Introduction
Danon disease was first described as a ‘lysosomal
glycogen storage disease with normal acid maltase’ by Danon et
al (1) in 1981. They reported
on two young, unrelated males with mental retardation,
cardiomyopathy and proximal myopathy, who exhibited apparently
generalized glycogenosis with no demonstrable enzyme defect. In
2000, Nishino et al (2)
concluded that a primary deficiency of lysosome-associated membrane
protein-2 (LAMP2) led to Danon disease. LAMP2 is a lysosomal
structural protein that is essential to the autophagy process.
LAMP2-deficient mice exhibit extensive accumulation of autophagic
vacuoles in several tissues (3),
and hepatocytes that lack LAMP2 exhibit similar accumulation of
autophagosomes (4). Therefore,
biopsies frequently contribute to the diagnosis of Danon disease;
however, they are neither necessary nor diagnostic. Similar
vacuoles may also be encountered in other metabolic diseases, such
as protein kinase AMP-activated non-catalytic subunit γ2 (PRKAG2)
syndrome and Fabry disease (5).
Danon disease is clinically characterized by the
triad of cardiomyopathy, skeletal myopathy and intellectual
impairment (1). As both skeletal
myopathy and intellectual impairment are usually mild, it is the
extent and severity of cardiomyopathy that predominates the
clinical progress and determines the outcome. Electric conduction
abnormalities are also common, followed by preexcitation with
Wolff-Parkinson-White (WPW) syndrome and arrhythmias (6). Other symptoms that are less prevalent
include hepatic disease (1,3), retinal disease (7,8) and
pulmonary disease (3).
Danon disease has an aggressive course; however, it
may be unnoticed or misdiagnosed in the early stages. Boucek et
al (6) reported that the
respective mean age at first symptoms, cardiac transplant and death
was 11.7, 20.8 and 20.1 years in males, and 26.8, 32.3, and 40.2
years in females. The most recent systematic review of Danon
disease revealed that 34.9% of patients experienced death or
received a heart transplant or ventricular assist devices (9). Therefore, patients with Danon disease
must be distinguished from patients with myocardial hypertrophy,
such as PRKAG2 syndrome and Fabry disease, which are glycogen
storage diseases that also clinically lead to cardiac hypertrophy
and electrophysiologic abnormalities (10), and hypertrophic cardiomyopathy (HCM)
with sarcomere protein mutations.
The objective of the present study was to identify
the clinical and molecular characteristics of Danon disease and to
compare the clinical, electrocardiographic (ECG) and
echocardiographic features of patients with Danon disease with
those of patients with the PRKAG2 syndrome, Fabry disease and
sarcomere protein mutation.
Materials and methods
Study population
An exon and boarding intron analysis of 96 cardio
disease-associated genes (Table
SI) was performed in 770 patients with HCM using
second-generation sequencing (11)
at the HCM clinic in the Department of Ultrasound, Xijing Hospital
(Xi'an, China) between May 2012 and February 2020. The identified
mutations were confirmed in family members of the probands and 300
healthy controls. A total of seven patients with LAMP2 mutation and
their families were included in the present study. Furthermore, 5
patients with the PRKAG2 mutations, 8 patients with galactosidase A
mutations and 12 patients with sarcomere protein mutations were
among the 770 patients with HCM, and they were compared with the
patients with Danon disease. All participants who were evaluated
provided written informed consent. All investigations were
conducted in compliance with the principles of the Declaration of
Helsinki and were approved by the ethics committee of Xijing
Hospital, Fourth Military Medical University (Xi'an, China). For
participants aged <16 years, written informed consent was
obtained from the parents.
All measurements were made to coincide with the
guidelines of the European Society of Cardiology (12). HCM was defined by a maximal left
ventricular (LV) wall thickness (MLVWT) of ≥15 mm in adults and ≥13
mm in first-degree relatives, excluding those that may be explained
solely by loading conditions or the patient being an athlete. If
the patient was aged <18 years, the diagnosis of HCM was made
based on MLVWT >2 SD than the predicted mean (z-score ≥2, where
a z-score is defined as the number of standard deviations from the
population mean). Patients with hypertension, coronary artery
disease, aortic coarctation, congenital or valvular heart disease,
metabolic disorder or athlete's heart were excluded.
ECG
Standard 12-lead ECG was recorded prior to or after
the echocardiographic examination. Ventricular preexcitation was
characterized by short PR intervals (PR <120 msec) and/or
initial QRS slurring (delta wave) during the sinus rhythm. LV
voltage was reported as the S wave in V1 plus the
maximal R wave in V5 or V6 (SV1 +
RV5 or RV6), according to the Sokolow-Lyon
criteria (13).
Transthoracic echocardiography
(TTE)
TTE was performed using the Philips iE33 ultrasound
system (Philips Medical Systems) and the EPIQ 7C Ultrasound System
(Philips Medical Systems). The patients were placed in the left
lateral position as if undergoing a routine echocardiographic
study. The ECG was recorded simultaneously. Hypertrophied LV
segments were identified from two-dimensional echocardiographic
images according to the American Heart Association 17-segment model
(14). Asymmetric hypertrophy
indicated that the ratio between the interventricular septum
thickness (IVS) and LV posterior wall thickness (LVPW) was >1.3.
MLVWT was defined as the greatest dimension at any site within the
LV myocardium. All echocardiographic measurements, including left
atrial (LA) dimensions, LV ejection fraction (LVEF) and LV outflow
tract (LVOT) gradient, were determined according to guidelines from
the American Society of Echocardiography and the European
Association of Cardiovascular Imaging (14). Furthermore, the early diastolic peak
velocities of mitral inflow (E) and the late diastolic peak
velocities of mitral inflow (A) and mitral annulus (e') were
measured. Then, the E/e' ratio and the E/A ratio were calculated to
evaluate LV diastolic function (15).
Cardiac magnetic resonance (CMR)
CMR examinations were performed using a 1.5-T CMR
imaging system (Magnetom Aera; Siemens AG). All images were
acquired with ECG-gating and during repeated breath-hold
conditions. The presence and amount of scarring were assessed using
phase-sensitive late gadolinium enhancement CMR (LGE-CMR).
