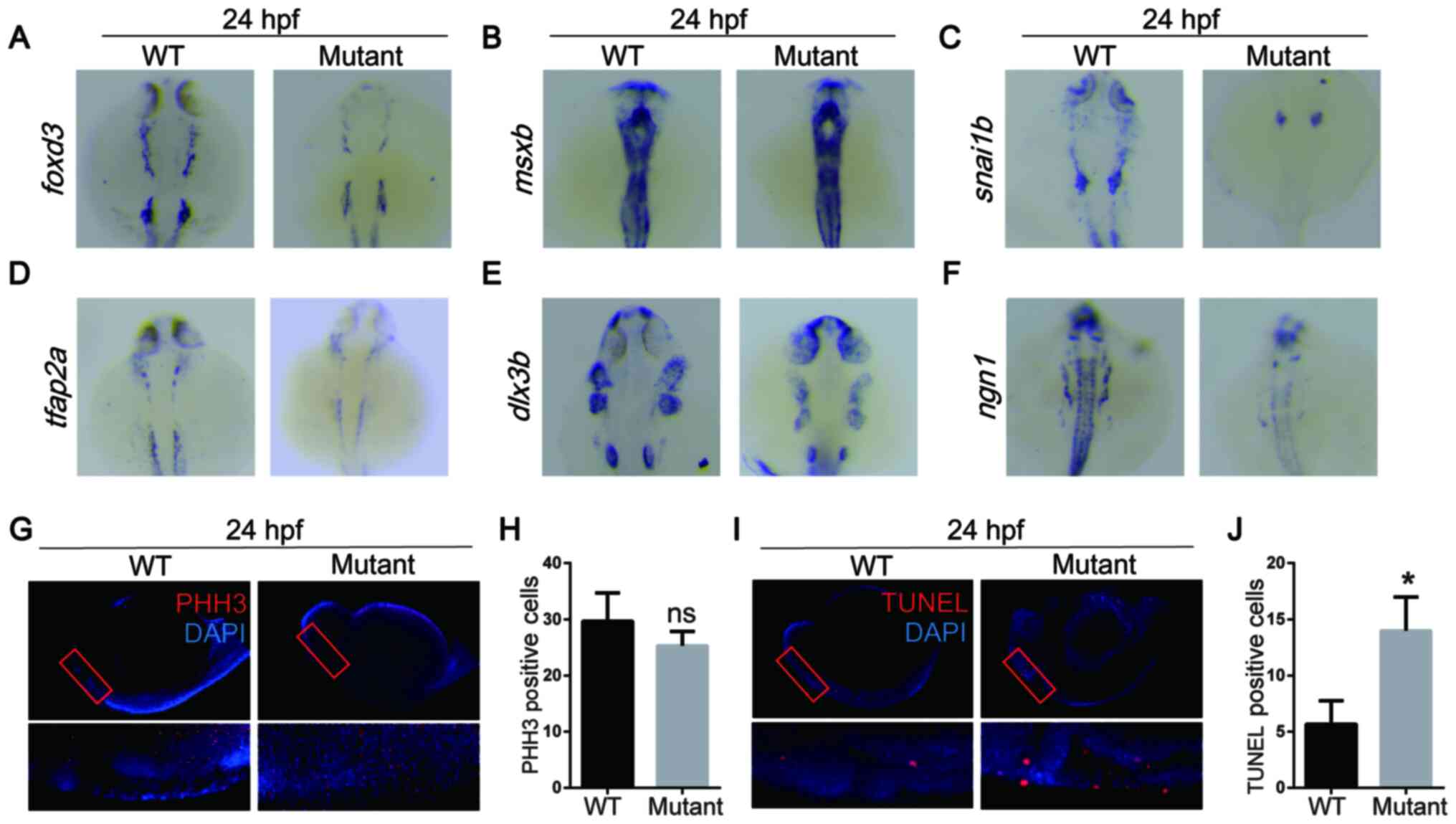 | Figure 2MicroRNA-1 knockout affects neural
crest cell apoptosis during the migration and differentiation
periods of embryonic development. In situ hybridization of
zebrafish at 24 hpf with (A) foxd3, (B) msxb, (C)
snai1b, (D) tfap2a, (E) dlx3b and (F)
ngn1 probes. (G) Whole mount immunostaining of zebrafish at
24 hpf with PHH3 (red) and DAPI (blue). (H) Quantitative analysis
of immunostaining. (I) TUNEL staining at 24 hpf with TUNEL (red)
and DAPI (blue). (J) Quantitative analysis of TUNEL staining. Data
are presented as the mean ± standard deviation (n=5).
