Introduction
Hypercapnia and hypercapnic acidosis have protective
effects during lung injury induced by mechanical ventilation,
ischemia-reperfusion, endotoxins and cecal ligations in numerous
experimental models (1). In
clinical treatment, hypercapnia has been linked to improvements in
the outcomes of patients with acute lung injury (ALI)/acute
respiratory distress syndrome (ARDS) (2,3).
Moreover, permissive or therapeutic hypercapnia also exerts
protective effects during ALI, which involve the inhibition of key
transcriptional activators in the NF-κB pathway during inflammation
(2-5).
Our previous study confirmed that implementing acidic
preconditioning (APC) by adjusting the respiratory frequency has a
protective effect on ALI during the peri-operative period of
mechanical ventilation in rat lung ischemia reperfusion (6). APC protocols are easy to operate and
make the levels of hypercapnia controllable, thus increasing their
applicability (6). Collectively,
these aforementioned studies suggested that APC may represent a
clinical application for the treatment of ALI. However, the
mechanism of action through which APC reduces ALI remains poorly
understood.
Angiotensin-converting enzyme (ACE) 2 exerts a
protective effect on the cardiovascular system, liver and lungs
(7-9).
ACE2 inhibits the activity of ACE carboxypeptidase, which produces
angiotensin II, the predominant active peptide of the
renin-angiotensin system (RAS) (10,11).
ACE2 catalyzes the hydrolysis of angiotensin II and thus functions
as a powerful negative regulator of the RAS, that balances the
functions of ACE (12).
Importantly, ACE2 has been identified as the receptor for severe
acute respiratory syndrome coronavirus and the novel coronavirus
disease-19 strain (13,14). Moreover, the downregulation of ACE2
plays an important role in the pathogenesis of severe lung failure
following viral infection (13-15).
Increasing evidence suggests that lipopolysaccharide
(LPS) can induce ALI by downregulating ACE2 and upregulating
angiotensin II (16,17). Interestingly, the pathogenesis
behind ALI/ARDS caused by viral and bacterial lung infection
appears to share a similar mechanism involving reduced levels of
ACE2 and increased levels of angiotensin II, which contribute to
lung damage (18). Although these
published studies established the relationship between LPS-induced
ALI and ACE2, the role of APC on ACE2 in LPS-induced ALI remains
unclear.
The aim of the present study was to examine the
mechanisms of action through which APC alleviates LPS-induced
ALI/ARDS. APC was implemented to both in vivo and in
vitro models of lung injury induced by LPS. The expression of
ACE2 and key inflammatory factors, as well as the severity of lung
injury were measured. In addition, bioinformatics data suggested
that microRNA (miRNA/miR)-200c-3p directly targeted the
3'unstranslated region (UTR) of ACE2. To test the hypothesis that
miR-200c-3p regulates ACE2 expression in APC to alleviate
LPS-induced ALI/ARDS, the effect of miR-200c-3p in LPS-induced lung
injury following APC was evaluated in A549 cells. The functional
role of miR-200c-3p on the regulation of ACE2 expression and the
associated effects on LPS-induced lung injury was examined. As
such, the present study identified a possible mechanism of action
underlying the effects of APC on LPS-induced ALI.
Materials and methods
Model establishment and grouping
The animals were used with approval from the ethics
committee of The First Affiliated Hospital of Nanchang University
and all experiments undertaken in accordance with the relevant
regulations. A total of 30 male Sprague-Dawley rats (Beijing
Huafukang Bioscience Co., Ltd.), weighing 200-280 g and aged 8-10
weeks were placed in a professional animal breeding room, at a
temperature of 18-26˚C and relative humidity of 40-70%, with a 12 h
light/dark cycle and ad libitum access to food and water.
The rats were anesthetized intraperitoneally with 50 mg/kg sodium
pentobarbital and orally intubated with a sterile plastic catheter.
The rats were randomly assigned into control, LPS and APC groups
(n=10 in each group). Rats in the control group were administered
50 µl saline. In the LPS group, ALI was induced by intratracheal
injections of 800 µg LPS dissolved in 50 µl saline (E. coli;
model no. 055:B5; Sigma-Aldrich; Merck KGaA) (19). Rats in the APC group were given
three 5-min cycles of inhalation of mixed gas consisting of 20%
CO2 in normal air, followed by 10 min of recovery
(20). After recovery, rats in this
group also received 800 µg LPS dissolved in 50 µl saline. After the
experiment, the rats were anesthetized intraperitoneally with 50
mg/kg pentobarbital sodium and the right atrium was opened to allow
for the blood to flow until the heart stopped beating and death was
confirmed.
Assessment of lung tissues
H&E staining was performed to observe lung
morphological injury. The right main bronchus was isolated and
cross-clamped 12 h after LPS injection. Under constant pressure of
25 cm H2O, the left lung was filled with 0.5%
low-melting agarose in 10% formalin through a tracheotomy to cause
homogenous expansion of lung parenchyma. Subsequently, 10% formalin
was used for fixation of the inflated lungs for 48 h in room
temperature. The fixed samples were dehydrated by gradient
concentrations of ethanol and paraffin blocks were used for
embedding. H&E (Thermo Fisher Scientific, Inc.) was used to
stain the 5-µm paraffin-embedded tissue sections. The sections were
stained in room temperature as follows: 70% ethanol for 10 sec,
diethyl pyrocarbonate-treated water for 5 sec and hematoxylin with
RNAase inhibitor for 20 sec. Subsequently, 70% ethanol was applied
for 30 sec, eosin Y in 100% ethanol for 20 sec, followed by
dehydration with a series of ethanol for 30 sec each, and finally
xylene for 2 min. Histopathological changes in lung tissues were
observed under light microscope (Olympus Corporation;
magnification, x100 and x400). The inflammatory cell infiltration
was evaluated as a count of the average number of neutrophils in 5
high power fields. The lung injury score was calculated by hyaline
membranes formatting, alveolar wall thickening and airspaces
filling with proteinaceous debris, as previously described
(6). Each item is scored from zero
to four, of which zero points represent normal, one point
represents a very small change, two points represent a slight
change, three points represents moderate change and four points
represent a serious change. The pathological score of lung tissue
was the average of each score. The wet/dry (W/D) lung tissue weight
ratio was used to assess the degree of lung edema (6).