T1-weighted inversion-recovery imaging was performed to assess LGE
10 min after the administration of 0.2 mmol/kg contrast medium
(Dotarem; Guerbet).
Genetic analysis
A genetic analysis was performed as previously
described (11). In brief,
peripheral blood leucocytes were extracted from patients. Primers
were designed to amplify the coding exons of 96 cardio
disease-associated genes. The identified mutation was further
confirmed by bidirectional Sanger sequencing among the remaining
family members and 300 healthy unrelated individuals with normal
ECG and echocardiographic findings and without any family history
of cardiovascular disease.
Statistical analysis
Statistical analysis was performed with SPSS version
20 (IBM Corp.). Categorical variables were expressed as n (%) and
Fisher's exact test was used to compare these variables between
groups. Continuous variables were expressed as the mean ± SD and
differences between groups were compared using Student's t-test or
analysis of variance. For multiple comparisons, Tamhane T2 or
Bonferroni's post hoc test were performed. P≤0.05 was considered to
indicate a statistically significant difference.
Results
Clinical presentation
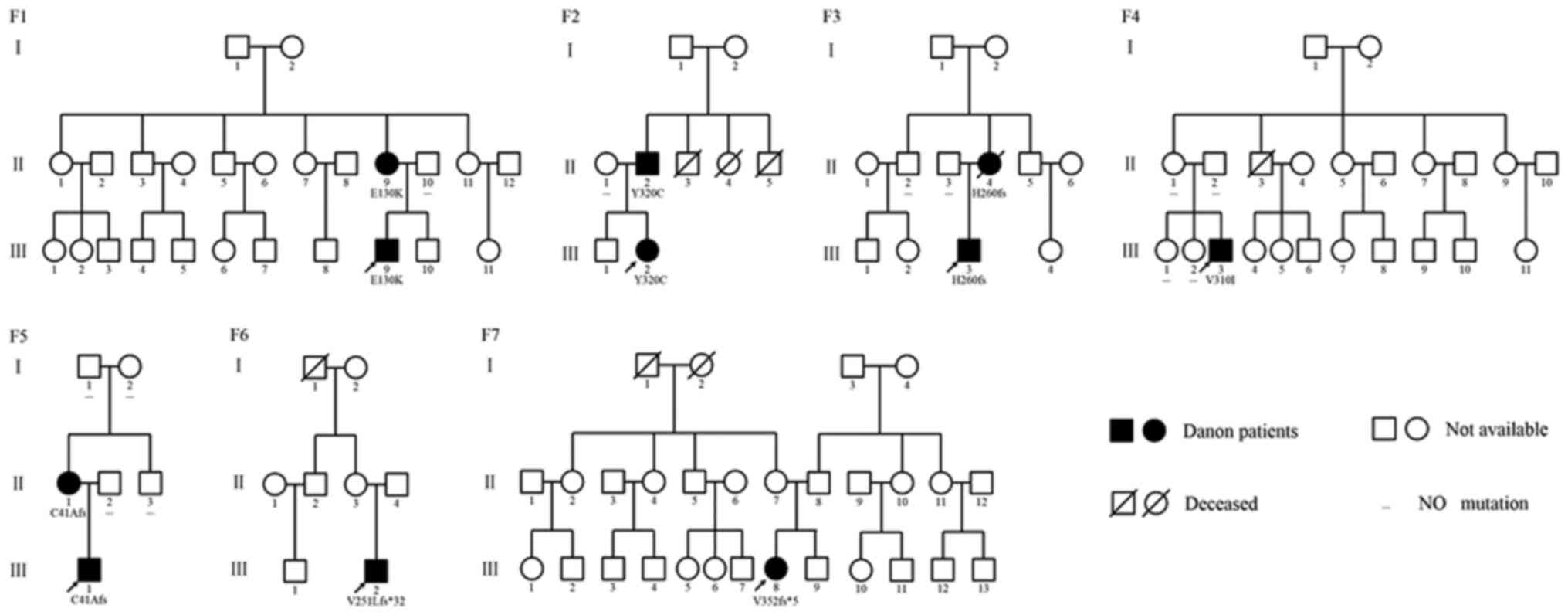
Among the 770 patients with HCM, seven Danon
patients were identified (0.9%) and 5 of them were male (71%;
Table I and Fig. 1). A total of three patients were
diagnosed at an early age (6 months, 1 and 3 years of age,
respectively). Of the patients with Danon disease, 2 patients were
asymptomatic and 4 had variable clinical presentations, including
chest pain, chest tightness, breathlessness and fatigue. The
proband (III-1) of family F5 was diagnosed incidentally when he was
in the hospital to cure a head hemangioma. Furthermore, one patient
had poor cardiac function and was classified as New York Heart
Association Class III (16) and the
cardiac function of other 6 patients were essentially normal.
 | Table IClinical, demographic and genetic
data of patients with Danon disease. |
Table I
Clinical, demographic and genetic
data of patients with Danon disease.