*P<0.05 vs. WT. WT, wild-type; foxd3, forkhead box
D3; msxb, muscle segment homeobox-B; snai1b, snail homolog 1b;
tfap2a, transcription factor AP2A; dlx3b, distal-less 3; ngn1,
neurog1; PHH3, phospho-histone H3; hpf, hours post fertilization;
ns, not significant. |
To examine migration, the expression of
snai1b and tfap2a was also examined in the NC area of
the head. In situ hybridization indicated that snai1b
and tfap2a expression appeared to be decreased in the mutant
group at 24 hpf compared with that in the WT group (Fig. 2C and D). These results suggested that migration
was inhibited in the mutant zebrafish.
During embryonic development in zebrafish, the first
and second pharyngeal arch develop into the mandible and hyoid
arches (1). The results of the
present study indicated that the expression of dlx3b in
miR-1 mutant embryos appeared to be decreased compared with that in
the WT group (Fig. 2E). This was
consistent with the mutant zebrafish phenotype, namely lower
mandibular arch length and mandibular retrognathia, which was
observed in the current study. Knockdown of miR-1 appeared to
decrease in ngn1 expression compared with that in the WT
group (Fig. 2F). This evidence
collectively suggested that miR-1 may serve an important role
during the migration and differentiation periods of NCCs.
Cell proliferation, which was assessed by
immunofluorescence staining of PHH3, exhibited no differences
between the two groups (Fig. 2G and
H). An assessment of apoptosis in
zebrafish embryos at 24 hpf was performed using a whole-mount TUNEL
assay. The results demonstrated that miR-1-knockout embryos
exhibited an increased level apoptosis compared with that in the WT
embryos (Fig. 2I and J). These results demonstrated the
importance of miR-1 in regulating apoptosis during NC
development.
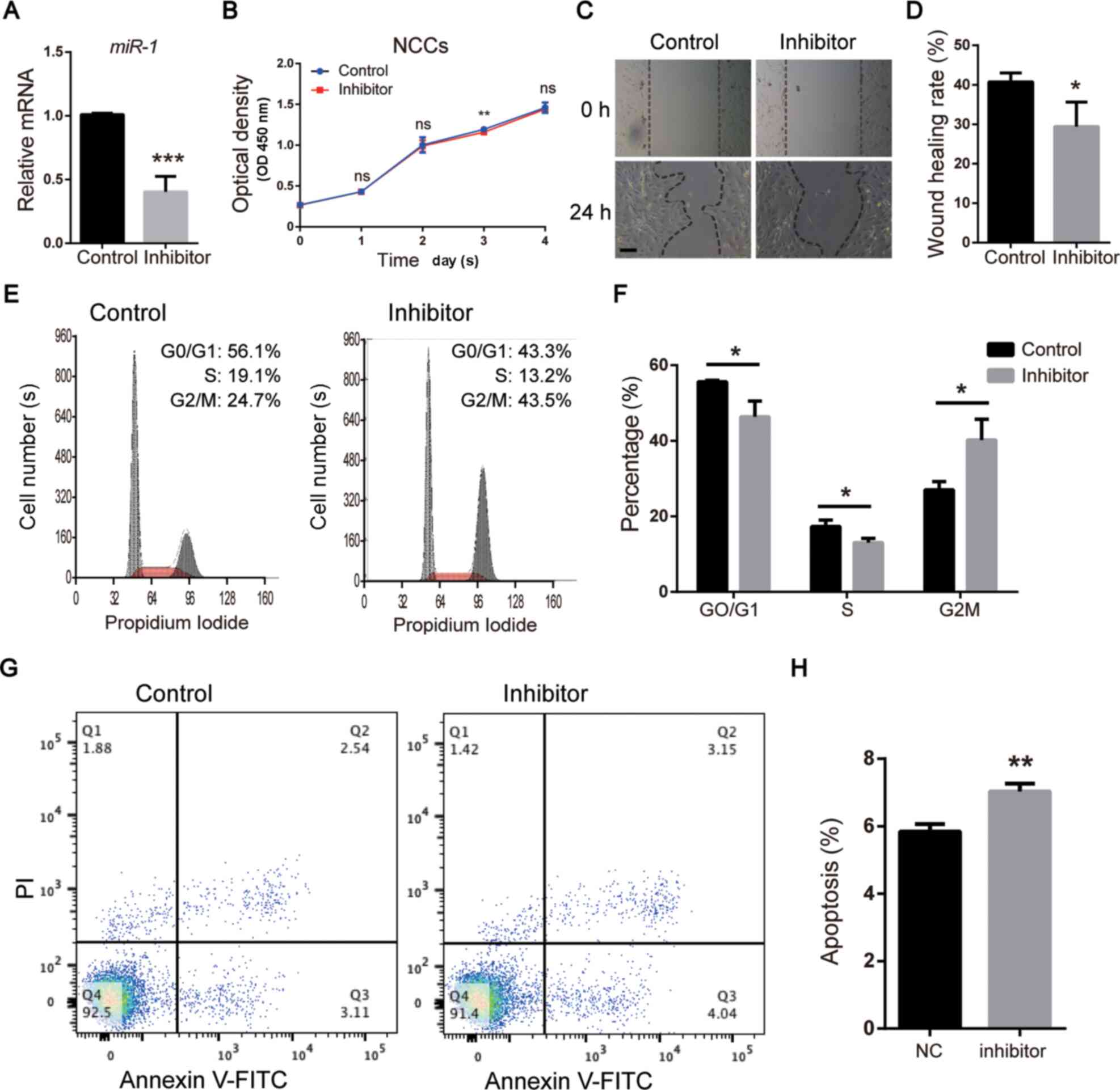
