Introduction
Sepsis is a life-threatening disease that leads to
organ dysfunction caused by a dysregulated host response to
infection (1). According to
statistical reports in 2016, 300-1,000 out of 10,000 individuals
suffer from sepsis per year in developed countries (2). The lung is one of the primary affected
organs (3). Due to a lack of
effective treatment methods, acute lung injury (ALI) caused by
sepsis is one of the main serious diseases which affect global
health (4). Therefore, further
investigation into the mechanism of ALI in sepsis is needed to
improve disease prognosis and to decrease the mortality rate. The
mechanism of ALI is highly complex as its pathophysiological
features involve inflammatory cells, inflammatory mediators and the
abnormal apoptosis of cells in the lung, including alveolar type II
epithelial cells, pulmonary vascular endothelial cells and alveolar
macrophages (AMs) (5-8).
In sepsis, endotoxin binds to the corresponding receptor on the
surface of alveolar macrophages, which induces the release of
inflammatory mediators (9). These
inflammatory mediators promote the chemotaxis of neutrophils into
the lung, leading to further increased release of inflammatory
mediators that finally results in damage to the alveolar epithelium
and vascular endothelium (10). The
disruption of the integrity of the respiratory membrane increases
permeability, promotes pulmonary edema, causes ALI and further
aggravates acute respiratory distress syndrome (ARDS) (10). A previous study has found that
macrophage inhibitory factor is positively associated with the
severity of ALI disease due to its presence in the serum of
patients with sepsis (11). This
indicates that proper functioning and maintaining the levels of
macrophages will help to reduce the degree of damage caused by
sepsis. Therefore, macrophages, including resident AMs and
macrophages recruited from the blood, are key factors in the
pathogenesis of ALI/ARDS. In addition, the occurrence of AM
apoptosis and dysfunction increases the risk of death in patients
with sepsis (12).
Programmed cell death-1 (PD-1) is a type I
transmembrane protein of the B7/CD28 superfamily with a relative
molecular weight of 50~52.5 kDa that functions as an
immunosuppressive molecule (13).
In addition, PD-1 contains an immunoreceptor tyrosine inhibitory
motif and immunoreceptor tyrosine-based switch motif (ITSM)
(14). Various immune cells,
including CD4+ and CD8+ T cells, B cells,
natural killer T cells, dendritic cells (DCs) and
monocytes/macrophages, can express high levels of PD-1 following
stimulation by inflammatory or tumor factors (15). PD-1 has two ligands, programmed cell
death-ligand 1 (PD-L1) also known as B7-H1 or CD274 and programmed
cell death-ligand 2 PD-L2 also known as B7-DC or CD273. PD-L1 is
expressed in several tissue and cell types and PD-L2 is expressed
on both DCs and macrophages (15).
After PD-1 binds to its corresponding ligand, Src homology 2
domain-containing protein tyrosine (SH)-1 and SHP-2 phosphatase
bind to the ITSM inducing the dephosphorylation of downstream T
cell and B cell receptor effector molecules, such as
phosphatidylinositol 3-kinase (PI-3K), protein kinase B (AKT/PKB),
and extracellular signal-regulated kinase 2 (ERK2) (16-18).
Previous studies have reported that the expression of PD-1 on
macrophages plays an important role in sepsis-induced ALI/ARDS. For
example, knockout of the pd-1 gene in mice increased the
ability of macrophages to clear bacteria in the blood and
peritoneal lavage fluid, as well as reduce the release of the
inflammatory factors TNF-α, IL-1β, IL-10 and C-C motif chemokine
ligand 2(19). Bao et al
(20) found that hemorrhagic
shock/sepsis increased the expression of PD-L1 in the lung tissue
of mice with ALI. In the aforementioned study, damage to the lung
tissue of pd-l1 gene-deficient ALI mice was mild and the
levels of proinflammatory cytokines interleukin (IL) 6 and tumor
necrosis factor (TNF)α in the bronchoalveolar lavage fluid were
also significantly reduced, which confirmed that PD-L1 may be
involved in the immune regulation of ALI by reducing the levels of
inflammatory factors.
The aims of the present study were to determine the
in vitro expression levels of PD-1 in alveolar macrophages
in ALI caused by sepsis, if this phenomenon contributes to the
acceleration of the alveolar macrophages apoptosis, and if the
decreased secretion of inflammatory factors attenuate the degree of
lung damage when the binding of PD-1 and PD-L1 is inhibited.
PD-1/PD-L1 inhibitors can inhibit the downstream molecular effects
and reduce apoptosis by blocking the binding of PD-1 to PD-L1. In
the present study, it was confirmed that BMS-1, a small molecular
PD-1/PD-L1 inhibitor, can be used not only for tumor research, but
also for research into inflammation (21).