| Item | Family F1
III-9 | Family F2
III-2 | Family F3
III-3 | Family F4
III-3 | Family F5
III-1 | Family F6
III-2 | Family F7
III-8 |
|---|
| Sex | Male | Female | Male | Male | Male | Male | Female |
| Age at
diagnosis | 11 years | 3 years | 12 years | 20 years | 6 months | 1 year | 23 years |
| History of sudden
deathc | - | + | - | - | - | - | - |
| History of
syncoped | + | - | - | - | - | - | - |
| SBP/DBP (mmHg) | 86/54 | a | a | 134/62 | a | a | 110/71 |
| Clinical
presentation | Chest
tightness | Tachypnea | Chest pain,
troponin | None | Angeioma | None | Fatigue, chest
tightness, breathlessness |
| NYHA class | I | I | I | I | I | I | III |
| Arrhythmia | Normal | Sinus
tachycardia | Normal | Second-degree AV
block and WPW | WPW | Normal | Premature
ventricular contraction |
| SV1 +
RV5 or RV6 (mV) | 4 | 2.1 | 10.4 | 6.3 | 9.3 | 7 | 1.20 |
| RV1 (mV) | 1.2 | 0.97 | 2.1 | 0.31 | 3.78 | 5.2 | 0.1 |
| Heart rate
(bpm) | 79 | 117 | 67 | 45 | 108 | 97 | 86 |
| LA (mm) | 22 | 24 | 30 | 43 | 13 | 18 | 40 |
| MLVWT (mm) | 14 | 17 | 25 | 18 | 16 | 15 | 8 |
| LVEF (%) | 67 | 86 | 67 | 40 | 61 | 76 | 20.6 |
| E/e' ratio | a | a | 15.4 | 39.3 | 16.3 | 20.9 | 41.3 |
| E/A ratio | >1 | >1 | >1 | >2 | >1 | >1 | >2 |
| SAM | - | + | - | - | + | - | - |
| LVOT-PG (mmHg) | 4 | 90 | 6 | 27 | 7 | 14 | 1 |
| Skeletal
myopathy | b | b | b | Yes | b | b | b |
| Ocular
manifestation | b | b | No | b | b | b | b |
| Cognitive
impairment | No | b | No | No | b | b | b |
| Genetic
analysis | c.388G>A
(p.E130K) | c.959A>G
(p.Y320C) | c.779-782del
(p.H260fs) | c.928G>A
(p.V310I) | c.121delT
(p.C41Afs) | c.750delA
(p.V251Lfs*32) | c.1054delG
(p.V352Ffs*5) |
| Treatment with
blocker | None | Metoprolol | Bisoprolol | None | Metoprolol | None | Bisoprolol |
| Pacemaker
implantation | None | None | None | CRT-P | None | None | None |
The surface 12-lead ECGs were abnormal with a high
voltage (average of SV1 + RV5 or
RV6 = 5.76 mV; Table I).
Furthermore, two patients had a ventricular preexcitation pattern
(WPW pattern) with a short PR interval and one patient had a
second-degree atrial ventricular block. Furthermore, four patients
presented with a fragmented QRS complex (fQRS) on their surface
ECG.
All patients had an HCM phenotype on presentation
with a mean MLVWT of 17 mm (range, 14-24 mm; Table I). Furthermore, one patient had mild
skeletal myopathy. A total of three patients underwent cognitive
assessment and no cognitive impairment was determined.
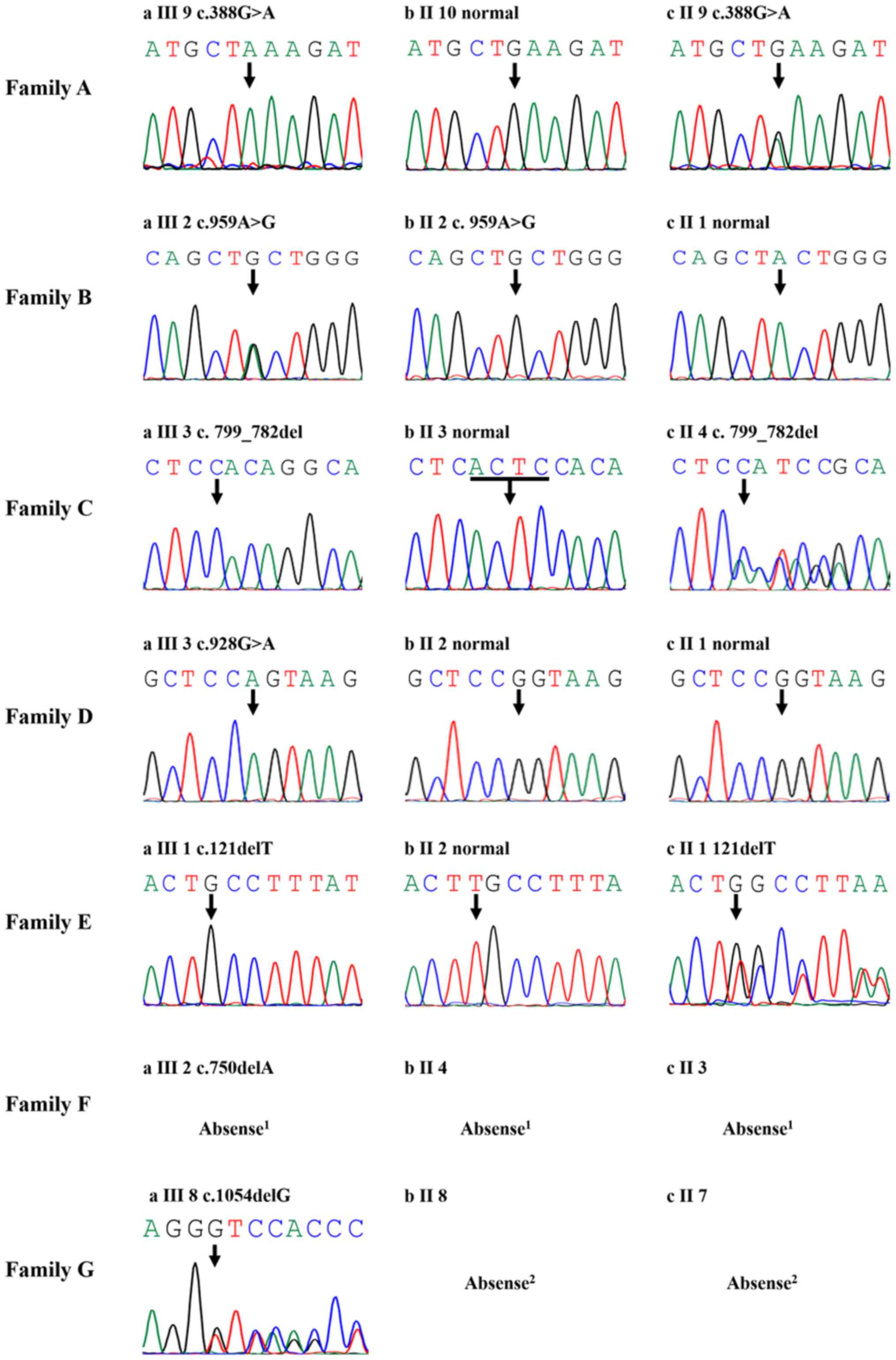
Genetic analysis
Each of the seven patients was identified with one
mutation in the LAMP2 gene (Table I
and Fig. 2). Among the mutations,
five were novel mutations, one mutation (c.928G>A) had
previously been reported (17) and
one mutation had been reported as a pathogenic mutation previously
ClinVar (Aug 09, 2013; https://www.ncbi.nlm.nih.gov/clinvar/variation/179062/)
but had not been reported in individuals or large population
studies. In total, three mutations were point mutations and four
were frameshift mutations. Regarding their occurrence in the
pedigrees, four mutations were detected in one of their parents,
one mutation was a de novo mutation and two mutations were
not confirmed in their pedigree because their parents were not
available.