miR-1 regulates NCC functions in
vitro
To examine the function of miR-1 in vitro,
miR-1 was knocked down in NCCs isolated from mice using a miR-1
inhibitor (Fig. 3A). Following
inhibition of miR-1, the proliferative ability of NCCs was slightly
reduced compared with that of the control group at day 3, whereas
no significant differences were observed at day 1, 2 and 4
(Fig. 3B). In addition, a wound
healing assay indicated that the migratory rate of the miR-1
inhibitor group was decreased compared with the control group
(Fig. 3C and D).
Subsequently, cell cycle and apoptosis analysis was
performed via flow cytometry. The results of the cell cycle
analysis suggested that knockdown of miR-1 increased the number of
cells at the G2/M phase compared with that in the
control group, indicating that cell cycle was arrested at
G2/M phase. By contrast, the numbers of NCCs at the
G0/G1 and S phases were reduced compared with
those in the control group (Fig. 3E
and F). In addition, following
transfection with the miR-1 inhibitor, the number of early and late
apoptotic cells was increased compared with that in the control
group (Fig. 3G and H). These in vitro results
demonstrated that miR-1 may be crucial for apoptosis in NCCs and
were consistent with the in vivo observations.
miR-1 affects NCC apoptosis via the
mitochondrial apoptosis pathway and caspase-3
Early and late apoptosis in mouse NCCs was detected
via AO/EB staining. The results demonstrated that the number of
early and late apoptotic cells was increased in the miR-1 inhibitor
group compared with that in the control group (Fig. 4A and B). In addition, TUNEL staining
demonstrated that compared with the control group, an increased
number of apoptotic cells was observed three days after
transfection with miR-1 inhibitor (Fig.
4C and D).