Materials and methods
Cell culture and treatment
A mouse alveolar macrophage cell line, MH-S, was
purchased from Wuhan Punosei Life Technology Co., Ltd. and was
routinely passaged in modified RPMI-1640 medium (Hyclone; GE
Healthcare Life Sciences) supplemented with 10% fetal bovine serum
(Cellmax Nutrients BV), 100 U/ml penicillin and 100 µg/ml
streptomycin in a humidified atmosphere of 5% CO2 at
37˚C. LPS and BMS-1 were at 4˚C. MH-S cells were passaged 3 times
and divided into three groups, a control group, LPS group and LPS +
BMS-1 group. The control group was provided the same amount of
RPMI-1640 medium as the LPS group and LPS + BMS-1 group. When the
LPS group and LPS + BMS-1 group cells had grown to 70-80%
confluence, they were stimulated with 10 ng/ml lipopolysaccharide
(LPS; Escherichia coli 055:B5; Sigma-Aldrich; Merck KGaA)
for 24 h in a humidified atmosphere of 5% CO2 at 37˚C,
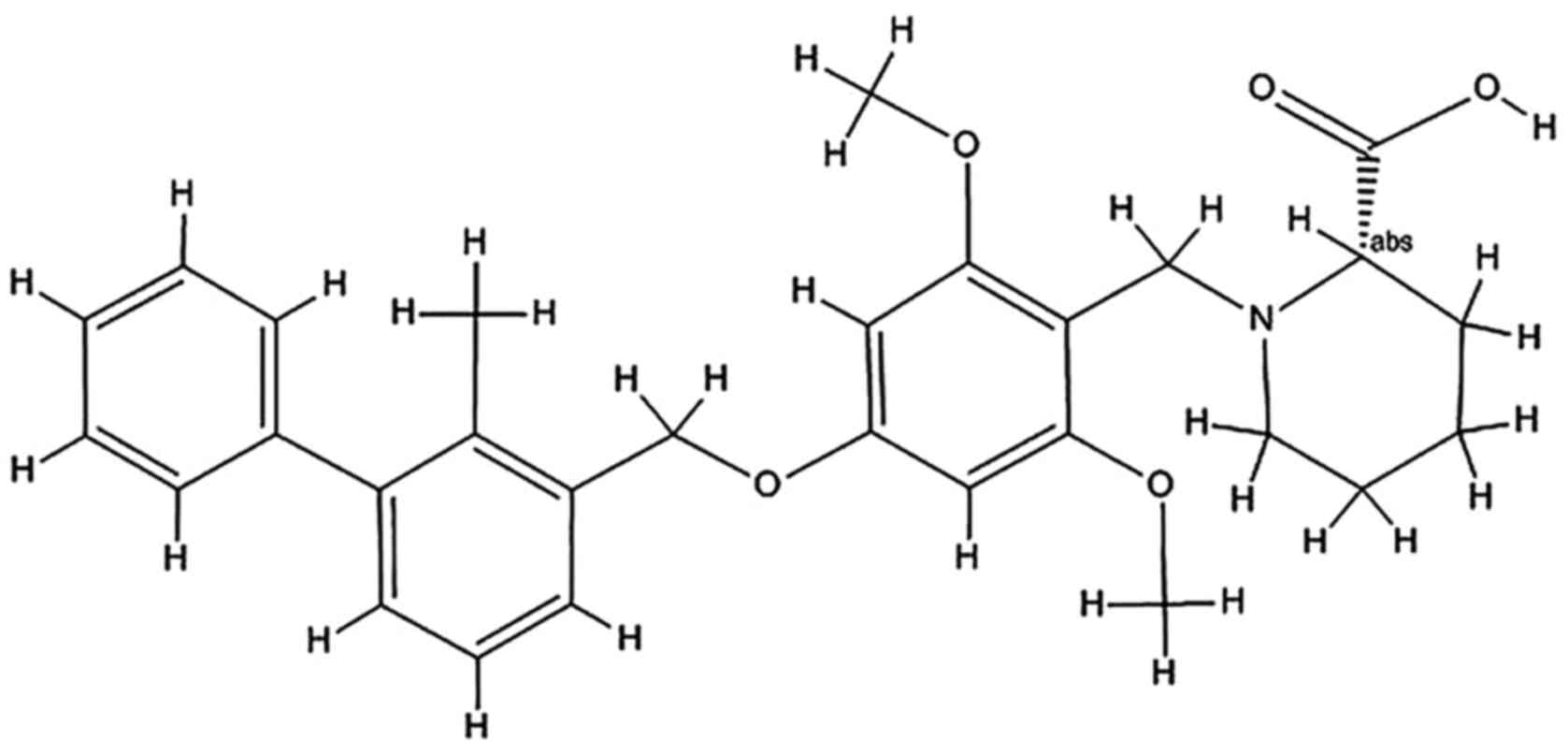
followed by treatment with 1 µmol/l BMS-1 for 72 h at 37˚C. BMS-1
is an inhibitor of the PD-1/PD-L1 protein/protein interaction with
an IC50 of 6-100 nM (Fig.