Immunofluorescence microscopy For ACE2
immunofluorescence assessment, a polyclonal rabbit anti-ACE2
antibody (1:1,000; Abcam; cat
no. ab108252) was used. Frozen lung tissues were
sectioned into 6 µm-thick slices, permeabilized (0.25% Triton
X-100), quenched and blocked (10% goat serum) for 1 h at room
temperature, after which samples were incubated with the polyclonal
rabbit anti-ACE2 antibody (1:100 diluted in PBS) overnight at room
temperature. Primary antibody-bound ACE2 was subsequently
visualized using fluorescein isothiocyanate-conjugated anti-rabbit
IgG antibodies (1:1,000; Abcam; cat. no. R330217) for 2 h at room
temperature. The distribution of the target protein in cells was
analyzed by fluorescence microscopy (Leica Microsystems, Inc.;
magnification, x400).
Potential miRNAs that target ACE2 in
ALI
To predict the potential miRNAs that regulate the
expression of the ACE2 protein, TargetScan (Release 7.2: March
2018; http://www.targetscan.org/vert_72/) was used to search
for predicted miRNA targets in the human genome. In addition, the
miR binding site on the ACE2 3'-UTR was identified using TargetScan
(http://www.targetscan.org/cgi-bin/targetscan/vert_72/view_gene.cgi?rs=ENST00000252519.3&taxid=9606&showcnc=0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1).
ACE2 serves an important role in virus-induced lung injury, which
is similar to LPS-induced lung injury (21). The reported top 20 highly expressed
miRNAs in H5N1 virus-infected A549 cells (18) were selected to predict the potential
miRNAs that target ACE2 in LPS-induced lung injury.
Cell culture
The human A549 lung adenocarcinoma epithelial cell
line was obtained from the American Type Culture Collection and
cultured in 5% CO2 at 37˚C in Ham's F12 nutrient medium
(HyClone; Cytiva) supplemented with 10% of FBS (Hyclone; Cytiva),
100 µg/ml of streptomycin and 100 IU/ml of penicillin. For LPS
stimulation, A549 cells were harvested, seeded (500 cells/well) in
6-well plates and incubated overnight at 37C. Samples were then
treated with 1 µg/ml LPS for 24 h. To replicate APC conditions,
cells were exposed to 20% CO2 for 4 h before receiving
LPS 1 µg/ml. The specific inhibitor of NF-κB, caffeic acid
phenethyl ester (CAPE; TargetMol) 75 µmol/l, was added to cells for
2 h before LPS exposure. The native group was cultured without LPS,
the control group was cultured with 1 µg/ml LPS for 24 h and the
CAPE group was cultured with CAPE 75 µmol/l for 2 h, after which 1
µg/ml LPS was applied for 24 h. The APC group was exposed to 20%
CO2 for 4 h, after which 1 µg/ml LPS was applied for 24
h. All treatment and culture incubations were performed at
37˚C.
Cell transfection
A549 cells were transfected with miR-200c-3p mimics
or miR mimics control 50 nmol/l (Shanghai GenePharma Co., Ltd.) at
37˚C for 36 h. Transfections were performed using Lipofectamine
RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. The sequences were as follows:
miR-200c-3p mimics, 5'-UAAUACUGCCGGGUAAUGAUGGA-3'; miR mimics
control, 5'-UUGUACUACACAAAAGUACUG-3'.
Western blot analysis
After transfections, the A549 cells, the cells were
treated as aforementioned (control, LPS and APC groups) and
collected in RIPA lysis buffer (Beijing BLKW Biotechnology Co.,
Ltd.). Protein expression levels were quantified using BCA Protein
Assay kit (Thermo Fisher Scientific, Inc.). Total protein (40 mg)
was loaded onto 10% SDS-PAGE gels. After electrophoresis, the
proteins were transferred to a PVDF supporting membrane. The
membrane was incubated for 12 h at 4˚C with primary antibodies
targeting ACE2 (1:1,000; Abcam; cat. no. ab108252) or β-actin
(1:1,000; Sigma-Aldrich; Merck KGaA). Following 15 min washes with
TBS-0.1% Tween-20. The membranes were blocked with 5% non-fat milk
at room temperature for 1 h and incubated with HRP-conjugated
secondary antibodies (1:10,000; Cell Signaling Technology, Inc.;
cat. no. 7074) for 1 h at room temperature. Blots were developed
using an ECL kit (cat. no. P0018; Beyotime Institute of
Biotechnology). The protein expression was subsequently analyzed
using ImageJ software 1.46 (National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was isolated from the cultured cells using
the Eaststep Total RNA Extraction kit (Promega Corporation)
according to the manufacturer's instructions. Subsequently, RNA was
reverse transcribed using a Transcriptor First-Stand cDNA Synthesis
kit (TransGen Biotech Co., Ltd.) according to the manufacturer's
protocol. qPCR was performed using the TransStart Green qPCR
SuperMix (TransGen Biotech Co., Ltd.) on a DA7600 real-time nucleic
acid amplification fluorescence detection system (Bio-Rad
Laboratories, Inc.). The thermocycling conditions were as follows:
98˚C for 15 min, and 96˚C for 5 min (initiation), 92˚C for 30 sec
(denaturation), 65˚C for 30 sec (annealing) and 62˚C for 1 min
(extension) for 40 cycles. The transcript of the housekeeping gene
GAPDH was quantified as a reference gene. All primers were designed
and optimized using Oligo 7.0 software (Molecular Biology Insights)
and synthesized by Sangon Biotech. The following primers were used
for amplification: i) ACE2 forward, 5'-CATTGGAGCAAGTGTTGGATCTT-3';
and reverse, 5'-GAGCTAATGCATGCCATTCTC-3'; ii) IL-1β forward,
5'-TGGACCTTCC-AGGATGAGGACA-3'; and reverse,
5'-GTTCATCTCGGAGCCTGTAGTG-3'; iii) IL-6 forward,
5'-AGTGAGGAACAAGCCAGAGC-3'; and reverse,
5'-CAGGGGTGGTTATTGCATCT-3'; and iv) GADPH forward,
5'-AGGGCTGCTTTTAACTCTGGT-3'; and reverse,
5'-CCCCACTTGAT-TTTGGAGGGA-3'. The primers for miR-200c-3p (forward,
5'-AGGGCTAATACTGCCGGGTAA-3'; reverse,
5'-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCCATC-3') were
purchased from GenoExplorerTM (GenoSensor Corporation). The U6
small nuclear ribonucleoprotein primer (forward,
5'-CGCTTCGGCAGCACATATAC-3'; reverse, 5'-TTCACGAATTTGCGTGTCAT-3')
was used to normalize miRNA levels. The target gene were
standardized to the Cq values of GAPDH (ΔCq) to calculate the
relative protein expression levels of the target genes, and the
value was determined as 2-ΔΔCq (22).