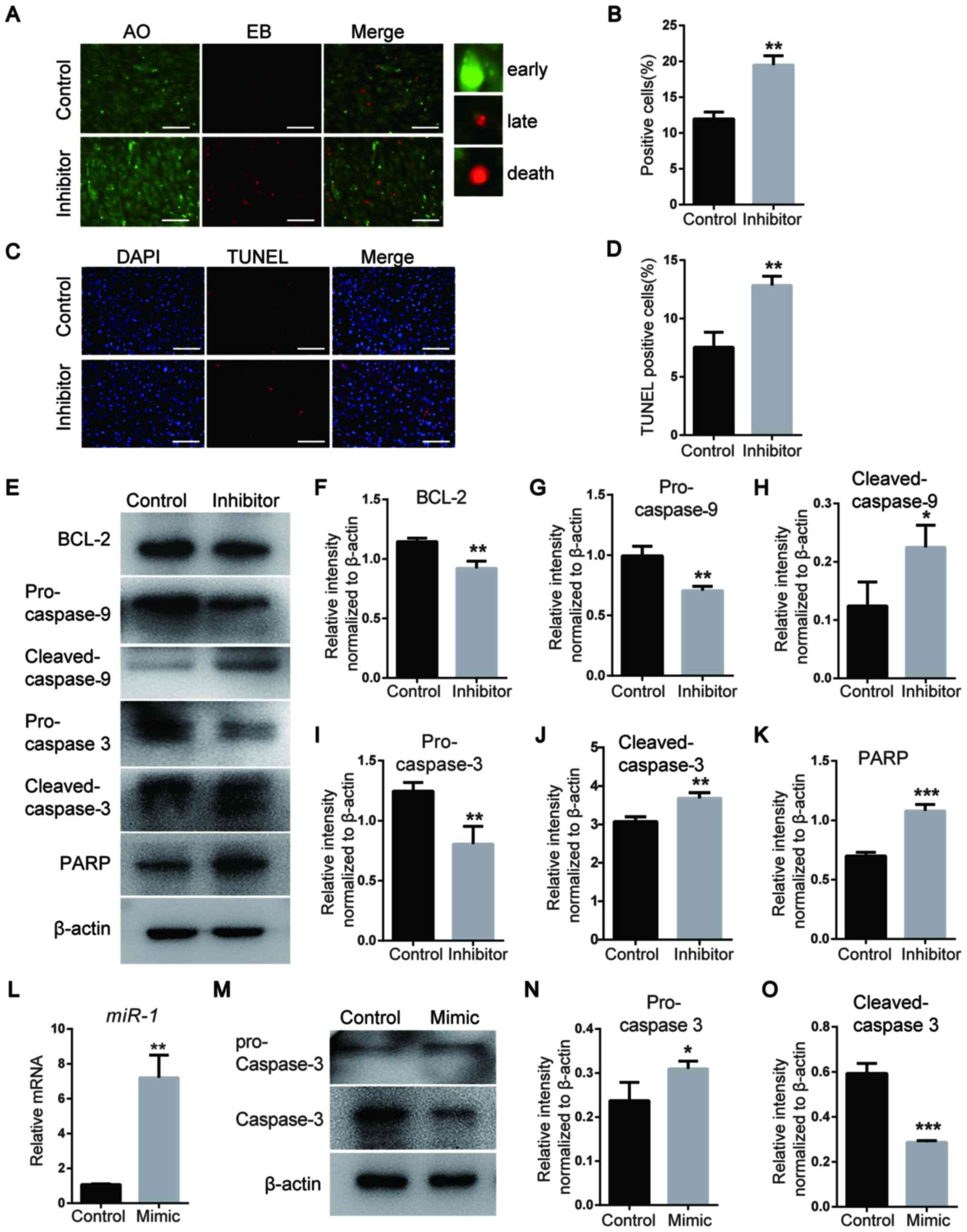 | Figure 4miR-1 affects the apoptosis of neural
crest cells via the mitochondrial apoptosis pathway and caspase-3.