1) (21). It has previously
been confirmed that LPS (0, 5, 10, 20 or 30 ng/ml) has no toxic
effect on MH-S cells and there is no statistically significant
difference of different concentrations of LPS on MH-S cells
(22). In the present study, MH-S
cells were treated with LPS at a concentration of 10 ng/ml
according to a previous study (22).
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from the treated MH-S cells
(~5x107) using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific Inc.). cDNA synthesis was performed from 2
µl of total RNA using a RevertAid First Strand cDNA Synthesis kit
(Thermo Fisher Scientific Inc.). The reverse transcription protocol
was as follows: 37˚C for 15 min and heated to 85˚C for 5 sec to
eliminate reverse transcriptase activity. The treated MH-S cells
were denatured at 95˚C for 30 sec, annealed at 95˚C for 5 sec and
extended at 60˚C for 30 sec, a total of 45 cycles. The levels of
PD-1 mRNA expression were assessed using a SYBR Green Mastermix kit
(Takara Bio, Inc.) with the CFX96 Touch™ Real-Time PCR
Detection system (Bio-Rad Laboratories, Inc.). The levels of mRNA
expression were calculated using the
2-ΔΔCq method and normalized to
GAPDH (23). The primer sequences
used are as follows: Mouse PD-1, forward,
5'-ATGACTTCCACATGAACATCCT-3', reverse,
5'-CTCCAGGATTCTCTCTGTTACC-3'; and GAPDH, forward,
GGCAAGTTCAACGGCACAGT, reverse, ATGACATACTCAGCACCGGG.
Western blotting
Following the different experimental treatments, the
MH-S cells (~5x107) cultured in 6-well plates, were
harvested and lysed in RIPA lysis buffer (Boster Biological
Technology) containing a 1% protease inhibitor (Boster Biological
Technology). The cellular supernatant was collected by
centrifugation at 12,000 x g for 15 min at 4˚C and the protein
concentrations were measured with a bicinchoninic protein assay kit
(Bio-Rad Laboratories, Inc.). The protein samples (20 µg/lane) were
separated by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidene
difluoride (PVDF) membranes (Zhangshu Zhenghe Biotechnology Co.,
Ltd.). The membranes were blocked in 5% non-fat milk proteins for 2
h at room temperature and then probed with the following primary
antibodies: PD-1 (cat. no. MB9410; 1:500; Bioworld Technology Inc.)
and β-actin (cat. no. BS6007; 1:3,000; Bioworld Technology Inc.)
overnight at 4˚C. The membranes were subsequently incubated with
horseradish peroxidase-conjugated secondary goat anti-mouse
antibodies (cat. no. BA1038; 1:1,000; Boster Biology Technology)
for 1 h at 37˚C. Finally, the protein bands were visualized using
an enhanced chemiluminescence (ECL) system (Thermo Fisher
Scientific, Inc.) and analyzed by ImageJ 1.50 (National Institutes
of Health.). β-actin was used as the loading control.
Flow cytometry
MH-S cells (~5x107) cultured in 6-well
plates, were harvested and washed three times with PBS and stained
with 5 µl Annexin V-FITC and 10 µl of propidium iodide for 15 min
in 500 ml binding buffer at room temperature. Surface exposure of
phosphatidylserine in the apoptotic cells was measured using an
Annexin V-FITC Apoptosis Detection kit (Shanghai Gensheng
Biotechnology Co., Ltd.) according to the manufacturer's
instructions. Apoptosis was analyzed using a flow cytometer (BD
FACSCalibur Cell Sorting System; BD Bioscience).
ELISA
Concentrations of IL-1β, IL-6, TNF-α and IL-10 in
the supernatants of treated MH-S cells were measured using
respective specific ELISA kits (R&D Systems, Inc.) in
accordance with the manufacturer's instructions (IL-1β, mouse
IL-1β, PicoKine ELISA kit, cat. no. EK0394; IL-6, mouse IL-6,
PicoKine ELISA kit, cat. no. EK0411; TNF-α, mouse TNF-α PicoKine
ELISA kit, cat. no. EK0527; IL-10, mouse IL-10 PicoKine ELISA kit,
cat. no. EK0417; all supplied from Boster Biological Technology,
Ltd.).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 7.0 (GraphPad Software, Inc.). The measurement data
are presented as the means ± standard deviation of three
independent experiments performed in triplicate. The comparison
between the 3 different groups was performed by one-way ANOVA
followed by the post hoc Student Newman Keule test. P<0.05 was
considered to indicate a statistically significant difference.
