Introduction
Pancreatic neuroendocrine tumor (PNET) is a
relatively rare type of neuroendocrine malignancy, originating from
the islets of Langerhans in the pancreas. PNETs comprise ~1-2% of
all pancreatic neoplasms, demonstrating an annual incidence of 1
per 100,000 and a mortality rate of 60% (1-4).
PNETs currently represent the second most common epithelial
neoplasm after pancreatic ductal adenocarcinoma (PDAC) and are
exhibiting a rising incidence and prevalence, particularly among
Caucasians and Asians (4-6).
In total, ~85% of PNETs are non-functional,
presenting with a worse prognosis compared with hormone-secreting
functional PNETs (7,8). PNETs represent a diverse group of
heterogeneous neoplasms with limited treatment options, which are
morphologically and genetically different from PDACs and have far
fewer mutations (6,9). Although biological classifications may
help define the various clinical behaviors and thus guide
treatment, to the best of our knowledge, the genetic background of
PNETs remains to be fully characterized.
Menin 1 (MEN1), death domain associated protein
(DAXX), ATRX chromatin remodeler (ATRX) and mTOR signaling pathway
genes, including phosphatase and tensin homolog (PTEN), TSC complex
subunit 1, TSC complex subunit 2, DEP domain containing 5, GATOR1
subcomplex subunit and phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit alpha (PIK3CA), were previously
identified to be commonly mutated in PNETs (8,10).
Additional novel germline mutations in the DNA repair genes, mutY
DNA glycosylase, checkpoint kinase 2 and BRCA2 DNA
repair-associated (BRCA2), and recurrent inactivating mutations or
chromosomal rearrangements in chromatin-remodeling genes, including
SET domain containing 2, histone lysine methyltransferase (SETD2),
AT-rich interaction domain 1A (ARID1A) and lysine methyltransferase
2C (KMT2C), have also been reported (4). At the time of diagnosis, ~60% of
patients with nonfunctional PNETs have developed liver metastases,
which results in a worse clinical outcome (11). Therefore, an improved understanding
of the genetic background in PNET liver metastases may pave the way
for the development of potential novel treatment strategies.
In the present study, 14 patients with PNETs were
recruited and target next-generation sequencing (NGS) was performed
on the primary fresh tumor tissues. The hybridization capture-based
NGS panel, Biotecan PanCancer Panoramic Detection (12), which was designed according to the
cancer-related database, clinical guidelines and high-quality
references of solid tumors, covered the exons and promoter areas of
the 612 cancer-associated genes (2.75M). Based on the sequencing
data, a mutational profile of 14 patients with PNET was constructed
and differences in the mutational profile between patients with and
without liver metastasis were further investigated. The study also
evaluated the utility of the NGS panel to guide the clinical
management of patients with PNET.
Materials and methods
Patient samples
Fresh primary tumors were collected from 14
pathologically confirmed sporadic patients with PNETs following
surgery at the Changhai Hospital (Shanghai, China) between February
2016 and September 2017. Images of H&E-stained tumor tissues of
two patients are exhibited in Fig.
S1A and B. The percentage of
Ki-67 nuclear staining (Ki-67 index; Fig. S1C-F) (13) was used to determine the grade of
PNET (14). The histological
diagnosis of tumors was performed and confirmed by two
pathologists. None of the patients had received any therapeutic
procedures, including chemotherapy or radiotherapy, prior to
surgery. Samples were immediately frozen in liquid nitrogen and
stored at -80˚C until further analysis. The clinicopathological
features of the 14 patients are presented in Table I.
 | Table ISummary of the baseline data of the
patients with pancreatic NET (n=14). |
Table I
Summary of the baseline data of the
patients with pancreatic NET (n=14).
| Variables | Value |
|---|
| Age (years) | 50 (28-65) |
| Male, sex | 7(50) |
| Functioning hormone
secretion status | 0 (0) |
| Location | |
|
Head | 4 |
|
Tail | 10 |
| Local invasion | |
|
Nerve
invasion | 4 |
|
Vascular
tumor thrombus | 4 |
| Lymph node
metastasis | |
|
Yes | 7 |
|
No | 7 |
| Liver
metastasis | |
|
Yes | 4 |
|
No | 10 |
| WHO
classification | |
|
NET G1 | 1 |
|
NET G2 | 11 |
|
NET G3 | 2 |
DNA extraction and quality control
(QC)
Genomic (g)DNA from the fresh frozen tumor tissues
was extracted using a QIAamp DNA Mini kit (Qiagen GmbH). The
quantity and purity of the gDNA were assessed using a
Qubit® 3.0 fluorometer (Invitrogen; Thermo Fisher
Scientific, Inc.) and a NanoDrop ND-1000 (Thermo Fisher Scientific,
Inc.). The fragmentation status was evaluated using the Agilent
2200 TapeStation system using the Genomic DNA ScreenTape assay
(Agilent Technologies, Inc.) to produce a DNA integrity number. An
additional QC step to determine the fresh frozen tissue DNA
integrity was performed using a multiplex PCR approach (15). In brief, 30 ng gDNA was amplified
using three different size sets of primers for the GAPDH gene
(200-400 bp), and the concentration of the PCR products was
determined using an Agilent 2100 Bioanalyzer instrument (Agilent
Technologies, Inc.). Then, to estimate the fresh frozen tissue gDNA
fragmentation, the average yield ratio value was determined by
calculating the yield ratio of each amplicon compared with the
Standard human genomic DNA (Promega Corproation; cat. no.
G3041).
Library preparation, hybridization
capture and amplification
A total of 300 ng of each gDNA sample, which was
based on Qubit quantification, was mechanically fragmented (duty
factor 10%; peak incident power 175 W; cycles per burst 200;
treatment time 240 sec; bath temperature 2-8˚C) on an E220 focused
ultrasonicator (Covaris, Inc.). The target DNA fragment size is
150-200 bp. Subsequently, 200 ng sheared gDNA was used to perform
end repair, A-tailing and adapter ligation with a KAPA library
preparation kit (Kapa Biosystems Inc.), according to the
manufacturer's protocol. Subsequently, the libraries were captured
using Agilent SureSelect XT custom 0.5-2.9M probes (Agilent
Technologies, Inc.) and amplified.