Comparison with other patients with
HCM
Compared with 5 patients with the PRKAG2 mutation
(Table II), patients with Danon
disease presented at an earlier age (10±9 years vs. 34±10 years,
P=0.001), had a smaller LA size (27±11 mm vs. 41±6 mm, P=0.033), a
thinner MLVWT (16±5 mm vs. 35±6 mm, P<0.001) and a lower
probability of pacemaker implantation (P=0.010). Compared with 8
patients with Fabry disease (Table
II), patients with Danon disease also presented at an earlier
age (10±9 years vs. 42±8 years, P<0.001), had a smaller LA size
(27±11 mm vs. 39±6 mm, P=0.036) and a thinner IVS (16±5 mm vs. 24±6
mm, P=0.036).
| Table IIClinical characteristics of patients
with Danon disease and PRKAG2. |
Table II
Clinical characteristics of patients
with Danon disease and PRKAG2.
| Item | Danon disease
(n=7) | PRKAG2 disease
(n=5) | P-value | Fabry disease
(n=8) | P-value |
|---|
| Age at diagnosis
(years) | 10±9 | 34±10 | 0.001 | 42±8 | <0.001 |
| Male sex (%) | 5 (71.4) | 5(100) | 0.470 | 7 (87.5) | 0.569 |
| History of SCD
(%) | 1 (14.3) | 1(20) | 1.000 | 1 (12.5) | 1.000 |
| History of syncope
(%) | 1 (14.3) | 4(80) | 0.072 | 2 (25.0) | 1.000 |
| Pacemaker
implantation (%) | 0 (0) | 4(80) | 0.010 | 1 (12.5) | 1.000 |
| LA size (mm) | 27±11 | 41±6 | 0.033 | 39±6 | 0.036 |
| MLVWT (mm) | 16±5 | 35±6 | <0.001 | 24±6 | 0.036 |
| LVEF (%) | 60±22 | 48±13 | 0.605 | 63±7 | 0.983 |
| LVOT-PG (mmHg) | 22±30 | 4±2 | 0.756 | 41±65 | 0.991 |
| LVEDD (mm) | 40±15 | 47±11 | 0.844 | 21±4 | 1.000 |
| SAM (mm) | 2 (28.6) | 1(20) | 1.000 | 4 (50.0) | 0.592 |
After excluding the 3 youngest patients with Danon
disease, the 4 remaining patients (Family F1 III-9, Family F3
III-3, Family F4 III-3, Family F7 III-8) were compared with sex-
and age-matched HCM patients with sarcomere-protein mutations by
one-to-three matching (Table
III). The 4 Danon patients exhibited a lower LVOT gradient
(12±12 mmHg vs. 47±35 mmHg, P=0.009) and worse diastolic function
(E/e': 32±14 vs. 16±7, P=0.013). Danon patients presented with
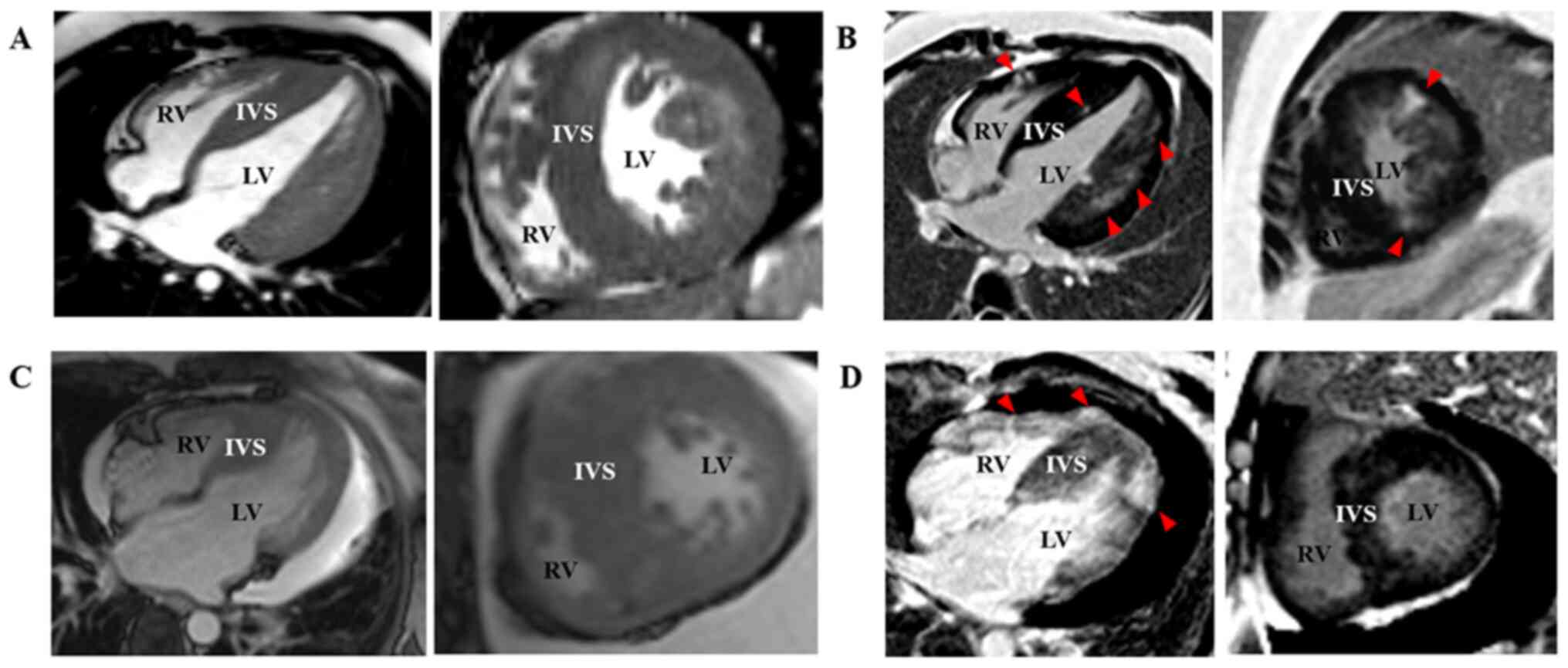
concentric LV hypertrophy on MRI and late gadolinium enhancement
revealed diffused abnormal enhancement (Fig. 3A and B). HCM patients with sarcomere-protein
mutations presented with asymmetric hypertrophy on MRI and late
gadolinium enhancement indicated spotted abnormal enhancement
(Fig. 3C and D).