(A) Detection of apoptosis on day 3 following miR-1 inhibitor
transfection using AO/EB staining. Scale bar, 100 µm. (B)
Quantitative analysis of AO/EB staining. (C) TUNEL staining of
NCCs. Scale bar, 100 µm. (D) Quantitative analysis of TUNEL
staining. (E) Protein levels of the mitochondrial apoptosis pathway
markers BCL-2, pro- and cleaved-caspase-3, pro- and
cleaved-caspase-9 and PARP detected via western blotting. (F-K)
Quantification of the protein levels of (F) BCL-2, (G)
pro-caspase-9, (H) cleaved-caspase-9, (I) pro-caspase-3, (J)
cleaved-caspase-3 and (K) PARP, which are presented in (E). (L)
miR-1 expression assessed by reverse transcription-quantitative
PCR. (M) Expression levels of pro- and cleaved-caspase-3 after
transfection with the miR-1 mimic. (N and O) Quantification of the
protein levels. Data are presented as the mean ± standard deviation
(n=3). *P<0.05, **P<0.01 and
***P<0.001 vs. Control. miR-1, microRNA-1; PARP, poly
ADP-ribose polymerase, AO, acridine orange; EB, ethidium
bromide. |
Alterations in the apoptotic pathway were examined
in mouse NCCs by detecting the expression levels of
apoptosis-associated proteins using western blotting (Fig. 4E-K). The expression levels of
cleaved-caspase-3 and -9 were increased in the miR-1 inhibitor
group compared with those in the control group (Fig. 4H and J), whereas the expression levels of
pro-caspase-3 were decreased in the miR-1 inhibitor group compared
with those in the control group (Fig.
4I). The expression levels of BCL-2, PARP and pro-caspase-9
were also examined. In the miR-1 inhibitor group, the expression
levels of PARP were upregulated, while those of BCL-2 and
pro-caspase-9 were decreased compared with the control group
(Fig. 4F, H and I).
Therefore, these results suggested that the mitochondrial apoptosis
pathway was activated following miR-1 inhibition. The increased
protein expression levels of cleaved caspase-3 in the miR-1
inhibitor group compared with the control group was consistent with
the increased apoptosis that was observed in the mutant zebrafish
compared with that in the WT group. To further analyze the
relationship between miR-1 and caspase-3, NCCs were transfected
with a miR-1 mimic (Fig. 4L). The
results indicated that the expression level of cleaved-caspase-3
was downregulated in cells transfected with miR-1 mimics compared
with that in the control group (Fig.
4M-O), suggesting that miR-1 inhibited the activation of
caspase-3 and affected the apoptosis of NCCs.
Collectively, these results suggested that miR-1
regulated the apoptosis of NCCs via the mitochondrial apoptosis
pathway.
Discussion
In a previous study, a latitudinal expression of
miR-1 was observed in WT zebrafish via in situ
hybridization, and it was demonstrated that the expression of miR-1
increased during embryonic development at 24 hpf (11).
In the present study, in situ hybridization
was used to detect the expression of miR-1 in WT zebrafish embryos.
The expression of miR-1 was demonstrated to be primarily localized
in the head, pharyngeal region and pectoral fin of zebrafish at 24
hpf. Previous findings have indicated that the injection of miR-1
morpholino oligomers in zebrafish resulted in impaired craniofacial
chondrogenesis, severe maxillofacial malformations and an abnormal
function of NCCs within 4 days post-fertilization. However, it was
not clear whether these defects were also affected by the injection
of the morpholino (11). Therefore,
a miR-1 knockout zebrafish model was generated in the present study
to elucidate the role of miR-1 in NC development.
NCCs have been reported to contribute to the
development of bones, cartilage, cranial ganglia and connective
tissues in the face and neck (1).
Previous studies have demonstrated that the abnormal expression of
genes associated with NC may result in craniofacial, bone and
cartilage dysplasia due to abnormal cell migration, proliferation
and differentiation (26-29).