Results
BMS-1 inhibits LPS-induced expression
of PD-1 mRNA and protein in MH-S cells
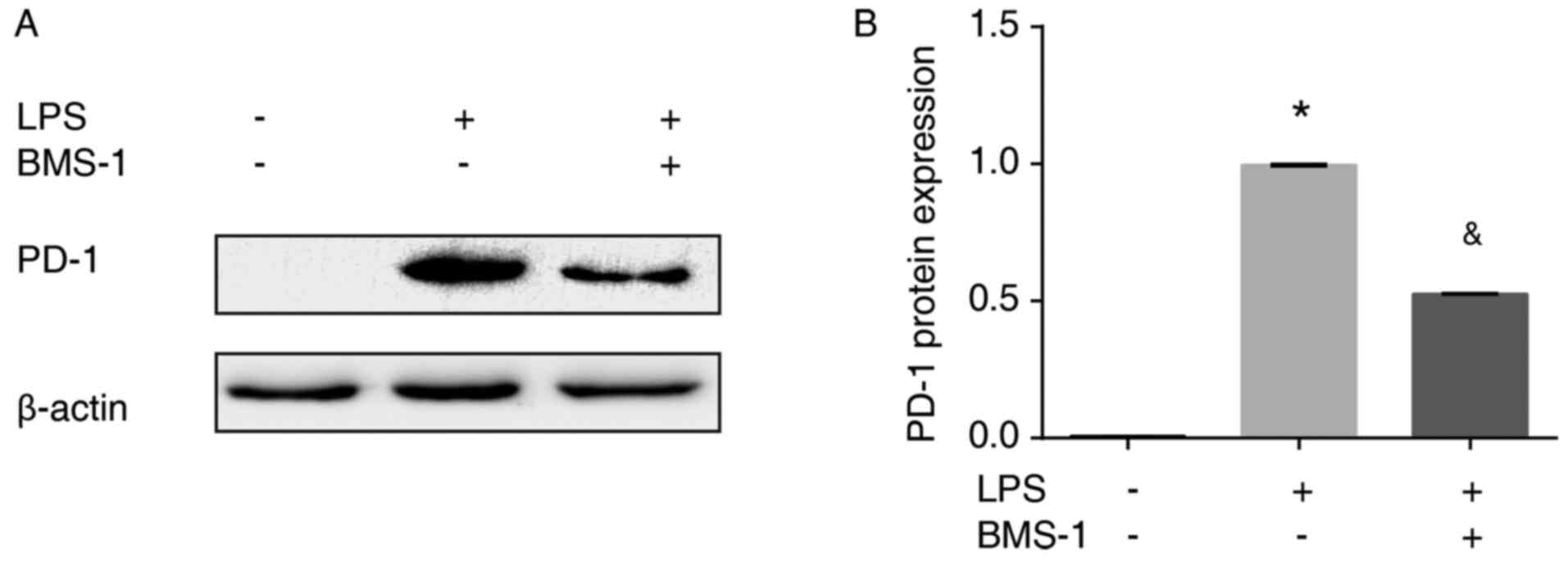
To confirm and explore the effects of BMS-1 on
LPS-induced MH-S cells, PD-1 mRNA and protein expression were
assessed using RT-qPCR and western blotting, respectively.
LPS-stimulated MH-S cells had increased levels of PD-1 mRNA and
protein expression compared with the control group (Figs. 2 and 3, P<0.01). PD-1 mRNA and protein
expression were reduced in MH-S cells treated with LPS+BMS-1
compared with cells just treated with LPS (Figs. 2 and 3, P<0.01). Taken together, these
results demonstrated that BMS-1 inhibited LPS-induced expression of
PD-1 in MH-S cells.
BMS-1 suppresses LPS-induced apoptosis
in MH-S cells
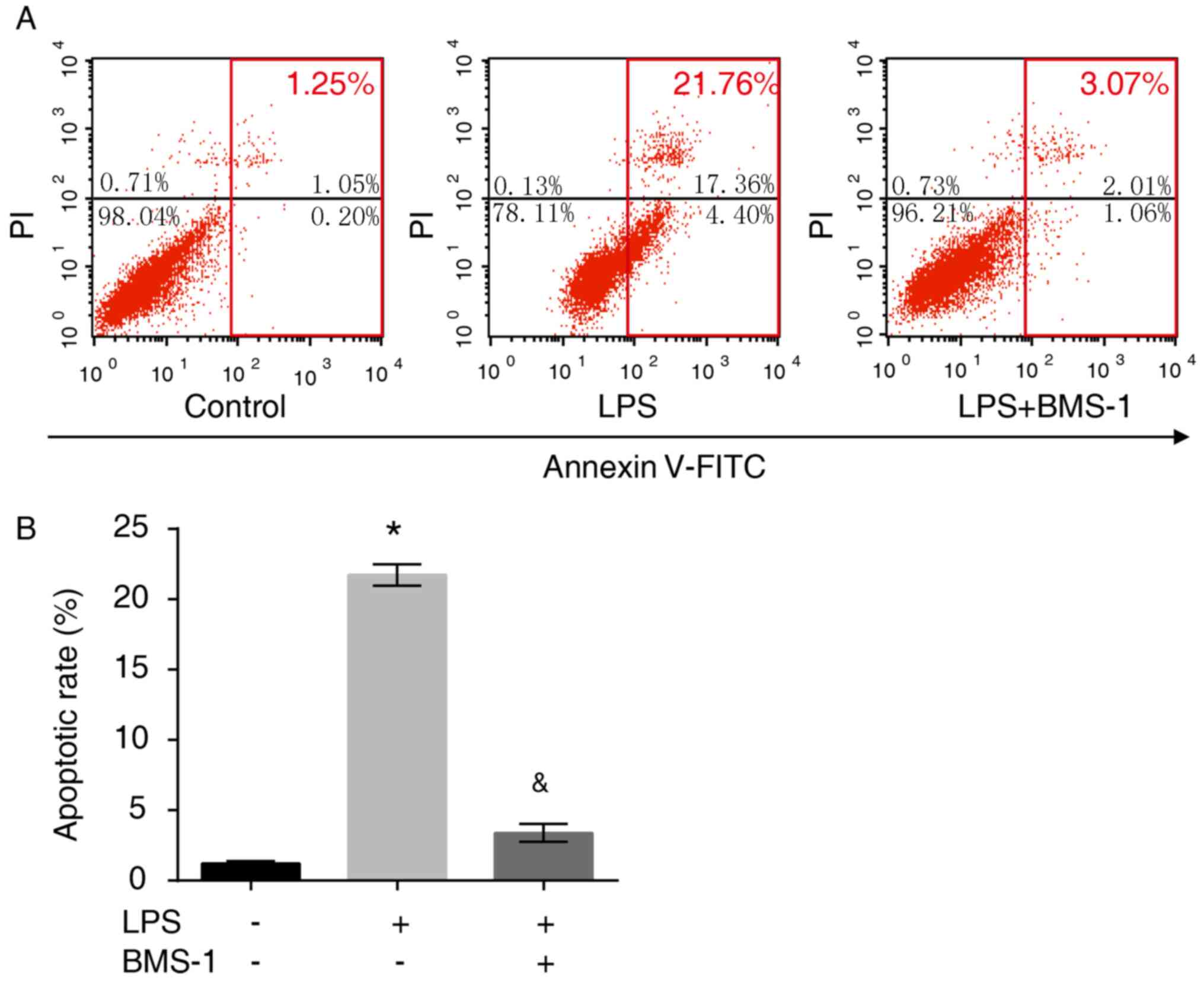
To investigate the apoptotic effect of BMS-1 in
LPS-stimulated MH-S cells, flow cytometry was performed and the
number of apoptotic cells were counted. LPS exposure resulted in
enhanced apoptosis compared with the control group (P<0.05),
which was attenuated by BMS-1 treatment (Fig. 4, P<0.05). These results
demonstrated that following LPS stimulation, alveolar macrophages
exhibit high levels of PD-1 expression and increased apoptosis.
Following treatment with a PD-1/PD-L1 inhibitor, both PD-1
expression and alveolar macrophage apoptosis were decreased.
BMS-1 exerts antiinflammatory activity
in LPS-treated MH-S cells
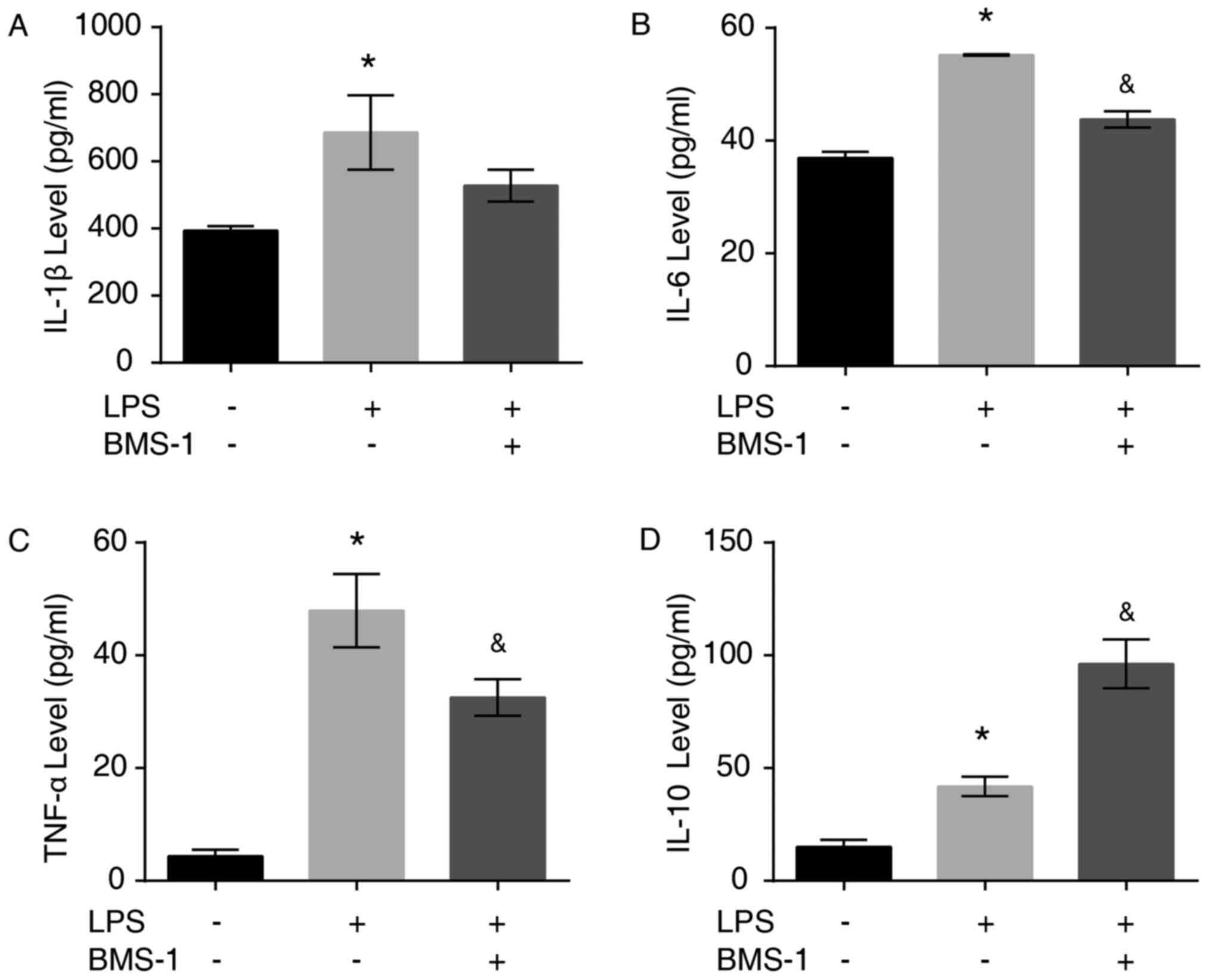
To investigate the antiinflammatory effects of
BMS-1, the levels of inflammatory factors IL-1β, IL-6, TNF-α and