| Table IIIClinical characteristics of patients
with Danon disease and patients with sarcomere-protein
mutations. |
Table III
Clinical characteristics of patients
with Danon disease and patients with sarcomere-protein
mutations.
| Item | Danon
(n=4)a | Sarcomere-protein
mutations (n=12) | P-value |
|---|
| Male sex | 3(75) | 9(75) | 1.000 |
| Age at diagnosis
(years) | 17±6 | 17±5 | 0.935 |
| History of
syncope | 1(25) | 4 (33.3) | 1.000 |
| Heart rate
(bpm) | 69±18 | 74±9 | 0.443 |
| QRS duration
(msec) | 116±36 | 96±11 | 0.353 |
| PR interval
(msec) | 149±37 | 132±25 | 0.301 |
| SV1 +
RV5 or RV6 (mV) | 5.48±3.88 | 4.25±1.32 | 0.577 |
| LA size (mm) | 34±10 | 32±7 | 0.701 |
| LVEF (%) | 49±23 | 65±5 | 0.250 |
| E/e' ratio | 32±14 | 16±7 | 0.013 |
| LVOT-PG (mmHg) | 12±12 | 47±35 | 0.009 |
| LVEDD (mm) | 50±13 | 38±3 | 0.194 |
| IVS (mm) | 16±7 | 25±8 | 0.063 |
| LVPW (mm) | 14±6 | 9±2 | 0.203 |
| SAM | 0 (0) | 8 (66.7) | 0.077 |
Case report
The proband (III-3) of family F3 came to our HCM
center (Xijing Hospital, Xi'an, China) at the age of 12 years (May,
2014) with complaints of chest tightness and pectoralgia during
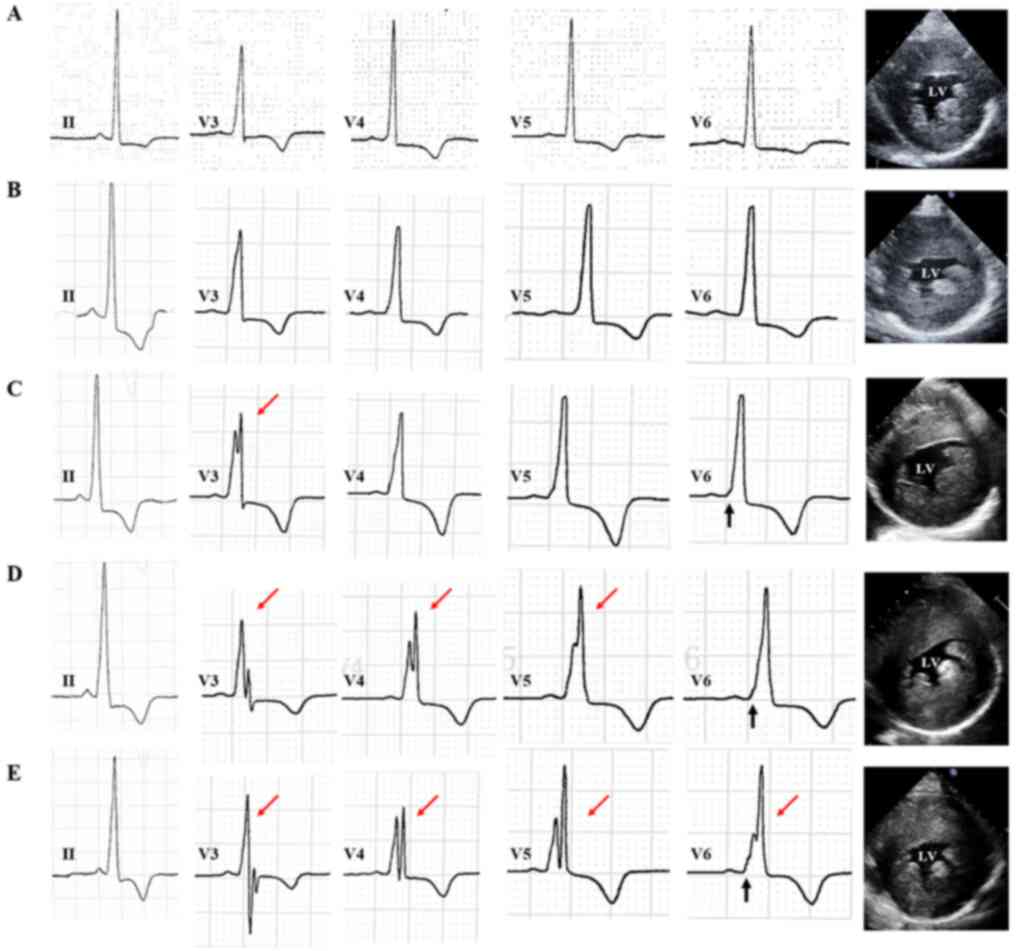
exercise along with elevated troponin (Trop). His ECG exhibited
sinus irregularity with a high voltage (RV5 + SV1=12.2 mV; Fig. 4A). Echocardiography indicated a
particularly increased IVS (25 mm) and LVPW (20 mm; Fig. 4A), preserved LVEF (65%), and normal
LVOT gradient (6 mmHg) and diastolic function (E/e'=15.4). The
proband's laboratory findings included myocardial damage [troponin
T (TropT), 15.93 ng/ml; myoglobin (Mb), 366.9 ng/ml; creatine
kinase isoenzyme MB (CK-MB), 7.9 ng/ml] and hepatic lesions
[alanine aminotransferase (ALT), 246 IU/; aspartate
aminotransferase (AST), 241 IU/l; alkaline phosphatase (ALP), 249
IU/l]. A further liver biopsy revealed mild fatty change and
lipofuscin in certain hepatic cells. The patient was advised to
continue hospitalization for medical treatment and was administered
irbesartan tablets, bisoprolol, polyene phosphatidylcholine
capsules and risuvastatin calcium tablets. Magnetic resonance
imaging assessment confirmed massive LV hypertrophy [LV anterior
wall (LVAW), 30 mm; LV lateral wall, 34 mm; IVS, 18-20 mm; Fig. 3A]. Late gadolinium enhancement
revealed focal changes in the thickened subendocardial IVS and
diffused abnormal enhancement in the subendocardial anterior and
lateral wall (Fig. 3B), which
suggested metabolic disease.