Statistical analysis
Data are presented as the mean ± SD. Statistical
analysis was carried out using unpaired Student's t-test. One-way
ANOVAs were used to evaluate the effect and pathological
characteristics of chemotherapy. Tukey's tests were used for the
post hoc tests to determine significant differences between groups
following a significant ANOVA result. Data that was not continuous
were analyzed using Kruskal-Wallis tests and Dunn's tests for the
post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
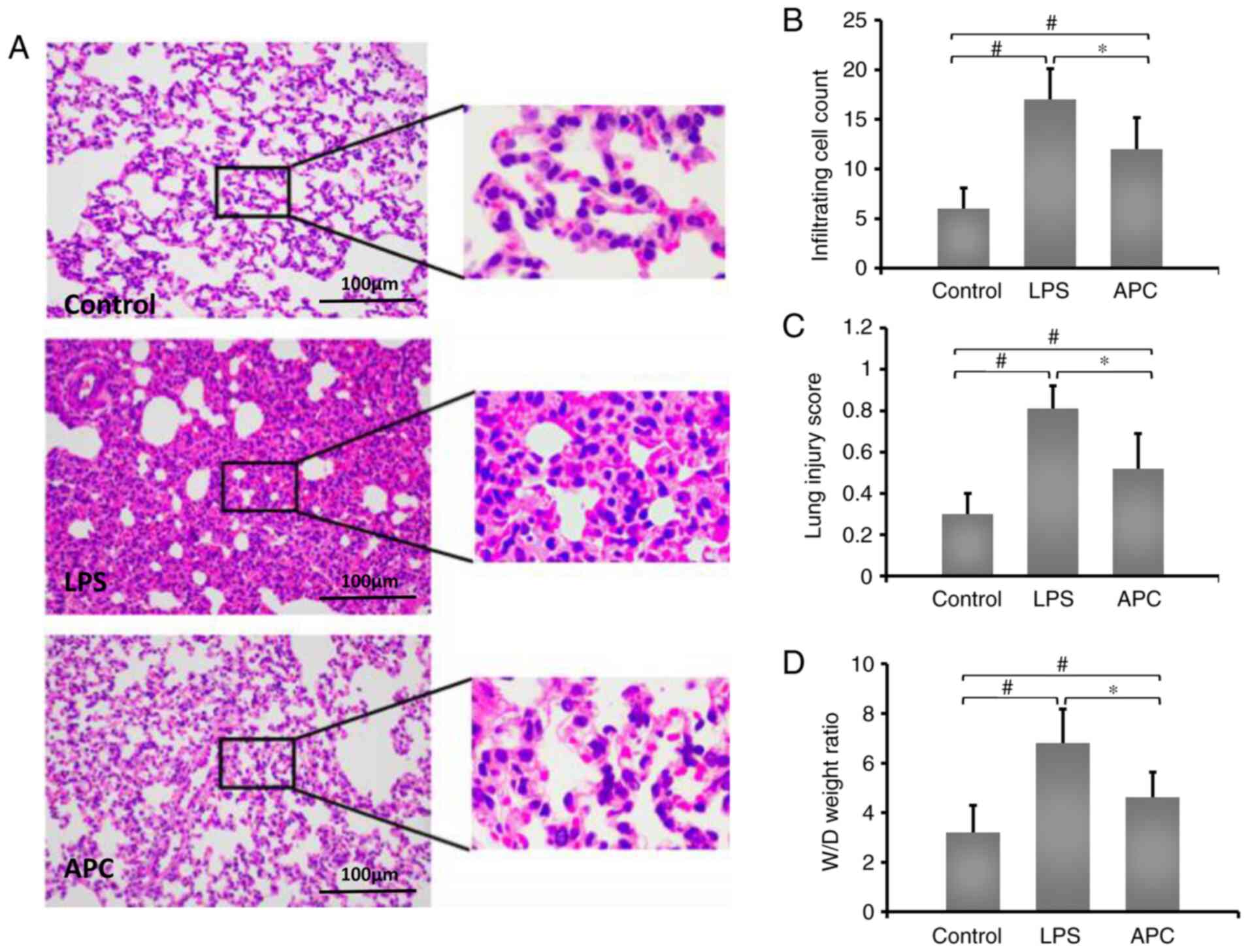
APC reduces acute lung injury induced
by LPS
To identify the effects of APC on LPC-induced acute
lung injury, LPS was administered to rats by intratracheal
injection. The lung tissue was obtained 12 h after injection.
Compared with the control group, LPS induced massive neutrophil
infiltration in alveoli and focal alveolar wall thickenings. In
addition, compared with the LPS group, APC reduced inflammatory
exudation (Fig. 1A). In addition,
the APC group displayed reduced inflammatory cell infiltration
compared with the LPS group (Fig.
1B). The lung injury score also decreased in the APC group
(Fig. 1C). Compared with the LPS
group (6.81±1.37), APC (5.62±1.72) significantly improved the W/D
tissue weight ratio in LPC-induced ALI (Fig. 1D).