Illumina sequencing
Following QC and quantification using the Agilent
2100 bioanalyzer (Agilent Technologies, Inc.) and a Qubit 2.0
fluorometer (Invitrogen; Thermo Fisher Scientific, Inc.), the
libraries were sequenced on the Illumina Next 500 platform
(Illumina, Inc.) in high-output mode, with 2x75 cycles.
Bioinformatics analysis
Clean data were obtained after filtering out the
adapters and reads with a proportion of N>10%, with N being the
unidentified bases in the sequencing process, using fastp (fastp
0.19.4) (16). Low-quality bases
(Phred score <15) were excised from the 3' ends of reads. Reads
with length of <36 bp after excision were removed. The clean
data were mapped to the reference human genome (University of
California Santa Cruz ID: hg19; GenBank accession no.
GCA_000001405.1) using the BWA alignment algorithm (BWA. 0.7.17)
(17). The alignment in the SAM
format was converted to BAM files using SAMtools (Samtools 1.9.0)
(18). Next, the genome analysis
toolkit (GATK; v4.0.2.1) (19) was
used for sorting, duplicate marking and base recalibration. The
final BAM files were analyzed using QualiMap v.2.2.1(20) to provide an overall overview of the
data, including mapped reads, mean mapping quality and mean
coverage. The variants [single nucleotide variants (SNV) and
insertion (Ins)-deletion (Del) mutations (InDels)] were called for
unpaired tumor sequences with 40 pooled blood samples (from healthy
individuals) using the GATK mutect2 tumor-only mode with the
parameter af-alleles-not-in-resource 0.00025%, and the germline
mutations and contaminations were filtered out using GATK
FilterMutectCalls with parameters (max-germline-posterior 0.995).
Somatic variants were annotated using the ANNOVAR software
tool.
The following filter conditions were used to
identify the candidate somatic alterations: i) all variations with
COSMIC evidence (http://cancer.sanger.ac.uk/cosmic) were retained; ii)
the variants with a mutational allele frequency (maf) >0.001 in
the 1,000 Genomes databases (1,000 Genomes Project Consortium;
https://www.internationalgenome.org/)
(21) were removed; iii) the
functional benign variant sites predicted by PolyPhen-2(22), SIFT (23), MutationTaster (24) and Combined Annotation-Dependent
Depletion (25) were removed; and
iv) only the following variant classifications were retained:
Missense_Mutation, Nonsense_Mutation, Splice_Site, Frame_Shift_Ins,
Frame_Shift_Del, In_Frame_Ins, In_Frame_Del.
Statistical analysis
The mutational landscape across the cohort was
created using the maftools package in R software (R 3.5.1, R Core
Team; https://www.R-Project.org). Kyoto
Encyclopedia of Genes and Genomes (KEGG) signaling pathway
enrichment analysis (https://www.kegg.jp/) was performed to investigate the
biological importance of the altered genes in all samples using the
clusterProfiler package (26) in R
software (R 3.5.1). A cut-off value of the adjusted P-value
(p.adjust) of <0.05 was used to identify significantly enriched
terms.
Results
Demographic and baseline
characteristics of the patients
The demographic and baseline characteristics of the
study participants are summarized in Table I. A total of 14 patients with PNET
were analyzed in the present study, including 7 males and 7
females, with a median age of 50 years (age range, 28-65 years). In
the study cohort, all tumors of patients were non-functional PNETs,
which do not make hormones, or make hormones that do not cause a
set of symptoms. Among them, 4 patients had tumors located in the
pancreas head and the other 10 patients had tumors located in the
pancreas tail. Regarding the World Health Organization grade
(27), 1 patient had a grade (G)1
classified NET, 11 patients had G2 classified NETs and 2 patients
had G3 classified NETs.
In 13 patients, the median follow-up time was 20
months (range, 5-38 months; Table
SI). One patient (case no. 11) was lost to follow-up. Among the
14 patients who were examined for lymph node status, 7 patients
were identified to have lymph node metastasis, while nerve invasion
and vascular tumor thrombus were present in 4 patients, and 4
patients demonstrated distant metastasis to the liver. Furthermore,
2 patients also presented with type II diabetes mellitus (T2DM).
The majority of the patients remained alive during the follow-up,
except for 1 patient (case no. 8), who died at 15 months following
surgery. In addition, 1 patient (case no. 13) developed liver
metastasis at 7 months following surgery. The percentage of Ki-67
of 14 patients varied between 1 and 40% (Table SI).
Recurrent mutated genes in PNET with
and without liver metastasis
Fig. S2 presents a
summary of the maf files (Table
SII) in the 14 patients with PNET. In total, 63 variations in
53 genes were identified, involving 5 variant classifications. For
the SNVs, C>T was the most frequent SNV class. The median number
of variants identified in the 14 samples was 5 (range, 1-8).
Fig. 1 presents the
mutational profile of 14 patients with PNET, including the top 20
recurrently mutated genes, according to the presence or absence of
liver metastasis. The mutated gene with the highest frequency was
MEN1 (4/14; 29%), which was consistent with the previously reported
mutation rate of MEN1 (23.1-56%) in PNET (5,6). The
other recurrent altered genes were revealed to be adrenoceptor
alpha 2B (ADRA2B; 3/14; 21%), ARVCF delta catenin family member
(ARVCF; 2/14; 14%), carbamoyl-phosphate synthetase 2, aspartate
transcarbamylase, and dihydroorotase (CAD; 2/14; 14%), neuregulin 1
(NRG1; 2/14; 14%), tumor protein p53 (TP53; 2/14; 14%) and
thioredoxin reductase 2 (TXNRD2; 2/14; 14%) (Fig. 1). ATRX was only identified to be
mutated in one case (1/14; 7%), which was lower than the reported
rate of somatic mutations (13-25%) (5,28).