IL-10 secreted by LPS-stimulated MH-S cells were detected using
ELISA. The secretion of the proinflammatory cytokines IL-1β, IL-6,
TNF-α and antiinflammatory cytokine IL-10 was increased following
LPS stimulation (Fig. 5A-D,
P<0.05). Treatment with BMS-1 reduced the LPS-induced increase
of TNF-α, IL-1β and IL-6 in MH-S cells, while IL-10 was increased
significantly (Fig. 5A-D, P=0.085
for IL-1β, P<0.05 for IL-6 TNF-α and IL-10).
Discussion
ALI caused by sepsis and ARDS remains the primary
cause of death among patients in the ICU (24). Alveolar macrophages serve an
important role in the pathophysiological process of ALI (25). In addition, a recent study confirmed
that the inhibition of alveolar macrophage apoptosis and excessive
inflammatory response can reduce the degree of ALI caused by sepsis
(26). The present study
investigated the apoptosis of MH-S cells and levels of inflammatory
mediators, such as IL-1, IL-6, TNF-α and IL-10, following the
administration of the PD-1/PD-L1 inhibitor BMS-1. RT-qPCR and
western blotting results confirmed that PD-1 was moderately
transcribed and expressed or expressed at basal levels in normal
cells; however, following LPS stimulation, alveolar macrophages
expressed high levels of PD-1. The flow cytometry findings of the
present study demonstrated that the apoptosis of alveolar
macrophages was significantly increased in the LPS group compared
with the control group. These findings indicated that PD-1 could be
expressed at high levels in alveolar macrophages in ALI, which may
cause an increase in the apoptosis of alveolar macrophages.