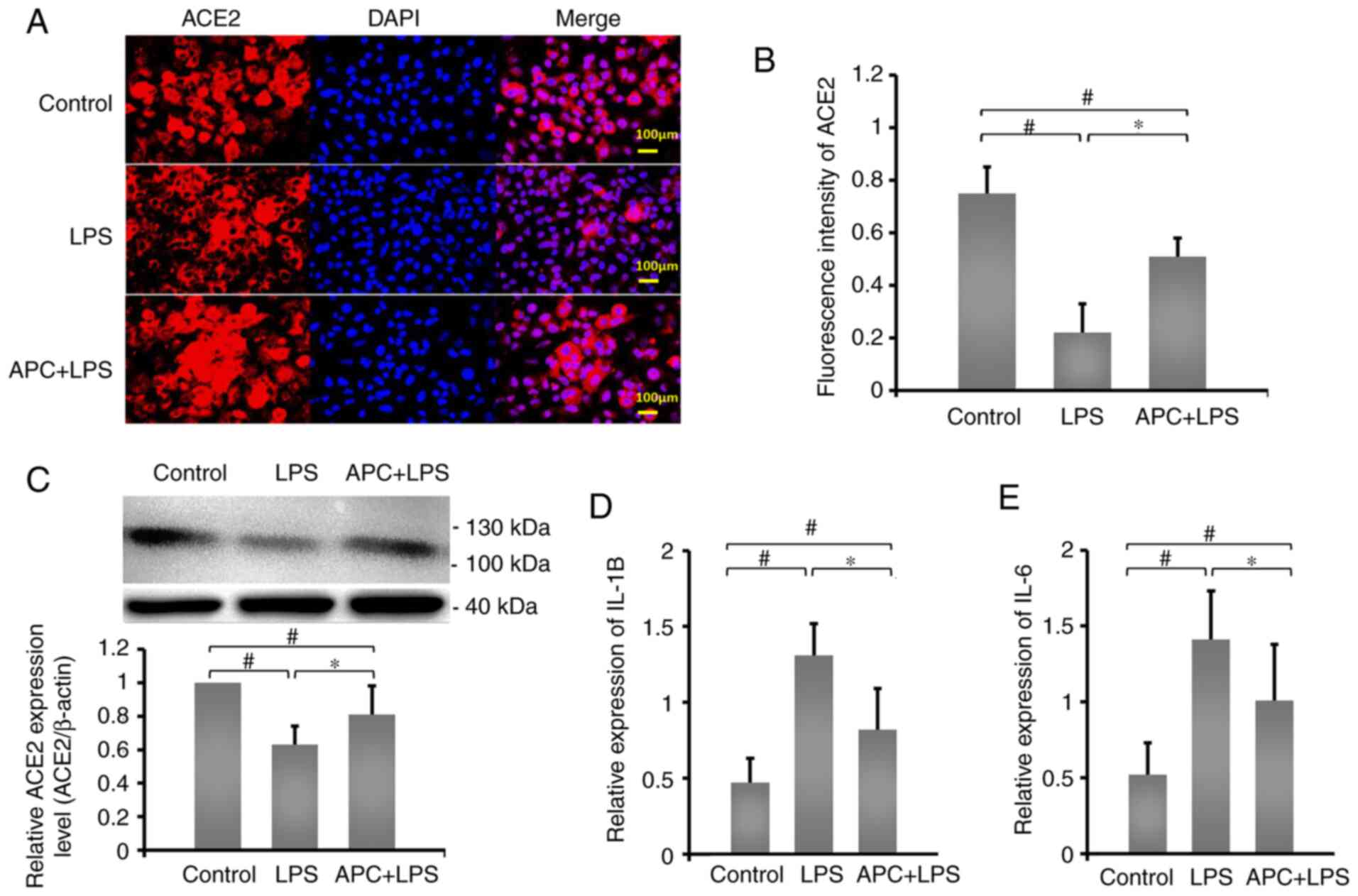
APC inhibits inflammation by reversing
LPS-induced downregulation of ACE2 expression
ACE2 plays an important role in LPS-induced lung
injury (23). In order to determine
the effect of ACE2 protein expression on APC inhibition of
LPS-induced ALI, immunofluorescence staining was used to examine
the expression of ACE2 protein in A549 cells (Fig. 2A). Fluorescence intensity results
demonstrated that, compared with the control group, ACE2 expression
in the LPS group (0.63±0.11) was significantly reduced, while APC
(0.81±0.17) reversed the LPS-induced downregulation of ACE2
expression (Fig. 2B). In addition,
compared with the control group, LPS significantly reduced the
expression levels of ACE2 (Fig.
2C), while IL-1β and IL-6 mRNA expression levels significantly
increased (Fig. 2D and E). The protein expression levels of ACE2
increased in the APC group, while the mRNA expression levels of
IL-1β and IL-6 decreased significantly, compared with the LPS
group. These results demonstrated that APC played a crucial
protective role in regulating the expression of ACE2 and inhibiting
LPS-induced ALI.
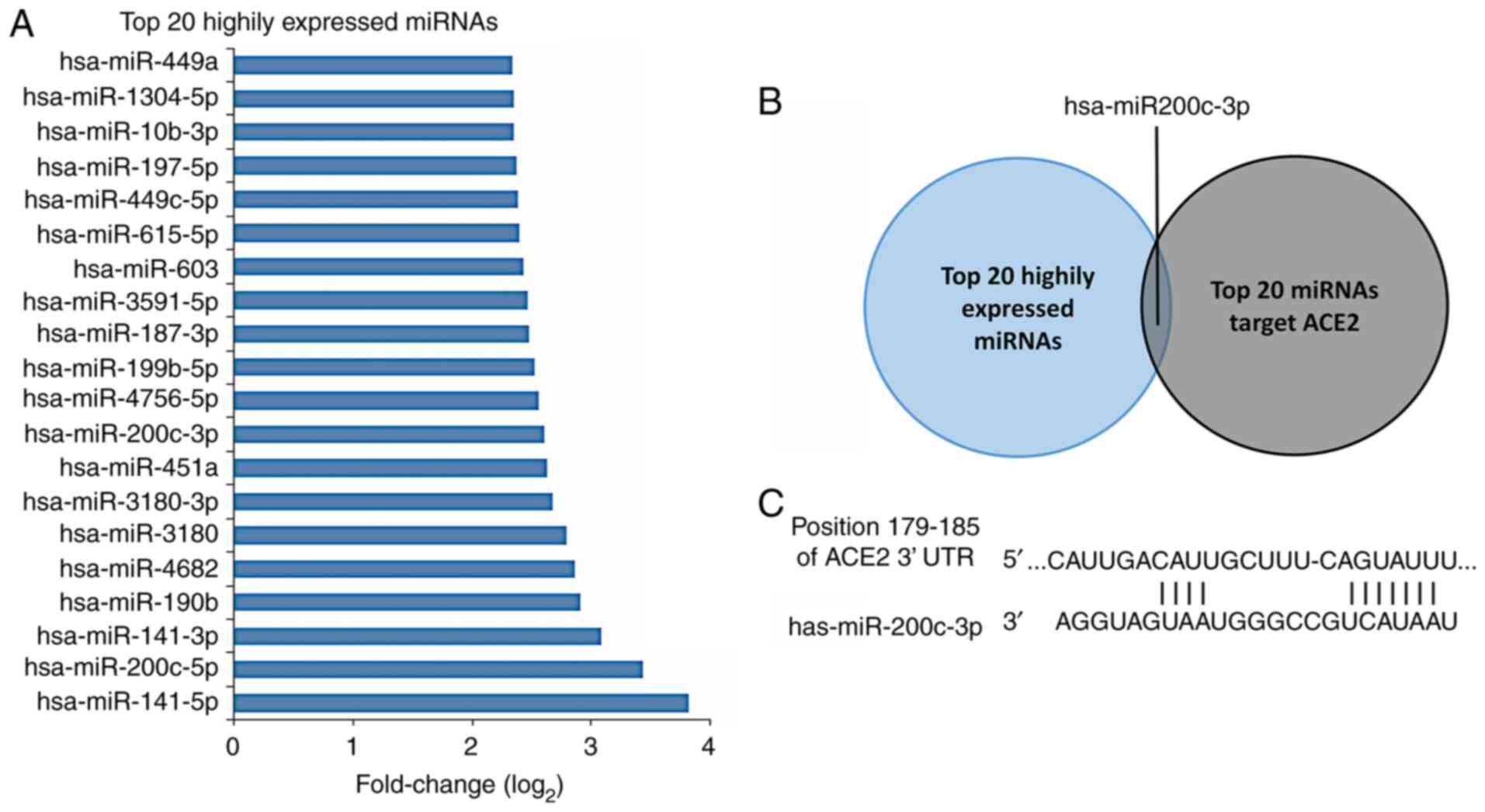
Potential miRNAs that target ACE2 in
ALI
Using TargetScan online tool, 281 miRNAs were
identified that may regulate ACE2 protein, which contained 3
conserved sites and 278 poorly conserved sites (Table S1). The top 20 expressed miRNAs
were chosen for the subsequent predictions. The reported top 20
highly expressed miRNAs in H5N1 virus-infected A549 cells were
selected (18) and the
log2 fold changes of miRNA expression were listed
(Fig. 3A). The intersection between
the top 20 highly expressed miRNAs in virus-infected A549 cells and
the top 20 miRNAs regulating the expression of ACE2 protein was
identified (Fig. 3C). Only one
miRNA matched this condition, namely miR-200c-3p, which was
partially similar to the miRNAs identified in a previous report
(18). miR-200c-3p and miR-141-3p
may have effects on the expression of ACE2 protein during H5N1
virus infection. The miR-200c-3p binding site on the ACE2 3'-UTR
was found and is displayed in Fig.
3C.
Effects of APC on the reduction of
LPS-induced ALI by miR-200c-3p/ACE2
To confirm the bioinformatics analysis, the relative
expression levels of miR-200c-3p and ACE2 were measured using
RT-qPCR (Fig. 4A and B). APC did not demonstrate a marked effect
on normal A549 cells. Following exposure to LPS, the relative
expression levels of miR-200c-3p increased significantly in A549
cells, while the relative expression of ACE2 decreased
significantly. Compared with the control LPS treated group, APC