Fig. 2 illustrates the frequency of
mutations and the resulting protein structure of MEN1 and ADRA2B,
while that of other genes (mutation frequency ≥2/14), including
ARVCF, CAD, NRG1, TP53 and TXNRD2, are presented in Fig. S3.
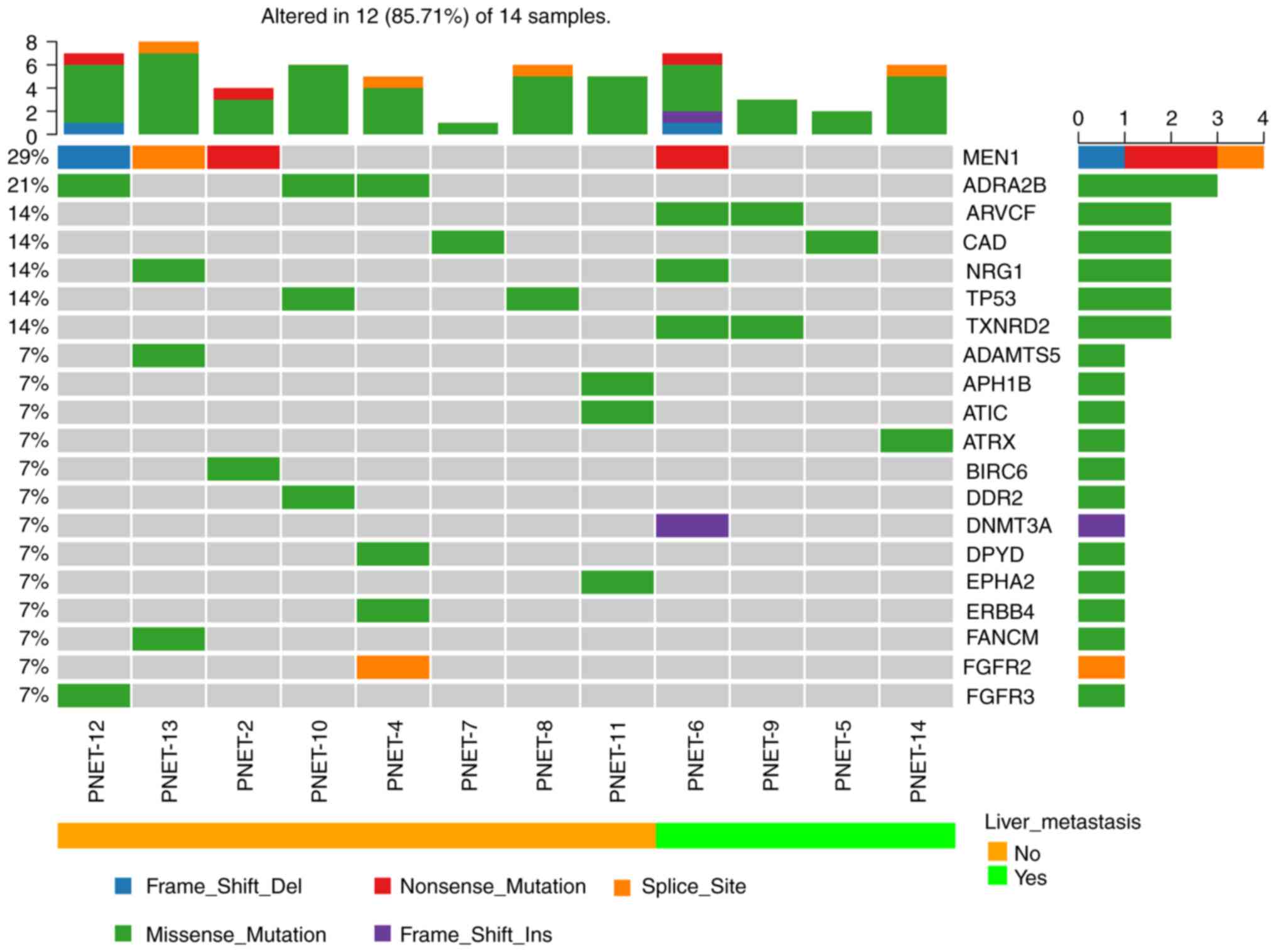 | Figure 1Mutational landscape and tumor
mutational burden in 14 patients with PNET was determined using
target next-generation sequencing. Patients were divided into liver
metastasis and non-liver metastasis groups. PNET, pancreatic
neuroendocrine tumor; MEN1, menin 1; ADRA2B, adrenoceptor alpha 2B;
ARVCF, ARVCF delta catenin family member; CAD, carbamoyl-phosphate
synthetase 2, aspartate transcarbamylase, and dihydroorotase; NRG1,
neuregulin 1; TP53, tumor protein p53; TXNRD2, thioredoxin
reductase 2; ADAMTS5, ADAM metallopeptidase with thrombospondin
type 1 motif 5; APH1B, aph-1 homolog B, gamma-secretase subunit;
ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide
formyltransferase/IMP cyclohydrolase; ATRX, ATRX chromatin
remodeler; BIRC6, baculoviral IAP repeat containing 6; DDR2,
discoidin domain receptor tyrosine kinase 2; DNMT3A, DNA
methyltransferase 3 alpha; DPYD, dihydropyrimidine dehydrogenase;
EPHA2, EPH receptor A2; ERBB4, erb-b2 receptor tyrosine kinase 4;
FANCM, FA complementation group M; FGFR2, fibroblast growth factor
receptor 2; FGFR3, fibroblast growth factor receptor 3. |
KEGG signaling pathway enrichment
analysis of all somatically mutated genes
To further investigate the biological functions of
the mutated genes, KEGG signaling pathway enrichment analyses were
performed. Table SIII presents all
of the significantly enriched signaling pathways associated with
the mutated genes. The top 15 significantly altered signaling
pathways based on p.adjust are presented in Fig. 3A. In total, 11 mutated genes were
identified to be enriched in the PI3K/AKT signaling pathway
(p.adjust=7.12x10-5), including fibroblast growth factor
receptor 3 (FGFR3), Janus kinase 3 (JAK3), integrin subunit alpha V
(ITGAV), EPH receptor A2 (EPHA2),
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
delta (PIK3CD), integrin subunit beta 1 (ITGB1), fibroblast growth
factor receptor 2 (FGFR2), phosphoinositide-3-kinase regulatory
subunit 3 (PIK3R3), erb-b2 receptor tyrosine kinase 4 (ERBB4), TP53
and PTEN. The PI3K/AKT signaling pathway is a well-established
driver of carcinogenesis and has been reported in various types of
cancer, such as gastric (29),
breast (30) and colorectal cancer
(31). Other well-known
cancer-associated signaling pathways were also identified,
including the focal adhesion, ErbB signaling pathway, Ras signaling
pathway and MAPK signaling pathway. Endocrine-related signaling
pathways, including the insulin resistance pathway, were also
identified.