According to the findings of the present study, when PD-1 was bound
to its ligand, PD-L1 was inhibited, the levels of PD-1 mRNA and
protein expression in the LPS+BMS-1 group were significantly lower
and the apoptosis of MH-S cells was decreased compared with the LPS
group. The ELISA results demonstrated that the expression of the
proinflammatory cytokines IL-1β, IL-6 and TNF-α were significantly
increased in the LPS group compared with the control group. In
addition, the expression of the antiinflammatory factor IL-10 was
markedly increased upon the stimulation of BMS-1, whereas
PD-1expression was inhibited. Following inhibition of PD-1/PD-L1
using BMS-1 in the present study, the levels of proinflammatory
cytokines IL-1β, IL-6 and TNF-α decreased significantly and the
level of the antiinflammatory cytokine IL-10 increased compared
with the LPS treated group. Thus, BMS-1 can inhibit proinflammatory
factors and promote the expression of antiinflammatory factors,
thereby reducing the inflammatory response associated with ALI.
The present study has several limitations. It has
only been shown in vitro that treatment with a PD-1/PD-L1
inhibitor can decrease the apoptosis of alveolar macrophages,
reduce inflammatory factor expression. In addition, the study did
not investigate if these effects were long lasting. The findings of
the present study were only assessed in alveolar macrophage MH-S
cells. In addition, the specific mechanism by which PD-1 and
PD-1/PD-L1 inhibitors impact the apoptosis of alveolar macrophages
and the release of inflammatory factors remains unclear.