downregulated the expression of miR-200c-3p and increased the
expression of ACE2 in LPS-treated A549 cells. To demonstrate the
interaction between APC and miR-200c-3p in LPS-induced lung injury,
the miR-200c-3p mimics were successful transfected into A549 cells.
It was found that the relative expression of miR-200c-3p increased
compared to the miR mimic controlled transfected cells (Fig. 4C), indicating that the transfection
was successful. In addition, following miR-200c-3p mimic
transfection in A549 cells, the protein expression levels of ACE2
were significantly downregulated (Fig.
4D), and compared with the native group, APC increased the
expression of ACE2 in LPS-treated A549 cells. This effect was
reversed by miR-200c-3p mimic transfection (Fig. 4E).
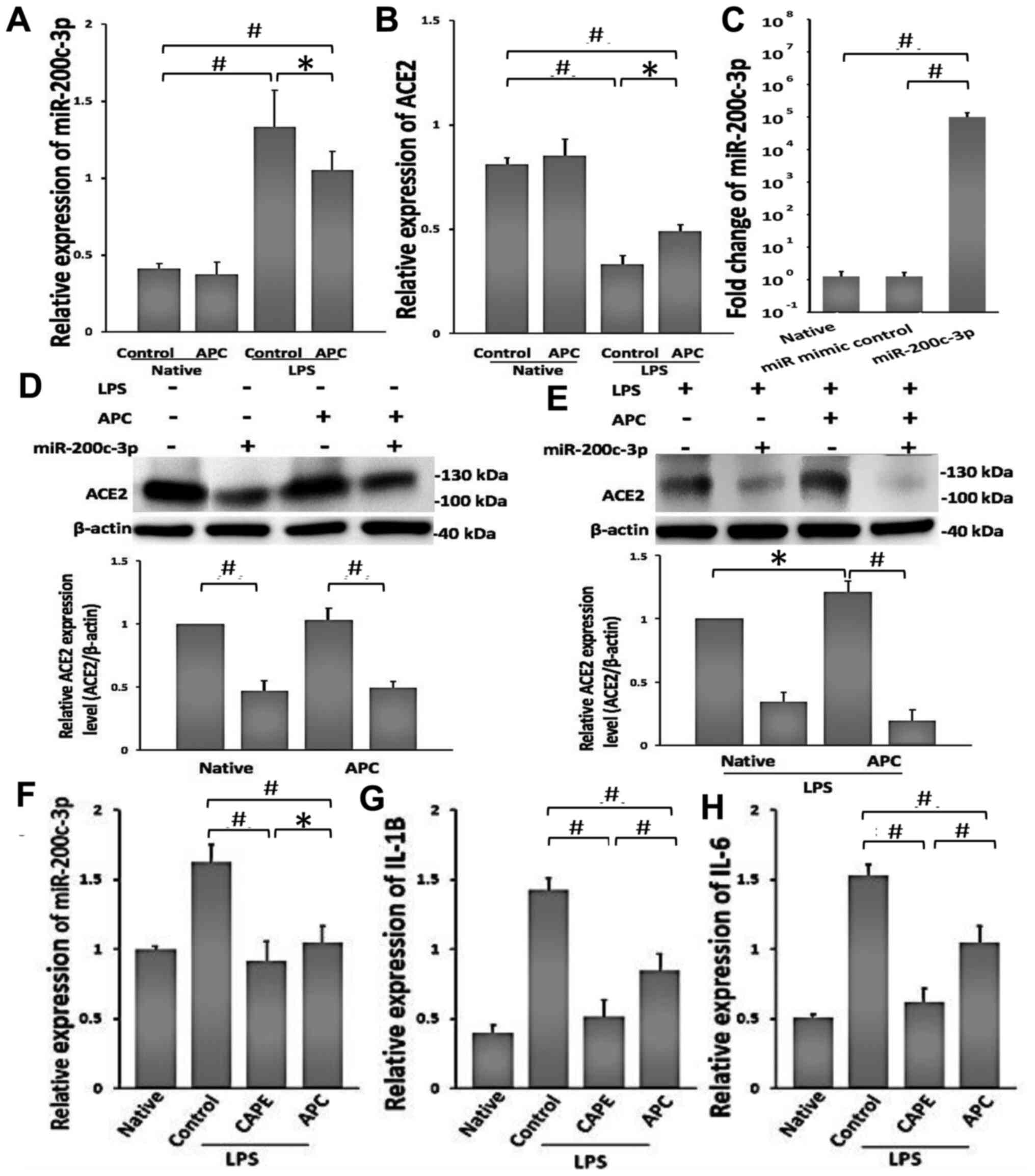 | Figure 4APC protects against LPS-induced lung
injury by downregulating miR-200c-3p and increasing ACE2
expression. (A and B) RT-qPCR analysis of the expression levels of
miR-200c-3p and ACE2 in A549 cells following 24-h treatment with 1
µg/ml LPS. Native, untreated with LPS in A549 cells. (C) The
transfection efficiency of miR-200c-3p mimics was detected using
RT-qPCR. A549 cells were transfected with mimics of miR-200c-3p for
36 h. ACE2 protein expression levels were analyzed following
further treatment (D) without LPS or (E) with LPS, using western
blotting and were normalized to β-actin. Native represents A549
cells without APC treatment. (F) RT-qPCR analysis of miR-200c-3p
levels following NF-κB inhibition. The specific inhibitor of NF-κB,
CAPE, was added to cells for 2 h before LPS exposure. Native
represents cells without LPS treatment. RT-qPCR analysis of the
expression of (G) IL-1β and (H) IL-6 in A549 cells after 2-h
treatment with CAPE. Native represents cells without LPS treatment.
Data are presented as the mean ± SD. n=3. *P<0.05 and
#P<0.01. ACE2, angiotensin-converting enzyme 2; APC,
acid preconditioning; CAPE, caffeic acid phenethyl ester; LPS,
lipopolysaccharide; miR, microRNA; RT-qPCR, reverse
transcription-quantitative PCR. |
Previous studies have demonstrated that NF-κB plays
a role in the regulation of miR-21, miR-125b-1, miR-21, miR-30b,
miR-23b-27b-24-1 and miR-200c-3p expression (18,24,25).
As such, CAPE significantly reduced the levels of miR-200c-3p,
compared with LPS-exposed cells. In addition, APC also resulted in
the inhibition of the upregulation of miR-200c-3p induced by LPS
(Fig. 4F). The expression levels of
inflammatory factors (IL-1β and IL-6) also significantly decreased
following treatment with CAPE in LPS-exposed cells compared with