From July 2015 to Oct. 2017, the patient's ECG
indicated gradually shortening PR intervals (from 134 to 104 msec)
and gradually wider QRS durations (from 100 to 144 msec) with delta
waves resembling the WPW pattern. The ECG also exhibited fQRS in
the V3 lead and in additional limb leads (V3-V5; Fig. 4B-D). Echocardiography exhibited an
increased thickness of the basal IVS (from 14 to 37 mm) and basal
LVAW (from 16 to 39 mm; Fig. 4B-D),
damaged diastolic function (E/e' from 15.4 to 21.0) and preserved
LVEF. The proband was prescribed valsartan, metoprolol,
hydrochlorothiazide and coenzyme Q10.
In Jan. 2018, the patient had palpitations and chest
tightness after eating rich food or overeating. ECG indicated
intraventricular block with fQRS in limb leads (V3-V6) and chest
leads (I, aVL; Fig. 4E). The
thickness of basal IVS had increased to 47 mm (Fig. 4E). Echocardiography revealed still
preserved LVEF (65%) but damaged E/e' (26.0), along with LVOT
obstruction (LVOTO) with a gradient of 20 mmHg. The patient's
laboratory findings also indicated increased levels of
transaminases, ALP, TropT, Mb and CK-MB (ALT, 230 IU/l; AST, 288
IU/l; ALP, 261 IU/l; TropT, 10.283 ng/ml; Mb, 279.80 ng/ml; CK-MB,
11.700 ng/ml). The patient was additionally administered with
carvedilol, nicorandil and silibinin.
Discussion
Danon disease is a rare clinical syndrome and its
true prevalence remains to be determined. In HCM patients, the
prevalence ranges from 1 to 6% (18-21).
In individuals with both unexplained LV hypertrophy (LVH) and
preexcitation, the prevalence increases to 17-30% (20,22).
The present study reported on 7 patients with a LAMP2 gene mutation
identified among 770 HCM patients (0.9%). The observed prevalence
was essentially in agreement with that reported by previous
studies, but there appeared to be a downward trend. Most of the
patients in the present study were under 18 years of age and
children may be more likely to visit a hospital specializing in
children's diseases rather than a general hospital such as ours.
This may partly contribute to the slightly reduced prevalence.
D'souza et al (23) reported that the average age of
symptom onset of frameshift and missense mutations in male patients
was 12.1±8.4 and 47.6±19.1 years, respectively, and the average age
of symptom onset of frameshift mutations in female patients was
21.6±11.9 years (no record for female patients with missense
mutation). In the present study, however, one male patient with a
missense mutation presented with chest tightness at 11 years of
age, which was much earlier than the average age of symptom onset
reported by the above study of 47.6 years. Furthermore, two
asymptomatic patients were coincidentally identified to have a
frameshift mutation prior to their first birthday. These
differences may be partly explained by screening at an earlier age.
However, patients with more severe pathogenic mutations may exhibit
symptoms at a younger age. It is noteworthy that genetic tests and
analyses should be provided to identify Danon patients at an
earlier age.
Danon disease is an X-linked dominant disorder with
a clinical profile that differs by gender. Boucek et al
(6) reported that the mean age of
first symptoms in males was 12.1±6.8 years, while that in females
was 15 years later at 27.9±14.5 years. It is difficult to determine
the age at onset in adult female patients due to the insidious
nature of the disease (24).
However, cases of female patients with childhood onset have also
been described in a number of studies (18,25-29).
In addition to a later symptom onset, females are thought to have a
relatively mild clinical phenotype (6,23). In
the present study, the proband of Family F2 exhibited tachypnea and
mitral regurgitation at the age of 3 years and had LVH and LVOTO
with systolic anterior motion. Of note, in the proband's father,
who was 36 years old, the disease was not as severe. Myocardial
biopsy and laboratory examination may be helpful to explain LAMP2
protein function, but they did not receive these relevant
examinations. The course of their disease will be further
monitored.
An fQRS on surface ECG reflects myocardial scarring
and abnormal conduction (30). It
may be used as an indirect marker to predict the presence of
fibrosis (31) and has been
associated with severe arrhythmic events and poor prognosis in HCM
(32,33). The patients of the present study who
presented with fQRS on surface ECG (57%) had an average age of ~11
years, which was much earlier than that reported in a previous
review on patients with HCM (32).
It may be hypothesized that hypertrophic muscle fibers and
increased interstitial collagen content due to glycogen deposition
may contribute to the earlier and more severe fibrosis present in
Danon disease. The histopathology of the myocardium and skeletal
muscle in rats with LAMP deficiency
(LAMP2y/-) displayed muscle fiber
disarray and interstitial fibrosis (34), supporting this theory.