A previous study demonstrated that the increase in
alveolar macrophages led to an increase in the secretion of
proinflammatory mediators, causing chemotactic central granulocytes
to accumulate in the lungs, which destroyed alveolar epithelial
cells and endothelial cells, resulting in increased permeability
and acute pulmonary edema (27).
Recently, a study has found that LPS increases PD-1 expression
through the NF-κB signaling pathway (28). This study demonstrated that
PD-1/PD-L1 inhibitor, BMS-1 reduced alveolar macrophage apoptosis
and decreased the release of inflammatory factors, leading to the
question of whether the treatment with BMS-1 has an effect on the
polarization of alveolar macrophages. Alveolar macrophages can be
divided into two subclasses: M1 and M2(29). M1s primarily serve a proinflammatory
role and are involved in immune responses to bacterial and viral
infections. M1 macrophages have high surface CD11c and CD16
expression. M2 macrophages are mainly involved in the
antiinflammatory response, which is associated with parasitic
infection, tissue remodeling, fibrosis and tumor development, and
exhibit high levels of CD206 and arginase 1 surface expression
(30). M2 macrophages are further
divided into 3 subtypes: A, b and c. Type a is induced by IL-4 and
IL-13, type b is induced by immune complexes and toll like
receptors or the IL-1 receptor and type c is induced by IL-10 and
glucocorticoids (30). In addition,
it has been confirmed that an increase in M1 alveolar macrophages
leads to an increase in inflammatory factors and an aggravation of
lung injury in rats (31). Thus,
inhibiting the polarization of alveolar macrophages to M1 minimizes
lung injury (31). Therefore, it
can be speculated that PD-1/PD-L1 inhibitors play a balancing role
in the polarization of M1 and M2 alveolar macrophages. The
antiapoptotic and antiinflammatory efficacy of PD-1/PD-L1
inhibitors in vivo should be assessed in future studies.
Further in vitro studies demonstrating whether such
PD-1/PD-L1 inhibitors polarize alveolar macrophages need to be
conducted.
To the best of our knowledge, the present study is
the first to demonstrate that BMS-1, an inhibitor of the
PD-1/PD-L1, pathway exerts antiapoptotic and antiinflammatory
effects on LPS-stimulated MH-S cells. The findings may provide
novel insight into the effects and molecular mechanisms of a
PD-1/PD-L1 inhibitor in sepsis-induced ALI. Thus, a PD-1/PD-L1
inhibitor may be a promising therapeutic strategy for treating
patients with ALI. Therefore, sepsis-induced ALI animal models are
required to further evaluate the effects of a PD-1/PD-L1 inhibitor
in vivo.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
Scientific Research Funding Project for Returnees in Shanxi
Province of China (grant no. 2011-105) and the Taiyuan Science and
Technology Project of Shanxi Province of China (grant no.
12016905).
Availability of data and materials
The datasets used and/or analyzed during the current
study are are available from the corresponding author on reasonable
request.
Authors' contributions
LJ designed the experiments, performed the reverse
transcription-quantitative PCR and western blotting experiments,
analyzed the data and wrote the manuscript. WZ and LH designed the
experiments and revised the manuscript for important intellectual
content. KL and TF cultured the cells and performed flow cytometric
analysis. QL and XZ assisted in completing western blot analysis.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kempker JA and Martin GS: The changing
epidemiology and definitions of sepsis. Clin Chest Med. 37:165–179.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang YM, Ji R, Chen WW, Huang SW, Zheng
YJ, Yang ZT, Qu HP, Chen H, Mao EQ, Chen Y and Chen EZ: Paclitaxel
alleviated sepsis-induced acute lung injury by activating MUC1 and
suppressing TLR-4/NF-κB pathway. Drug Des Devel Ther. 13:3391–3404.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Johnson ER and Matthay MA: Acute lung
injury: Epidemiology, pathogenesis, and treatment. J Aerosol Med
Pulm Drug Deliv. 23:243–252. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zahar JR, Timsit JF, Garrouste-Orgeas M,
Français A, Vesin A, Descorps-Declere A, Dubois Y, Souweine B,
Haouache H, Goldgran-Toledano D, et al: Outcomes in severe sepsis
and patients with septic shock: Pathogen species and infection
sites are not associated with mortality. Crit Care Med.
39:1886–1895. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Huang C, Zheng H, He W, Lu G, Li X, Deng Y
and Zeng M: Ghrelin ameliorates the human alveolar epithelial A549
cell apoptosis induced by lipopolysaccharide. Biochem Biophys Res
Commun. 474:83–90. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gill SE, Rohan M and Mehta S: Role of
pulmonary microvascular endothelial cell apoptosis in murine
sepsis-induced lung injury in vivo. Respir Res.
16(109)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li B, Fang J, Zuo Z, Yin S, He T, Yang M,
Deng J, Shen L, Ma X, Yu S, et al: Activation of porcine alveolar
macrophages by actinobacillus pleuropneumoniae lipopolysaccharide
via the toll-like receptor 4/NF-κB-mediated pathway. Infect Immun.