control group. Moreover, APC similarly decreased the expression
levels of IL-1 β and IL-6 (Fig. 4G
and H), suggesting that inhibition
of NF-κB signaling can downregulate miR-200c-3p, and APC had the
same effect as CAPE, which decreased the expression of miR-200c-3p,
IL-1 β and IL-6. This may represent a mechanism of action
underlying the effects of APC on LPS-induced lung injury.
Discussion
In the present study, APC reduced LPS-induced lung
injury by upregulating ACE2 expression levels and miR-200c-3p
expression was downregulated in this process. These findings
provide insight into the underlying mechanisms of APC in the
treatment of ALI/ARDS, which could present a potential therapeutic
approach for severe pneumonia caused by bacterial LPS or influenza
virus.
The protein encoded by the ACE2 gene belongs to the
ACE family of dipeptidyl carboxydipeptidases and presents homology
with ACE1(26). The function of
ACE2 is to hydrolyze angiotensin I to angiotensin (1-9) or
angiotensin II to angiotensin (1-7). Angiotensin II is a
pro-inflammatory molecule that directly binds to molecules of the
NF-κB pathway and promotes the expression of inflammatory cytokines
(27). Angiotensin (1-7)
specifically binds with the Mas receptor to regulate the PIP3/AKT
and ERK signaling pathways and therefore, exert anti-inflammatory
effects (28,29). In the lung injury model induced by
ACE/angiotensin II, with the activation of the NF-κB pathway, the
expression levels of intercellular cell adhesion molecule-1
increased, resulting in increased vascular permeability and an
increased degree of pulmonary edema (27,30).
The combination of coronavirus and ACE2 leads to a decrease in the
number of available ACE2 molecules, which inhibits the conversion
of angiotensin II to angiotensin (1-7). the angiotensin II produced
by angiotensin I through ACE continues to increase, resulting in
the accumulation of angiotensin II and aggravating inflammation
(31). In the present study, in
acute lung injury models exposed to LPS, the levels of ACE2
decreased and inflammatory factor levels increased, with these
effects being inhibited by APC.
ACE2 has been shown to prevent acute lung injury
caused by coronavirus, sepsis and influenza virus (32-34).
Moreover, these studies demonstrated that avian influenza virus and
bacterial LPS use a similar pathway to reduce ACE2 expression
levels (21,35). miRNAs can regulate gene expression
by binding to the 3'-UTR of target genes (36). In addition, miRNA dysfunction has
been implicated in several diseases, such as lung diseases, heart
disease and diabetes (37-40).