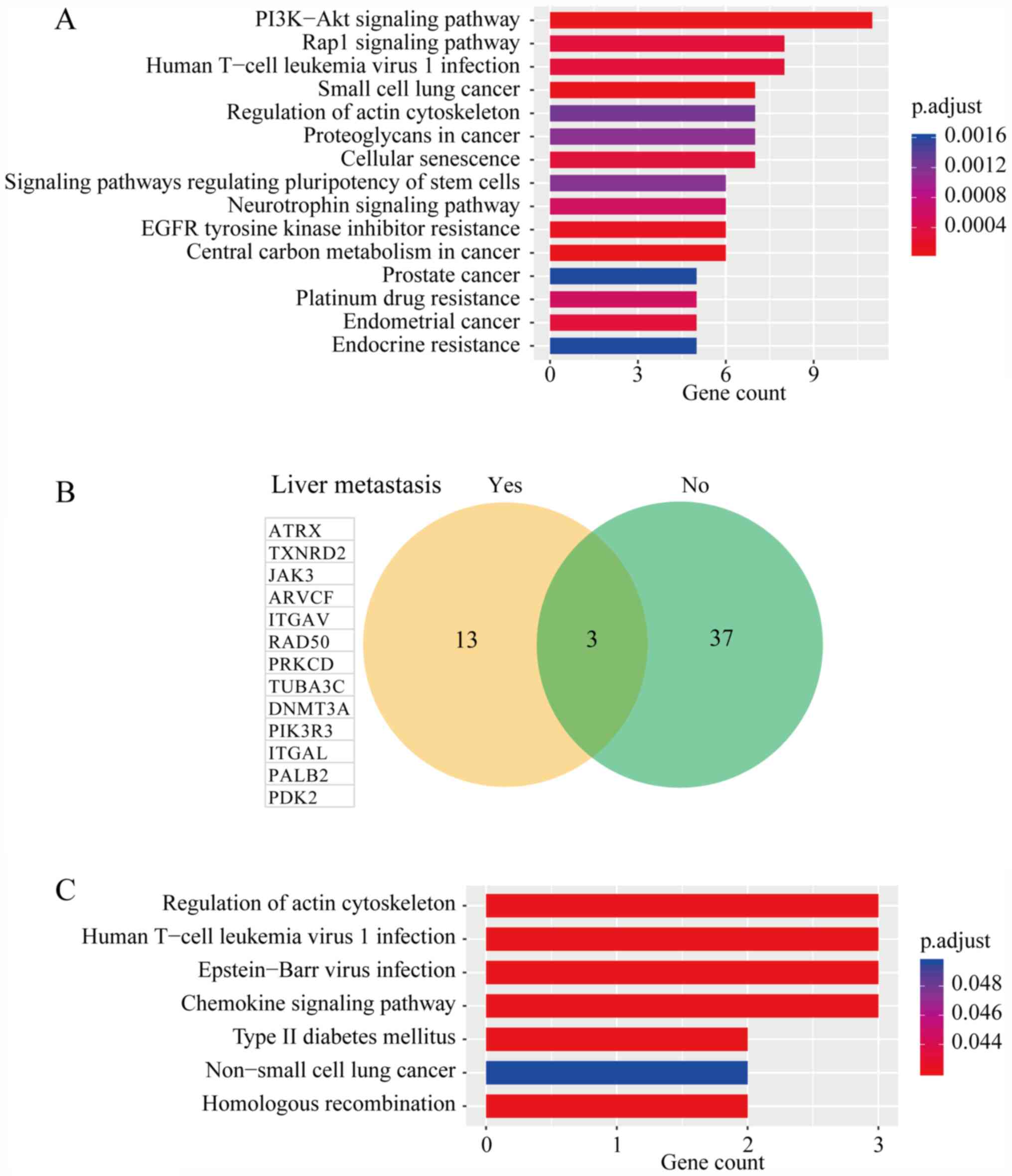 | Figure 3(A) Kyoto Encyclopedia of Genes and
Genomes signaling pathway enrichment analysis of all somatically
mutated genes. (B) Venn diagram of mutated genes between patients
with PNET with or without liver metastasis. (C) Signaling pathways
in which the 13 genes exclusively mutated in patients with PNET who
had developed liver metastasis were enriched. PNET, pancreatic
neuroendocrine tumor; p.adjust, adjusted P-value. ATRX, ATRX
chromatin remodeler; TXNRD2, thioredoxin reductase 2; JAK3, Janus
kinase 3; ARVCF, ARVCF delta catenin family member; ITGAV, integrin
subunit alpha V; RAD50, RAD50 double strand break repair protein;
PRKCD, protein kinase C delta; TUBA3C, tubulin alpha 3c; DNMT3A,
DNA methyltransferase 3 alpha; PIK3R3, phosphoinositide-3-kinase
regulatory subunit 3; ITGAL, integrin subunit alpha L; PALB2
partner and localizer of BRCA2; PDK2, pyruvate dehydrogenase kinase
2. |
Differences in the mutational profile
between two groups with/without liver metastasis
Three genes were discovered to be altered in the two
groups, including MEN1, CAD and NRG1 (Fig. 3B). In addition, 13 genes were
exclusively mutated in those patients with PNET who developed liver
metastasis, including ATRX, TXNRD2, JAK3, ARVCF, ITGAV, RAD50
double strand break repair protein (RAD50), protein kinase C delta
(PRKCD), tubulin alpha 3c (TUBA3C), DNA methyltransferase 3 alpha
(DNMT3A), PIK3R3, integrin subunit alpha L (ITGAL), partner and
localizer of BRCA2 (PALB2) and pyruvate dehydrogenase kinase 2
(PDK2). The results of the KEGG signaling pathway enrichment
analysis of these 13 genes is presented in Table SIV, and it was discovered that the
genes were involved in ‘Homologous recombination’, ‘Chemokine
signaling pathway’ and ‘Type II diabetes mellitus’.