Arad et al (20) reported that Danon patients had a
thicker MLVWT compared with patients with PRKAG2 and patients with
HCM and sarcomere protein mutation. However, in the present study,
Danon patients presented with a thinner MLVWT than patients with
PRKAG2. Furthermore, patients with Danon disease also had a thinner
MLVWT than patients with Fabry disease. This may be owing to
diagnosis of Danon patients at a younger age, compared with patient
with PRKAG2 syndrome and Fabry disease. They may also be linked to
the clinical features of Danon disease. Danon disease has an
aggressive progression in males and the present case report also
supports this notion. The MLVWT of the proband of Family F3
increased from 25 to 47 mm over a 4-year period. In normal Chinese
adolescents with BSA similar to this patient (BSA, 1.30-1.66), the
MLVWT is stable and ~7 mm (35). In
addition, the average age of the seven Danon patients was 10 years
and the data when they first came to our center were selected to
make comparisons with patients with PRKAG2 syndrome and Fabry
disease. At that time, their LV thickness may not have progressed
to a severe stage. In addition, the number of patients with Danon
disease in the present study was relatively small, which may have
affected the results. Patients with Danon disease and patients with
HCM were also compared and it was indicated that patients with
Danon disease had a worse diastolic function than patients with
HCM. However, the difference in IVS was not statistically
significant.
There are four limitations to the present study.
First, only a small number of Danon patients were included. This
may have influenced the estimation of the prevalence of Danon
disease in HCM. Furthermore, the study did not examine the
association between the type of mutation and the clinical
presentation. Secondly, three Danon patients in our study were too
young and two of them had been discovered coincidentally. This may
influenced the results of comparison between Danon patients and
other disease. Thirdly, the present study focused on the clinical
symptoms of cardiomyopathy and failed to completely evaluate
extracardiac manifestations, e.g. by cognitive evaluation and
laboratory examinations. At last, certain patients with Danon
disease did not receive muscle biopsy. Our group will continue to
recruit further Danon patients and add comprehensive examinations
in the future.
In conclusion, the present study reported on
early-onset attack of Danon disease in 5 males and 2 females with
characteristic preexcitation and fQRS on ECG. The present study
provided a comprehensive assessment to distinguish between patients
with Danon disease and other patients with HCM may provide valuable
information for early diagnosis and treatment.
Supplementary Material
Full list of 96 cardiac-associated
genes screened.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by International Cooperation
Funding of the China Science and Technology Ministry (grant no.
2014DFA31980), the National Natural Science Foundation of China
(grant nos. 81471197 and 81901755), the Key Program of Shaanxi
Province (grant no. 2017ZDXM-SF-058), the Key R&D Project of
Shaanxi Province (grant no. 2019KW-076), the Xijing Funded Project
for New Technologies and Services (grant no. 417432A) and the Key
Science and Technology Innovation Team Project of Shaanxi Province
(grant no. 2014KCT-20).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XYW, BW and XLZ conceived and designed the study.
XYW, ZLM, YL, CHL QLY and XLZ were responsible for the acquisition
and analysis of the data. DH, ZRL and LWL were responsible for the
analysis and interpretation of the data. All authors substantially
contributed to the revision of the manuscript, and read and
approved the final manuscript.
Ethics approval and consent to
participate
All study procedures in this study were approved by
the Ethics Committee of Xijing Hospital, Fourth Military Medical
University (Xi'an, China). All patients provided written informed
consent form prior to participation in the study. For participants
aged <16 years, written informed consent was obtained from the
parents.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Danon MJ, Oh SJ, DiMauro S, Manaligod JR,
Eastwood A, Naidu S and Schliselfeld LH: Lysosomal glycogen storage
disease with normal acid maltase. Neurology. 31:51–57.
1981.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nishino I, Fu J, Tanji K, Yamada T,
Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al: Primary
LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and
myopathy (Danon disease). Nature. 406:906–910. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Tanaka Y, Guhde G, Suter A, Eskelinen EL,
Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K and
Saftig P: Accumulation of autophagic vacuoles and cardiomyopathy in
LAMP-2-deficient mice. Nature. 406:902–906. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Eskelinen E, Illert AL, Tanaka Y,
Schwarzmann G, Blanz J, Von Figura K and Saftig P: Role of LAMP-2
in lysosome biogenesis and autophagy. Mol Biol Cell. 13:3355–3368.
2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Arad M: Cardiac Danon disease: Insights
and challenges. Int J Cardiol. 245:211–212. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Boucek D, Jirikowic J and Taylor M:
Natural history of Danon disease. Genet Med. 13:563–568.
2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lee JJ, Ishihara K, Notomi S, Efstathiou
NE, Ueta T, Maidana D, Chen X, Iesato Y, Caligiana A and Vavvas DG:
Lysosome-associated membrane protein-2 deficiency increases the
risk of reactive oxygen species-induced ferroptosis in retinal
pigment epithelial cells. Biochem Biophys Res Commun. 521:414–419.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thompson DA, Constable PA, Liasis A,
Walters B and Esteban MT: The Physiology of the retinal pigment
epithelium in Danon disease. Retina. 36:629–638. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Brambatti M, Caspi O, Maolo A, Koshi E,
Greenberg B, Taylor MRG and Adler ED: Danon disease: Gender
differences in presentation and outcomes. Int J Cardiol. 286:92–98.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sweet ME, Mestroni L and Taylor MRG:
Genetic infiltrative cardiomyopathies. Heart Fail Clin. 14:215–224.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang L, Zuo L, Hu J, Shao H, Lei C, Qi W,
Liu Y, Miao Y, Ma X, Huang CL, et al: Dual LQT1 and HCM phenotypes
associated with tetrad heterozygous mutations in KCNQ1, MYH7,
MYLK2, and TMEM70 genes in a three-generation Chinese family.
Europace. 18:602–609. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Authors/Task Force members, Elliott PM,
Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege
AA, Lafont A, Limongelli G, et al: 2014 ESC Guidelines on diagnosis
and management of hypertrophic cardiomyopathy: The task force for
the diagnosis and management of hypertrophic cardiomyopathy of the
European Society of Cardiology (ESC). Eur Heart J. 35:2733–2779.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sokolow M and Lyon TP: The ventricular
complex in left ventricular hypertrophy as obtained by unipolar
precordial and limb leads. Am Heart J. 37:161–186. 1949.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lang RM, Badano LP, Mor-Avi V, Afilalo J,
Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA,
Kuznetsova T, et al: Recommendations for cardiac chamber
quantification by echocardiography in adults: An update from the
American Society of Echocardiography and the European Association
of Cardiovascular Imaging. J Am Soc Echocardiogr. 281–39.