For example, dlx3b has been associated with the differentiation of
the pharyngeal arch in zebrafish (30). Also, Ngn1 has been reported to
regulate the differentiation of the dorsal root ganglion in
zebrafish development (31). In the
current study, in situ hybridization evidence collectively
suggested that miR-1 serve an important role during the migration
and differentiation periods of NCCs.
miRNAs are a class of highly conserved, noncoding
RNA molecules with a length of 18-25 nucleotides. The mature
sequence of miR-1 is highly conserved among different vertebrates,
including zebrafish, mice and humans, which suggests that miR-1 may
exhibit similar functions among vertebrates (32). The role of miR-1 in regulating the
development of heart, cartilage and liver, among other organs, has
been reported (33-35).
In the present study, the following abnormal phenotypes were
observed in the miR-1-knockout model: Mandibular retraction,
shortening of the inferior dental arch and lingual arch, and
atypical neural crest-derived tissues, such as abnormal pericardial
edema and pigment cells.
Li et al (36) reported that miR-1 decreased
cardiomyocyte apoptosis via mediating the expression of
apoptosis-related genes in the heart. In the current study, TUNEL
staining revealed an increase in apoptosis of pharyngeal NCCs in
miR-1 mutant compared with WT zebrafish, which was consistent with
previous reports (36). By
contrast, PHH3 immunofluorescence staining did not demonstrate any
differences between the WT and mutant groups; additional time
points may be required to determine whether alterations in NCC
proliferation were caused by miR-1 knockout.
Caspase-3 has been reported to be a key protein in
the mitochondrial apoptosis pathway, and RARP is one of the
principal targets of caspase-3(37). Also, pro-caspase-9 requires to be
activated to subsequently cleave and activate caspase-3(38). Western blotting results suggested
that miR-1 may regulate the mitochondrial apoptosis pathway.
The biogenesis of miRNAs is a complex process. After
several processing steps (39),
mature single-stranded miRNAs have been indicated to enter the
miRNA-induced silencing complex, which silences the expression of
target genes primarily at the post-transcriptional level (40). The target genes to be silenced are
selected via base-pairing interactions between the miRNA and the
target mRNA that contains a partial or complete complementary
sequence, generally localized in 3'-untranslated region (41). The results of the current study
suggested that caspase-3 may be targeted by miR-1. However, the
regulatory pathways and mechanisms of this interaction require
additional studies.
In conclusion, the present study demonstrated an
important role of miR-1 in regulating the apoptosis of NCCs during
embryonic development via modulating the mitochondrial apoptosis
pathway. A normal expression of miR-1 was identified to be
essential for the development of NC derivatives, including the
pharyngeal cartilage, mandible and hyoid bone. A thorough
understanding of molecular pathway by which miR-1 regulates its
target genes may aid the prevention and treatment of
miR-1-associated developmental malformations.
Supplementary Material
Genotypic identification of mutant
zebrafish via PCR amplification and subsequent electrophoresis.
Each pair of two adjacent lanes (from left to right) represents the
PCR identification results of one specimen. The first lane
indicates the amplification product of the primer pair Mir1-1-11bp
wild-type F1/Mir1-1 R, and the second lane represents the
amplification product of the primer pair Mir1-1-11bp mutant
F1/Mir1-1 R. The results indicated that all specimens were
homozygous mutants. F, forward; R, reverse; Mir1, microRNA-1.
Genotype of microRNA-1 knockout mutant
in zebrafish.
Primers used in the identification of
genotype by PCR.
The sense and antisense probes used in
whole mount in situ hybridization.
Acknowledgements
Not applicable.
Funding
Funding: The current study was funded by the National Natural
Science Foundation of China (grant no. 81771029), the Natural
Science Fund for Colleges and Universities in Jiangsu Province,
China (grant no. 18KJA320004), the Southeast University-Nanjing
Medical University Cooperative Research Project (grant no.
JX218GSP20180705) and the Priority Academic Program Development of
Jiangsu Higher Education Institutions (grant no. 2014-037).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
NZ, WQ, DW, AGR and LY performed the experiments and
analyzed the data. NZ, YM, CM, ZX and JM designed the study. NZ,
AGR, ZX and JM wrote the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the School of Stomatology of Nanjing Medical
University (approval no. 1403049), and all procedures were
performed according to the guidelines of the Animal Care Committee
of Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mayor R and Theveneau E: The neural crest.