Actionable genomic alterations in
PNETs
To determine the clinical value of these genomic
alterations, the OncoKB-annotator (https://github.com/oncokb/oncokb-annotator) was used
to group all alterations, without filtering the conditions, into
various levels according to the evidence of clinical actionability.
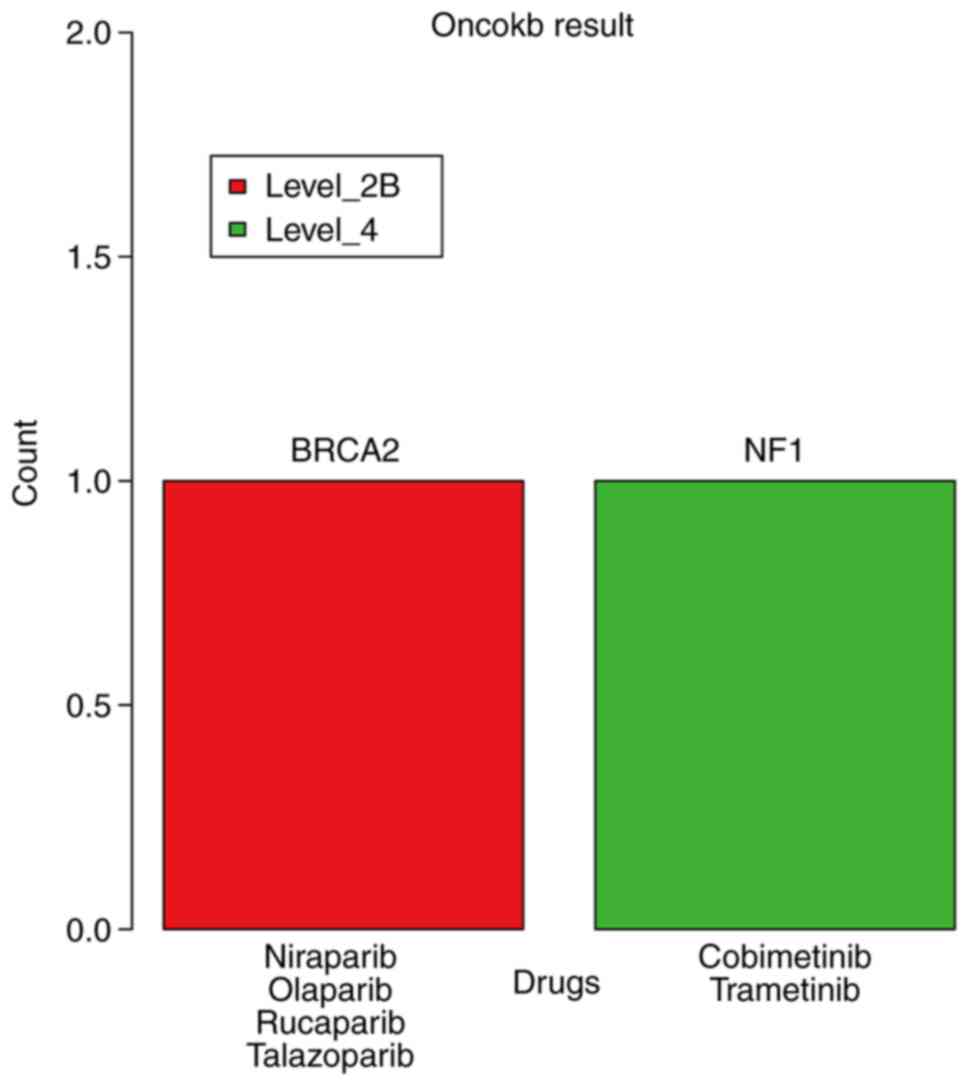
Altogether, two potentially actionable genomic alterations in BRCA2
(p.Q548Q) and neurofibromin 1 (NF1; p.Q1188X) were detected in
patient nos. 14 and 8, respectively (Fig. 4). The evidence level for BRCA2
(p.Q548Q) for patients with PNET was LEVEL_2B, which was defined by
OncoKB as a standard care biomarker recommended by the National
Comprehensive Cancer Network (NCCN, https://education.nccn.org/) or other expert panels as
predictive of a response to a Food and Drug Administration-approved
drug; the corresponding drugs were niraparib, olaparib, rucaparib
and talazoparib. The evidence level for NF1 (p.Q1188X) was LEVEL_4,
which was defined as compelling biological evidence supporting the
biomarker as being predictive of a response to a drug, and the
corresponding drugs were cobimetinib and trametinib.