(e14)2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nagueh SF, Appleton CP, Gillebert TC,
Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka
PA and Evangelista A: Recommendations for the evaluation of left
ventricular diastolic function by echocardiography. J Am Soc
Echocardiogr. 22:107–133. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Martin D, Arthur C, Richard G, Levin RI;
Criteria Committee, Devereaux RP, et al: Nomenclature and Criteria
for Diagnosis of Diseases of Heart and great vessels. 9th edition.
Little Brown, Boston, MA. 1994.
|
|
17
|
Gourzi P, Pantou MP, Gkouziouta A,
Kaklamanis L, Tsiapras D, Zygouri C, Constantoulakis P, Adamopoulos
S and Degiannis D: A new phenotype of severe dilated cardiomyopathy
associated with a mutation in the LAMP2 gene previously known to
cause hypertrophic cardiomyopathy in the context of Danon disease.
Eur J Med Genet. 62:77–80. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang Z, McMahon CJ, Smith LR, Bersola J,
Adesina AM, Breinholt JP, Kearney DL, Dreyer WJ, Denfield SW, Price
JF, et al: Danon disease as an underrecognized cause of
hypertrophic cardiomyopathy in children. Circulation.
112:1612–1617. 2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Charron P, Villard E, Sébillon P, Laforêt
P, Maisonobe T, Duboscq-Bidot L, Romero N, Drouin-Garraud V,
Frébourg T, Richard P, et al: Danon's disease as a cause of
hypertrophic cardiomyopathy: A systematic survey. Heart.
90:842–846. 2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Arad M, Maron BJ, Gorham JM, Johnson WH
Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ,
Seidman CE and Seidman JG: Glycogen storage diseases presenting as
hypertrophic cardiomyopathy. N Engl J Med. 352:362–372.
2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cheng Z, Cui Q, Tian Z, Xie H, Chen L,
Fang L, Zhu K and Fang Q: Danon disease as a cause of concentric
left ventricular hypertrophy in patients who underwent
endomyocardial biopsy. Eur Heart J. 33:649–656. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu Y, Chen X, Wang F, Liang Y, Deng H,
Liao H, Zhang Q, Zhang B, Zhan X, Fang X, et al: Prevalence and
clinical characteristics of Danon disease among patients with left
ventricular hypertrophy and concomitant electrocardiographic
preexcitation. Mol Genet Genomic Med. 7(e638)2019.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
D'souza RS, Levandowski C, Slavov D, Graw
SL, Allen LA, Adler E, Mestroni L and Taylor MR: Danon disease:
Clinical features, evaluation, and management. Circ Heart Fail.
7:843–849. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sugie K, Komaki H, Eura N, Shiota T, Onoue
K, Tsukaguchi H, Minami N, Ogawa M, Kiriyama T, Kataoka H, et al: A
nationwide survey on Danon disease in Japan. Int J Mol Sci.
19(3507)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hedberg Oldfors C, Máthé G, Thomson K,
Tulinius M, Karason K, Östman-Smith I and Oldfors A: Early onset
cardiomyopathy in females with Danon disease. Neuromuscul Disord.
25:493–501. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Maron BJ, Roberts WC, Arad M, Haas TS,
Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP and Ho CY:
Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy.
JAMA. 301:1253–1259. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Miani D, Taylor M, Mestroni L, D'Aurizio
F, Finato N, Fanin M, Brigido S and Proclemer A: Sudden death
associated with Danon disease in women. Am J Cardiol. 109:406–411.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dara BS, Rusconi PG and Fishman JE: Danon
disease: Characteristic late gadolinium enhancement pattern on
cardiac magnetic resonance imaging. Cardiol Young. 21:707–709.
2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kim H, Cho A, Lim BC, Kim MJ, Kim KJ,
Nishino I, Hwang YS and Chae JH: A 13-year-old girl with proximal
weakness and hypertrophic cardiomyopathy with Danon disease. Muscle
Nerve. 41:879–882. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ogura S, Nakamura K, Morita H, Toh N,
Nakagawa K, Yoshida M, Watanabe A, Nishii N, Miyoshi T and Ito H:
New appearance of fragmented QRS as a predictor of ventricular
arrhythmic events in patients with hypertrophic cardiomyopathy.
Circ J. 84:487–494. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ratheendran AC, Subramanian M, Bhanu DK,
Prabhu MA, Kannan R, Natarajan KU, Saritha Sekhar S, Thachathodiyil
R, Harikrishnan MS and Pai PG: Fragmented QRS on
electrocardiography as a predictor of myocardial scar in patients
with hypertrophic cardiomyopathy. Acta Cardiol. 75:42–46.
2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Rattanawong P, Riangwiwat T, Kanitsoraphan
C, Chongsathidkiet P, Kanjanahattakij N, Vutthikraivit W and Chung
EH: Baseline fragmented QRS increases the risk of major arrhythmic
events in hypertrophic cardiomyopathy: Systematic review and
meta-analysis. Ann Noninvasive Electrocardiol.
23(e12533)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Özyılmaz S, Akgül Ö, Uyarel H, Pusuroğlu
H, Karayakalı M, Gül M, Çetin M, Satılmışoğlu H, Yıldırım A and
Bakır İ: Assessment of the association between the presence of
fragmented QRS and the predicted risk score of suddena cardiac
death at 5 years in patients with hypertrophic cardiomyopathy.
Anatol J Cardiol. 18:54–61. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ma S, Zhang M, Zhang S, Wang J, Zhou X,
Guo G, Wang L, Wang M, Peng Z, Guo C, et al: Characterisation of
Lamp2-deficient rats for potential new animal model of Danon
disease. Sci Rep. 8(6932)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu N, Bei X, Zhou W, He X-Z, Tao H-W and
Liu X: New reference values for echocardiography dimensions of 800
healthy Chinese infants and children. Chinese J Med Ultrasound
(electronic edition). 1:40–49. 2012.PubMed/NCBI View Article : Google Scholar
|