86:e00642–17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Moldoveanu B, Otmishi P, Jani P, Walker J,
Sarmiento X, Guardiola J, Saad M and Yu J: Inflammatory mechanisms
in the lung. J Inflamm Res. 2:1–11. 2009.PubMed/NCBI
|
|
11
|
Pahuja M, Tran C, Wang H and Yin K:
Alveolar macrophage suppression in sepsis is associated with high
mobility group box 1 transmigration. Shock. 29:754–760.
2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yamazaki T, Akiba H, Iwai H, Matsuda H,
Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, et al:
Expression of programmed death 1 ligands by murine T cells and APC.
J Immunol. 169:5538–5545. 2002.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shinohara T, Taniwaki M, Ishida Y,
Kawaichi M and Honjo T: Structure and chromosomal localization of
the human PD-1 gene (PDCD1). Genomics. 23:704–706. 1994.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hassan SS, Akram M, King EC, Dockrell HM
and Cliff JM: PD-1, PD-L1 and PD-L2 gene expression on T-cells and
natural killer cells declines in conjunction with a reduction in
PD-1 protein during the intensive phase of tuberculosis treatment.
PLoS One. 10(e0137646)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dong H, Zhu G, Tamada K and Chen L: B7-H1,
a third member of the B7 family, co-stimulates T-cell proliferation
and interleukin-10 secretion. Nat Med. 5:1365–1369. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Latchman Y, Wood CR, Chernova T, Chaudhary
D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, et al:
PD-L2 is a second ligand for PD-1 and inhibits T cell activation.
Nat Immunol. 2:261–268. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Taube JM, Klein A, Brahmer JR, Xu H, Pan
X, Kim JH, Chen L, Pardoll DM, Topalian SL and Anders RA:
Association of PD-1, PD-1 ligands, and other features of the tumor
immune microenvironment with response to anti-PD-1 therapy. Clin
Cancer Res. 20:5064–5074. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huang X, Venet F, Wang YL, Lepape A, Yuan
Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS and Ayala A:
PD-1 expression by macrophages plays a pathologic role in altering
microbial clearance and the innate inflammatory response to sepsis.
Proc Natl Acad Sci USA. 106:6303–6308. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bao X, Sun H and Yang Q: The role and
mechanism of costimulatory molecular programmed death ligand-1 in
acute lung injury. Zhonghua Crisis Emerg Med. 28:498–503. 2016.
|
|
21
|
Zhao Y, Jia Y, Shi T, Wang W, Shao D,
Zheng X, Sun M, He K and Chen L: Depression promotes hepatocellular
carcinoma progression through a glucocorticoids mediated
up-regulation of PD-1 expression in tumor infiltrating NK cells.
Carcinogenesis: Feb 4, 2019 (Epub ahead of print). doi:
10.1093/carcin/bgz017.
|
|
22
|
Zhou Z, Su Y and Fa XE: Isorhynchophylline
exerts anti-inflammatory and anti-oxidative activities in
LPS-stimulated murine alveolar macrophages. Life Sci. 223:137–145.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Niesler U, Palmer A, Radermacher P and
Huber-Lang MS: Role of alveolar macrophages in the inflammatory
response after trauma. Shock. 42:3–10. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kubota Y, Iwasaki Y, Harada H, Yokomura I,
Ueda M, Hashimoto S and Nakagawa M: Role of alveolar macrophages in
Candida-induced acute lung injury. Clin Diagn Lab Immunol.
8:1258–1262. 2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang L, Zhang Z, Zhuo Y, Cui L, Li C, Li
D, Zhang S, Cui N, Wang X and Gao H: Resveratrol alleviates
sepsis-induced acute lung injury by suppressing inflammation and
apoptosis of alveolar macrophage cells. Am J Transl Res.
10:1961–1975. 2018.PubMed/NCBI
|
|
27
|
Fu Y, Zhou J, Liu K, Hou L and Zhang W:
Effects of miR-155 on IL-6, IL-10 and MIP-2 in alveolar macrophages
of acute lung injury induced by sepsis. Chin J Crit Care Med.
5:159–164. 2019.
|
|
28
|
Nam S, Lee A, Lim J and Lim JS: Analysis
of the expression and regulation of PD-1 protein on the surface of
myeloid-derived suppressor cells (MDSCs). Biomol Ther (Seoul).
27:63–70. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mitsi E, Kamng'ona R, Rylance J, Solórzano
C, Jesus Reiné J, Mwandumba HC, Ferreira DM and Jambo KC: Human
alveolar macrophages predominately express combined classical M1
and M2 surface markers in steady state. Respir Res.
19(66)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Italiani P and Boraschi D: From monocytes
to M1/M2 macrophages: Phenotypical vs functional differentiation.
Front Immunol. 5(514)2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yin D, Wang W, Han W and Fan C: Targeting
Notch-activated M1 macrophages attenuate lung tissue damage in a
rat model of ventilator induced lunginjury. Int J Mol Med.
44:1388–1398. 2019.PubMed/NCBI View Article : Google Scholar
|