Discussion
The current treatments for PNET include medical
therapy, surgery and radiotherapy, which are frequently
unsuccessful, as the median survival time for patients with PNET is
as low as ~3.6 years (2). Targeted
therapies may provide a potential benefit for the patients;
however, in order to develop these therapies, an improved
understanding of the genetic landscape of PNET is required. In the
present study, 14 PNET samples were analyzed in-depth by NGS-based
gene panel sequencing; the study identified several novel recurrent
genomic alterations in ADRA2B, ARVCF, CAD and NRG1, as well as the
enrichment of altered genes in the PI3K/AKT signaling pathway.
Mutations in MEN1, DAXX/ATRX and mTOR signaling
pathway genes have been frequently observed in ~55-65% of patients
with non-functional PNET (28). In
the present study, a hybridization capture-based NGS panel was
constructed to detect the genomic alterations in 612
cancer-associated genes. With this method, 14 cases of
nonfunctional PNET with or without liver metastasis were sequenced.
The highest frequency of the altered driver gene, MEN1, was also
identified in the present study cohort, which was altered in four
patients (4/14; 29%). MEN1 encodes menin, a tumor suppressor
associated with a syndrome known as multiple endocrine neoplasia
type 1(32). Its inactivation
drives various phenotypes (4) which
involves widespread transcriptional dysregulation via histone
modifications (33), the activation
of mTOR through AKT expression (34), the suppression of homologous
recombination DNA damage response genes (35) and the dysregulation of telomerase
reverse transcriptase (36). In
addition, the altered genes were discovered to be highly enriched
in the PI3K/AKT signaling pathway; this is consistent with other
previous studies, which reported that mTOR pathway genes were
frequently altered in PNETs and suggested the potential benefits of
mTOR inhibitors, including everolimus, for Chinese patients with
PNET.
In addition, the results of the present study
revealed that the patients with PNETs that developed liver
metastasis had distinct mutational profiles in the primary tumors
compared with cases without liver metastasis. Lawrence et al
(37) reported that there was a
high consistency in the genome sequence, structure and expression
between the primary tumors and hepatic metastases in one patient
with PNET; however, the metastatic tumor lost certain variations or
expression features observed in the primary tumor, whilst gaining
certain de novo changes. In the present study, 13 genes were
identified to be exclusively mutated in the patients who had
developed liver metastasis, which were significantly enriched in 7
signaling pathways. The present study paid close attention to T2DM
(p.adjust=0.030), which was enriched for 2 mutated genes, PRKCD and
PIK3R3. Diabetes mellitus is an endocrine disease, which frequently
occurs in patients with PNET. Long-standing diabetes mellitus has
been discovered as a risk factor for PNET development (38,39),
and has been suggested to be associated with poor prognosis
(40). Fan et al (41) determined that patients with PNET
with T2DM were at an increased risk for tumor metastasis (odds
ratio=2.81; P=0.001) through investigating the clinicopathological
characteristics of 299 patients with PNET. However, in the present
study, the two patients with PNET and T2DM did not demonstrate
liver metastasis. These results may reflect the substantial
research in a large group of patients, which is further required to
determine the relationship between T2DM and liver metastasis in
PNETs.
In the present study, two potentially actionable
genomic alterations in BRCA2 (p.Q548Q) and NF1 (p.Q1188X) were
identified. BRCA2 is one of the most well-described cancer
susceptibility genes, which serves critical roles in homologous DNA
repair. High-risk germline or somatic mutations in BRCA2 usually
lead to the defective repair of double-strand DNA breaks. BRCA2
mutations have been associated with multiple types of cancer,
including breast, ovarian, pancreatic and prostate cancer (42). Several studies or case reports have
reported the existence of BRCA2 germline mutations in PNET
(4,6,43),
which is consistent with the results of the present study and
highlights the germline contribution to clinically sporadic PNETs.
NF1 resides on chromosome 17 and encodes neurofibromin, a 220 kDa
cytoplasmic protein, which is both a negative regulator of Ras and
a positive regulator of adenylyl cyclase, the enzyme responsible
for the generation of intracellular cyclic AMP (cAMP). Therefore,
the loss of NF1 promotes the hyperactivation of downstream effector
proteins of Ras signaling, including mTOR and MAPK kinase (MEK),
and decreased intracellular cAMP levels. Germline mutations in NF1
were discovered to result in neurofibromatosis type 1, a complex
autosomal-dominant disorder that affects multiple organ systems
(44,45). PNET is an uncommon clinical
manifestation of the NF1 syndrome and is reported to be present in
<10% of cases (46). Previously,
in an anecdotal report, the heterozygous germline mutation c.499
del TGTT was identified in a patient with NF1 syndrome and a
well-differentiated pancreatic endocrine carcinoma (47). To the best of our knowledge, the
presence of the NF1 mutant p.Q1188X has not been previously
reported in nonfunctional PNET, which may be due to the difficulty
to identify causative mutations in NF1 due to the large size of the
gene (~60 exons) and the diversity of clinical manifestations.
However, whether this novel alteration of NF1 identified in the
present study is targetable remains elusive. Further functional
annotation is required to guide the clinical application of the MEK
inhibitors, namely cobimetinib and trametinib.
Although the present study provided novel results,
there are several limitations worthy of consideration. One
limitation was the small sample size used in the study; only 14
patients with PNET were included and the present study was a
retrospective study performed at a single center, which limits the
generalizability of the results to the overall population of
patients with PNET. Gene Ontology enrichment also should be used to
investigate the biological functions of the mutated genes in the
future large cohort study. Furthermore, functional studies in
vitro were not performed to determine the biological effects of
these mutated genes, which may also limit the significance of the
results of the present study.
In conclusion, the present study identified several
novel recurrent genomic alterations and confirmed the enrichment of
gene alterations in the PI3K/AKT signaling pathway in a small
cohort of Chinese patients with PNET. These results may shed light
on opportunities for the personalized treatment of sporadic PNETs.