In the present study, the reported top 20 miRNAs in H5N1
virus-infected A549 cells were compared to the top 20 miRNAs
targeting ACE2 and only one miRNA (miR-200c-3p) was identified that
met these two conditions. In addition, following transfection with
miR-200c-3p mimics, the expression levels of ACE2 were
significantly downregulated in A549 cells. Notably, APC induced the
downregulation of miR-200c-3p which was increased following
LPS-induced injury to A549 cells. Moreover, ACE2 expression levels
were also increased in the APC group. These results are consistent
with previous studies (17,22,23),
suggesting that overexpression of ACE2 protects against LPS-induced
lung injury in rats. As such, the present data showed that
miR-200c-3p downregulated the expression of ACE2, and APC
upregulated ACE2 expression by reducing the expression levels of
miR-200c-3p, leading to protective effects against LPS-induced lung
injury.
NF-κB is a critical regulator of inflammation in
ALI. Bacteria and viruses can induce activation of NF-κB, thereby
promoting inflammation (41).
Previous studies have suggested that LPS activates NF-κB through
the activation of Toll-like receptors (41). In LPS-induced lung injury models and
during the process of ARDS, ACE2 mitigates lung injury, likely by
inhibiting NF-κB activation (17).
In the present study, APC reduced the levels of miR-200c-3p and
upregulated the expression of ACE2. Furthermore, the levels of
miR-200c-3p were upregulated following LPS treatment, which was
suppressed by CAPE, an inhibitor of NF-κB (5). In addition, APC has the same
inhibitory effects on NF-κB as CAPE. It has been reported that
hypercapnia has a protective effect on the inhibition of key
transcriptional activators in ALI inflammation through the NF-κB
pathway (3,5) and the present study was consistent
with these findings. As such, the present results may provide a
possible mechanism of action underlying the inhibition of ALI by
APC.
The present study has its limitations. Firstly,
virus-infected cell models were not used in this study. Secondly,
it has been suggested that the LPS-induced ALI model cannot fully
recapitulate the pathology observed in patients with ALI/ARDS
(42,43). Therefore, more clinical experiments
are needed to further clarify the role of APC. In addition, in
clinical practice, not all patients with ALI/ARDS require
ventilator support.
In conclusion, the present study demonstrated that
LPS could inhibit the expression of ACE2 by upregulating the levels
of miR-200c-3p and by suppressing the NF-κB pathway. APC inhibited
this effect, thus reducing the inflammation in LPS-induced ALI. For
patients with ALI who need ventilator support, APC may represent a
novel therapeutic approach with potential beneficial effects in
clinical practice.
Supplementary Material
The miRNAs regulate the ACE2
protein
Acknowledgements
The authors would like to thank Dr Duan Shun and Dr
Lan Xuekai (School of Medicine, Virginia Commonwealth University,
Richmond, Virginia, USA) for their writing assistance.
Funding
This study was supported by the Foundation of Science and
Technology of Jiangxi Province Plan (grant nos. 20202BABL206071 and
20181BBG78022).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
This study conception and design were performed by
LQ. Acquisition of data was performed by GY, YJ, JJH, MDF and YCH.
JZH performed the histological examination of the lung tissues. GY
and LQ confirm the authenticity of all the raw data. Critical
revision of the manuscript was given by all authors. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ijland MM, Heunks LM and van der Hoeven
JG: Bench-to-bedside review: Hypercapnic acidosis in lung
injury-from ‘permissive’ to ‘therapeutic’. Crit Care.
14(237)2010.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Tan J, Liu Y, Jiang T, Wang L, Zhao C,
Shen D and Cui X: Effects of hypercapnia on acute cellular
rejection after lung transplantation in rats. Anesthesiology.
128:130–139. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Shigemura M, Lecuona E and Sznajder JI:
Effects of hypercapnia on the lung. J Physiol. 595:2431–2437.
2017.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Laffey JG and Kavanagh BP: Hypocapnia. N
Engl J Med. 347:43–53. 2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Contreras M, Masterson C and Laffey JG:
Permissive hypercapnia: What to remember. Curr Opin Anesthesiol.
28:26–37. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Qu LC, Jiao Y, Jiang ZJ, Song ZP and Peng
QH: Acidic preconditioning protects against ischemia-reperfusion
lung injury via inhibiting the expression of matrix
metalloproteinase 9. J Surg Res. 235:569–577. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Crackower MA, Sarao R, Oudit GY, Yagil C,
Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang
L, Pei Y, et al: Angiotensin-converting enzyme 2 is an essential
regulator of heart function. Nature. 417:822–828. 2002.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Huentelman MJ, Grobe JL, Vazquez J,
Stewart JM, Mecca AP, Katovich MJ, Ferrario CM and Raizada MK:
Protection from angiotensin II-induced cardiac hypertrophy and
fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp
Physiol. 90:783–790. 2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Abdul-Hafez A, Mohamed T, Omar H, Shemis M
and Uhal BD: The renin angiotensin system in liver and lung: Impact
and therapeutic potential in organ fibrosis. J Lung Pulm Respir
Res. 5(00160)2018.PubMed/NCBI
|
|
10
|
Oudit GY and Penninger JM: Recombinant
human angiotensin-converting enzyme 2 as a new renin-angiotensin
system peptidase for heart failure therapy. Curr Heart Fail Rep.
8:176–183. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chappell MC, Marshall AC, Alzayadneh EM,
Shaltout HA and Diz DI: Update on the Angiotensin converting enzyme
2-Angiotensin (1-7)-MAS receptor axis: Fetal programing, sex
differences, and intracellular pathways. Front Endocrinol
(Lausanne). 4(201)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kuba K, Imai Y, Ohto-Nakanishi T and
Penninger JM: Trilogy of ACE2: A peptidase in the renin-angiotensin
system, a SARS receptor, and a partner for amino acid transporters.
Pharmacol Ther. 128:119–128. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cheng H, Wang Y and Wang GQ:
Organ-protective effect of angiotensin-converting enzyme 2 and its
effect on the prognosis of COVID-19. J Med Virol. 92:726–730.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Contini C, Di Nuzzo M, Barp N, Bonazza A,
De Giorgio R, Tognon M and Rubino S: The novel zoonotic COVID-19
pandemic: An expected global health concern. J Infect Dev Ctries.