Development. 140:2247–2251. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Trainor PA: Specification and patterning
of neural crest cells during craniofacial development. Brain Behav
Evol. 66:266–280. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Keyte A and Hutson MR: The neural crest in
cardiac congenital anomalies. Differentiation. 84:25–40.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jones NC, Lynn ML, Gaudenz K, Sakai D,
Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al:
Prevention of the neurocristopathy Treacher Collins syndrome
through inhibition of p53 function. Nat Med. 14:125–133.
2008.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Etchevers HC, Amiel J and Lyonnet S:
Molecular bases of human neurocristopathies. Adv Exp Med Biol.
589:213–234. 2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chai Y and Maxson RE Jr: Recent advances
in craniofacial morphogenesis. Dev Dyn. 235:2353–2375.
2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ying SY and Lin SL: Current perspectives
in intronic micro RNAs (miRNAs). J Biomed Sci. 13:5–15.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Townley-Tilson WH, Callis TE and Wang D:
MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac
muscle development, function, and disease. Int J Biochem Cell Biol.
42:1252–1255. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li P, Wei X, Guan Y, Chen Q, Zhao T, Sun C
and Wei L: MicroRNA-1 regulates chondrocyte phenotype by repressing
histone deacetylase 4 during growth plate development. FASEB J.
28:3930–3941. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Samal E, Evangelista M, Galang G,
Srivastava D, Zhao Y and Vedantham V: Premature microRNA-1
expression causes hypoplasia of the cardiac ventricular conduction
system. Front Physiol. 10(235)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang D, Weng Y, Guo S, Qin W, Ni J, Yu L,
Zhang Y, Zhao Q, Ben J and Ma J: MicroRNA-1 regulates NCC migration
and differentiation by targeting sec63. Int J Biol Sci.
15:2538–2547. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Fishman MC: Genomics. Zebrafish - the
canonical vertebrate. Science. 294:1290–1291. 2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu QY, Wu ZL, Lv WJ, Yan YC and Li YP:
Developmental expression of cyclin H and Cdk7 in zebrafish: The
essential role of cyclin H during early embryo development. Cell
Res. 17:163–173. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Schulte-Merker S, Ho RK, Herrmann BG and
Nüsslein-Volhard C: The protein product of the zebrafish homologue
of the mouse T gene is expressed in nuclei of the germ ring and the
notochord of the early embryo. Development. 116:1021–1032.
1992.PubMed/NCBI
|
|
15
|
de Peralta MS, Mouguelar VS, Sdrigotti MA,
Ishiy FA, Fanganiello RD, Passos-Bueno MR, Coux G and Calcaterra
NB: Cnbp ameliorates Treacher Collins syndrome craniofacial
anomalies through a pathway that involves redox-responsive genes.
Cell Death Dis. 7(e2397)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ning G, Liu X, Dai M, Meng A and Wang Q:
MicroRNA-92a upholds Bmp signaling by targeting noggin3 during
pharyngeal cartilage formation. Dev Cell. 24:283–295.
2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Babb-Clendenon S, Shen YC, Liu Q, Turner
KE, Mills MS, Cook GW, Miller CA, Gattone VH II, Barald KF and
Marrs JA: Cadherin-2 participates in the morphogenesis of the
zebrafish inner ear. J Cell Sci. 119:5169–5177. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo S, Zhang Y, Zhou T, Wang D, Weng Y,
Chen Q, Ma J, Li YP and Wang L: GATA4 as a novel regulator involved
in the development of the neural crest and craniofacial skeleton
via Barx1. Cell Death Differ. 25:1996–2009. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guo S, Zhang Y, Zhou T, Wang D, Weng Y,
Wang L and Ma J: Role of GATA binding protein 4 (GATA4) in the
regulation of tooth development via GNAI3. Sci Rep.