Of note, patients with PNET who developed liver metastasis revealed
distinct mutational profiles compared with cases without
metastasis. Furthermore, two potentially clinically actionable
genomic alterations were identified in BRCA2 and NF1, suggesting
further clinical options of treatment targets in the future.
Supplementary Material
(A and B) Images of H&E-stained
tumor tissues (scale bar, 200 μm). Ki-67 expression levels
were determined using immunohistochemistry. Representative images
with (C) 1%, (D) 5%, (E) 15% and (F) 25% Ki 67 (scale bars, 100
μm).
Summary and visualization of maf files
using the maftools package in R software. (A) Variant
classification. (B) Variant type. (C) SNV class. (D) The count of
variants per sample. (E) Variant classification summary. (F) Top 10
mutated genes. The colors of variant classification in subfigure D,
E and F are in accordance with subfigure A. Del, deletion; Ins,
insertion; SNP, single nucleotide polymorphism; SNV, single
nucleotide variant.
Proportion of mutated genes which were
mutated in ≥2 samples. Del, deletion; MEN1, menin 1; ADRA2B,
adrenoceptor alpha 2B; ARVCF, ARVCF delta catenin family member;
CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase,
and dihydroorotase; NRG1, neuregulin 1; TP53, tumor protein p53;
TXNRD2, thioredoxin reductase 2.
Clinicopathological features and
outcomes of the 14 PNET cases.
MAF file summarising the coding
substitutions and indels present in the 14 pancreatic
neuroendocrine tumors, after filtering conditions.
Signaling pathways enriched by KEGG
analysis of all somatic mutated genes in 14 PNETs patients.
Signaling pathways enriched by KEGG
analysis of specific somatic mutated genes in 4 PNETs patients with
liver metastasis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Special Fund for
Transformation and Application of Precision Medicine of the Second
Military Medical University (grant no. 2017JZ41) and the Major
Projects of Special Development Funds in Zhangjiang National
Independent Innovation Demonstration Zone, Shanghai (grant no.
ZJ2017-ZD-012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the BioProject database (BioProject ID:
PRJNA657158; http://www.ncbi.nlm.nih.gov/bioproject/657158).
Authors' contributions
KZ, TL, JZ, SW, HJ and GJ conceived and designed the
study. YB, CN, HW and YP recruited the patients, performed the
clinical examination, collected and organized the data. KZ, JZ, PM
and SW analyzed and interpreted the data. KZ, TL and JZ wrote the
manuscript. SW, HJ and GJ reviewed and edited the manuscript. All
authors read and approved the manuscript.
Ethics approval and consent to
participate
Ethical approval for the recruitment of human
subjects was obtained from the Ethics Committee of Shanghai
Changhai Hospital (Shanghai, China) and the protocol was in
accordance with the ethical guidelines provided by the Declaration
of Helsinki (1975). Written informed consent was obtained from each
patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hopper AD, Jalal M and Munir A: Recent
advances in the diagnosis and management of pancreatic
neuroendocrine tumours. Frontline Gastroenterol. 10:269–274.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dasari A, Shen C, Halperin D, Zhao B, Zhou
S, Xu Y, Shih T and Yao JC: Trends in the incidence, prevalence,
and survival outcomes in patients with neuroendocrine tumors in the
United States. JAMA Oncol. 3:1335–1342. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dumlu EG, Karakoc D and Özdemir A:
Nonfunctional pancreatic neuroendocrine tumors: Advances in
diagnosis, management, and controversies. Int Surg. 100:1089–1097.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Scarpa A, Chang DK, Nones K, Corbo V,
Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et
al: Whole-genome landscape of pancreatic neuroendocrine tumours.
Nature. 543:65–71. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Raj N, Shah R, Stadler Z, Mukherjee S,
Chou J, Untch B, Li J, Kelly V, Saltz LB, Mandelker D, et al:
Real-time genomic characterization of metastatic pancreatic
neuroendocrine tumors has prognostic implications and identifies
potential germline actionability. JCO Precis Oncol 2018:
PO.17.00267, 2018.
|
|
6
|
Ji S, Yang W, Liu J, Zhao J, Chen L, Ni Q,
Long J and Yu X: High throughput gene sequencing reveals altered
landscape in DNA damage responses and chromatin remodeling in
sporadic pancreatic neuroendocrine tumors. Pancreatology.
18:318–327. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang J, Francois R, Iyer R, Seshadri M,
Zajac-Kaye M and Hochwald SN: Current understanding of the
molecular biology of pancreatic neuroendocrine tumors. J Natl
Cancer Inst. 105:1005–1017. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Batukbhai BDO and De Jesus-Acosta A: The
molecular and clinical landscape of pancreatic neuroendocrine
tumors. Pancreas. 48:9–21. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chan CS, Laddha SV, Lewis PW, Koletsky MS,
Robzyk K, Da Silva E, Torres PJ, Untch BR, Li J, Bose P, et al:
ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a
distinct alpha-cell signature subgroup. Nat Commun.
9(4158)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pipinikas CP, Berner AM, Sposito T and
Thirlwell C: The evolving (epi)genetic landscape of pancreatic
neuroendocrine tumours. Endocr Relat Cancer. 26:R519–R544.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
<gutjnl-2018-insulinomas and
non-functional PNETs.pdf>.
|
|
12
|
Cai H, Jing C, Chang X, Ding D, Han T,
Yang J, Lu Z, Hu X, Liu Z, Wang J, et al: Mutational landscape of
gastric cancer and clinical application of genomic profiling based
on target next-generation sequencing. J Transl Med.
17(189)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rindi G, Bordi C, La Rosa S, Solcia E and
Fave GD: Gruppo Italiano Patologi Apparato Digerente (GIPAD);
Società Italiana di Anatomia Patologica e Citopatologia
Diagnostica/International Academy of Pathology, Italian division
(SIAPEC/IAP). Gastroenteropancreatic (neuro)endocrine neoplasms:
The histology report. Dig Liver Dis. 43 (Suppl 4):S356–S360.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Halfdanarson TR, Strosberg JR, Tang L,
Bellizzi AM, Bergsland EK, O'Dorisio TM, Halperin DM, Fishbein L,
Eads J, Hope TA, et al: The North American neuroendocrine tumor
society consensus guidelines for surveillance and medical
management of pancreatic neuroendocrine tumors. Pancreas.
49:863–881. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Clavé S, Gimeno J, Muñoz-Mármol AM, Vidal
J, Reguart N, Carcereny E, Pijuan L, Menéndez S, Taus Á, Mate JL,
et al: ROS1 copy number alterations are frequent in non-small cell
lung cancer. Oncotarget. 7:8019–8028. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li H and Durbin R: Fast and accurate
long-read alignment with burrows-wheeler transform. Bioinformatics.
26:589–595. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup. The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
García-Alcalde F, Okonechnikov K,
Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF and
Conesa A: Qualimap: Evaluating next-generation sequencing alignment
data. Bioinformatics. 28:2678–2679. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet Chapter.
7(Unit7.20)2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kircher M, Witten DM, Jain P, O'Roak BJ,
Cooper GM and Shendure J: A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet.
46:310–315. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Nagtegaal ID, Odze RD, Klimstra D, Paradis
V, Rugge M, Schirmacher P, Washington KM, Carneiro F and Cree IA:
WHO Classification of Tumours Editorial Board. The 2019 WHO
classification of tumours of the digestive system. Histopathology.
76:182–188. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hong X, Qiao S, Li F, Wang W, Jiang R, Wu
H, Chen H, Liu L, Peng J, Wang J, et al: Whole-genome sequencing
reveals distinct genetic bases for insulinomas and non-functional
pancreatic neuroendocrine tumours: Leading to a new classification
system. Gut. 69:877–887. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cancer Genome Atlas Research Network.
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature. 513:202–209. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cancer Genome Atlas Network. Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cancer Genome Atlas Network. Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Beijers HJBH, Stikkelbroeck NML,
Mensenkamp AR, Pfundt R, van der Luijt RB, Timmers HJLM, Hermus
ARMM and Kempers MJE: Germline and somatic mosaicism in a family
with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur J
Endocrinol. 180:K15–K19. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Matkar S, Thiel A and Hua X: Menin: A
scaffold protein that controls gene expression and cell signaling.
Trends Biochem Sci. 38:394–402. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wang Y, Ozawa A, Zaman S, Prasad NB,
Chandrasekharappa SC, Agarwal SK and Marx SJ: The tumor suppressor
protein menin inhibits AKT activation by regulating its cellular
localization. Cancer Res. 71:371–382. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fang M, Xia F, Mahalingam M, Virbasius CM,
Wajapeyee N and Green MR: MEN1 is a melanoma tumor suppressor that
preserves genomic integrity by stimulating transcription of genes
that promote homologous recombination-directed DNA repair. Mol Cell
Biol. 33:2635–2647. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lin SY and Elledge SJ: Multiple tumor
suppressor pathways negatively regulate telomerase. Cell.
113:881–889. 2003.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lawrence B, Blenkiron C, Parker K, Tsai P,
Fitzgerald S, Shields P, Robb T, Yeong ML, Kramer N, James S, et
al: Recurrent loss of heterozygosity correlates with clinical
outcome in pancreatic neuroendocrine cancer. NPJ Genom Med.
3(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Leoncini E, Carioli G, La Vecchia C,
Boccia S and Rindi G: Risk factors for neuroendocrine neoplasms: A
systematic review and meta-analysis. Ann Oncol. 27:68–81.
2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ben Q, Zhong J, Fei J, Chen H, Yv L, Tan J
and Yuan Y: Risk factors for sporadic pancreatic neuroendocrine
tumors: A case-control study. Sci Rep. 6(36073)2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Gallo M, Ruggeri RM, Muscogiuri G, Pizza
G, Faggiano A and Colao A: NIKE Group. Diabetes and pancreatic
neuroendocrine tumours: Which interplays, if any? Cancer Treat Rev.
67:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Fan Z, Gong Y, Huang Q, Yang C, Cheng H,
Jin K, Fan K, Ni Q, Yu X, Luo G and Liu C: Diabetes is associated
with the metastasis of pancreatic neuroendocrine tumors. Pancreas.
49:751–756. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Maxwell KN and Domchek SM: Cancer
treatment according to BRCA1 and BRCA2 mutations. Nat Rev Clin
Oncol. 9:520–528. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Sharma MB, Carus A, Sunde L,
Hamilton-Dutoit S and Ladekarl M: BRCA-associated
pancreatico-biliary neoplasms: Four cases illustrating the emerging
clinical impact of genotyping. Acta Oncol. 55:377–381.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lin AL and Gutmann DH: Advances in the
treatment of neurofibromatosis-associated tumours. Nat Rev Clin
Oncol. 10:616–624. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gutmann DH, Ferner RE, Listernick RH, Korf
BR, Wolters PL and Johnson KJ: Neurofibromatosis type 1. Nat Rev
Dis Primers. 3(17004)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Frost M, Lines KE and Thakker RV: Current
and emerging therapies for PNETs in patients with or without MEN1.
Nat Rev Endocrinol. 14:216–227. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Perren A, Wiesli P, Schmid S, Montani M,
Schmitt A, Schmid C, Moch H and Komminoth P: Pancreatic endocrine
tumors are a rare manifestation of the neurofibromatosis type 1
phenotype: Molecular analysis of a malignant insulinoma in a NF-1
patient. Am J Surg Pathol. 30:1047–1051. 2006.PubMed/NCBI View Article : Google Scholar
|