7(1534)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lu M, Guo S, Hong F, Zhang Y, Yuan L, Ma C
and Ma J: Pax2 is essential for proliferation and osteogenic
differentiation of mouse mesenchymal stem cells via Runx2. Exp Cell
Res. 371:342–352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ono W, Sakagami N, Nishimori S, Ono N and
Kronenberg HM: Parathyroid hormone receptor signalling in
osterix-expressing mesenchymal progenitors is essential for tooth
root formation. Nat Commun. 7(11277)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Barrallo-Gimeno A, Holzschuh J, Driever W
and Knapik EW: Neural crest survival and differentiation in
zebrafish depends on mont blanc/tfap2a gene function. Development.
131:1463–1477. 2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nelms BL, Pfaltzgraff ER and Labosky PA:
Functional interaction between Foxd3 and Pax3 in cardiac neural
crest development. Genesis. 49:10–23. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Medeiros DM and Crump JG: New perspectives
on pharyngeal dorsoventral patterning in development and evolution
of the vertebrate jaw. Dev Biol. 371:121–135. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lake JI, Avetisyan M, Zimmermann AG and
Heuckeroth RO: Neural crest requires Impdh2 for development of the
enteric nervous system, great vessels, and craniofacial skeleton.
Dev Biol. 409:152–165. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lei R, Zhang K, Wei Y, Chen M, Weinstein
LS, Hong Y, Zhu M and Li H and Li H: G-protein α-subunit Gsα is
required for craniofacial morphogenesis. PLoS One.
11(e0147535)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Shao R, Liu J, Yan G, Zhang J, Han Y, Guo
J, Xu Z, Yuan Z, Liu J, Malumbres M, et al: Cdh1 regulates
craniofacial development via APC-dependent ubiquitination and
activation of Goosecoid. Cell Res. 26:699–712. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tu CT, Yang TC, Huang HY and Tsai HJ:
Zebrafish arl6ip1 is required for neural crest development during
embryogenesis. PLoS One. 7(e32899)2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Alexander C, Piloto S, Le Pabic P and
Schilling TF: Wnt signaling interacts with bmp and edn1 to regulate
dorsal-ventral patterning and growth of the craniofacial skeleton.
PLoS Genet. 10(e1004479)2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cornell RA and Eisen JS: Delta/Notch
signaling promotes formation of zebrafish neural crest by
repressing Neurogenin 1 function. Development. 129:2639–2648.
2002.PubMed/NCBI
|
|
32
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297.
2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Fleissner F, Jazbutyte V, Fiedler J, Gupta
SK, Yin X, Xu Q, Galuppo P, Kneitz S, Mayr M, Ertl G, et al: Short
communication: Asymmetric dimethylarginine impairs angiogenic
progenitor cell function in patients with coronary artery disease
through a microRNA-21-dependent mechanism. Circ Res. 107:138–143.
2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Karp X and Ambros V: Developmental
biology. Encountering microRNAs in cell fate signaling. Science.
310:1288–1289. 2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu P, Guo M and Hay BA: MicroRNAs and the
regulation of cell death. Trends Genet. 20:617–624. 2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li W, Liu M, Zhao C, Chen C, Kong Q, Cai Z
and Li D: miR-1/133 attenuates cardiomyocyte apoptosis and
electrical remodeling in mice with viral myocarditis. Cardiol J.
27:285–294. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Perchellet EM, Wang Y, Weber RL,
Sperfslage BJ, Lou K, Crossland J, Hua DH and Perchellet JP:
Synthetic 1,4-anthracenedione analogs induce cytochrome c release,
caspase-9, -3, and -8 activities, poly(ADP-ribose) polymerase-1
cleavage and internucleosomal DNA fragmentation in HL-60 cells by a
mechanism which involves caspase-2 activation but not Fas
signaling. Biochem Pharmacol. 67:523–537. 2004.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J,
Lee J, Provost P, Rådmark O, Kim S, et al: The nuclear RNase III
Drosha initiates microRNA processing. Nature. 425:415–419.
2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lin SL and Ying SY: Gene silencing in
vitro and in vivo using intronic microRNAs. Methods Mol Biol.
1733:107–126. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Huntzinger E and Izaurralde E: Gene
silencing by microRNAs: Contributions of translational repression
and mRNA decay. Nat Rev Genet. 12:99–110. 2011.PubMed/NCBI View Article : Google Scholar
|