14:254–264. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Danser AJ, Epstein M and Batlle D:
Renin-angiotensin system blockers and the COVID-19 pandemic: At
present there is no evidence to abandon renin-angiotensin system
blockers. Hypertension. 175:1382–1385. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen Q, Liu J, Wang W, Liu S, Yang X, Chen
M, Cheng L, Lu J, Guo T and Huang F: Sini decoction ameliorates
sepsis-induced acute lung injury via regulating ACE2-Ang (1-7)-Mas
axis and inhibiting the MAPK signaling pathway. Biomed
Pharmacother. 115(108971)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang
M, Xue Y, Xi C, Zhou M and Jiang W: Angiotensin-converting enzyme 2
prevents lipopolysaccharide-induced rat acute lung injury via
suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep.
6(27911)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu Q, Du J, Yu X, Xu J, Huang F, Li X,
Zhang C, Li X, Chang J, Shang D, et al: miRNA-200c-3p is crucial in
acute respiratory distress syndrome. Cell Discov.
3(17021)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang X, Huang H, Yang T, Ye Y, Shan J,
Yin Z and Luo L: Chlorogenic acid protects mice against
lipopolysaccharide-induced acute lung injury. Injury. 41:746–752.
2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang CH, Fan YY, Wang XF, Xiong JY, Tang
YY, Gao JQ, Shen Z, Song XH, Zhang JY, Shen Y, et al: Acidic
preconditioning protects against ischemia-induced brain injury.
Neurosci Lett. 523:3–8. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gu H, Xie Z, Li T, Zhang S, Lai C, Zhu P,
Wang K, Han L, Duan Y, Zhao Z, et al: Angiotensin-converting enzyme
2 inhibits lung injury induced by respiratory syncytial virus. Sci
Rep. 6(19840)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ye R and Liu Z: ACE2 exhibits protective
effects against LPS-induced acute lung injury in mice by inhibiting
the LPS-TLR4 pathway. Exp Mol Pathol. 113(104350)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Niu J, Shi Y, Tan G, Yang CH, Fan M,
Pfeffer LM and Wu ZH: DNA damage induces NF-κB-dependent
microRNA-21 up-regulation and promotes breast cancer cell invasion.
J Biol Chem. 287:21783–21795. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhou R, Hu G, Liu J, Gong AY, Drescher KM
and Chen XM: NF-kappaB p65-dependent transactivation of miRNA genes
following Cryptosporidium parvum infection stimulates epithelial
cell immune responses. PLoS Pathog. 5(e1000681)2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Minato T, Nirasawa S, Sato T, Yamaguchi T,
Hoshizaki M, Inagaki T, Nakahara K, Yoshihashi T, Ozawa R, Yokota
S, et al: B38-CAP is a bacteria-derived ACE2-like enzyme that
suppresses hypertension and cardiac dysfunction. Nat Commun.
11(1058)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Celec P: Nuclear factor kappa B-molecular
biomedicine: The next generation. Biomed Pharmacother. 58:365–371.
2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kvandova M and Dovinova I: Functioning of
the PPAR gamma and its effect on cardiovascular and metabolic
Diseases. In: Metabolic Syndrome, edition: 160 Greentree Drive,
Suite 101, Dover, DE 19904, USA, pp1-41, 2017 http://smgebooks.com/metabolic-syndrome/index.php.
|
|
29
|
Passos-Silva DG, Verano-Braga T and Santos
RA: Angiotensin-(1-7): Beyond the cardio-renal actions. Clin Sci.
124:443–456. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rodrigues Prestes TR, Rocha NP, Miranda
AS, Teixeira AL and Simoes-E-Silva AC: The anti-inflammatory
potential of ACE2/angiotensin-(1-7)/mas receptor axis: evidence
from basic and clinical research. Current Drug Targets.
18:1301–1313. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kuba K, Imai Y, Rao S, Jiang C and
Penninger JM: Lessons from SARS: Control of acute lung failure by
the SARS receptor ACE2. J Mol Med. 84:814–820. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zou Z, Yan Y, Shu Y, Gao R, Sun Y, Li X,
Ju X, Liang Z, Liu Q, Zhao Y, et al: Angiotensin-converting enzyme
2 protects from lethal avian influenza A H5N1 infections. Nat
Commun. 5(3594)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wang J, Zhao S, Liu M, Zhao Z, Xu Y, Wang
P, Lin M, Xu Y, Huang B, Zuo X, et al: ACE2 expression by colonic
epithelial cells is associated with viral infection, immunity and
energy metabolism. medRxiv, Feb 7, 2020 doi: https://doi.org/10.1101/2020.02.05.20020545.
|
|
34
|
Gralinski LE, Sheahan TP, Morrison TE,
Menachery VD, Jensen K, Leist SR, Whitmore A, Heise MT and Baric
RS: Complement activation contributes to severe acute respiratory
syndrome coronavirus pathogenesis. MBio. 9:e01753–18.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan
B, Yang P, Sarao R, Wada T, Leong-Poi H, et al:
Angiotensin-converting enzyme 2 protects from severe acute lung
failure. Nature. 436:112–116. 2005.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ebert MS and Sharp PA: Emerging roles for
natural microRNA sponges. Curr Biol. 20:R858–R861. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Christopher AF, Kaur RP, Kaur G, Kaur A,
Gupta V and Bansal P: MicroRNA therapeutics: Discovering novel
targets and developing specific therapy. Perspect Clin Res.
7:68–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Magenta A, Greco S, Gaetano C and Martelli
F: Oxidative stress and microRNAs in vascular diseases. Int J Mol
Sci. 14:17319–17346. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhang HN, Xu QQ, Thakur A, Alfred MO,
Chakraborty M, Ghosh A and Yu XB: Endothelial dysfunction in
diabetes and hypertension: Role of microRNAs and long non-coding
RNAs. Life Sci. 213:258–268. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Caballero AE: Endothelial dysfunction in
obesity and insulin resistance: A road to diabetes and heart
disease. Obes Res. 11:1278–1289. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Alexopoulou L, Holt AC, Medzhitov R and
Flavell RA: Recognition of double-stranded RNA and activation of
NF-κB by Toll-like receptor 3. Nature. 413:732–738. 2001.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Uchida T, Shirasawa M, Ware LB, Kojima K,
Hata Y, Makita K, Mednick G, Matthay ZA and Matthay MA: Receptor
for advanced glycation end-products is a marker of type I cell
injury in acute lung injury. Am J Respir Criti Care Med.
173:1008–1015. 2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Proudfoot AG, McAuley DF, Griffiths MJ and
Hind M: Human models of acute lung injury. Dis Models Mech.
4:145–153. 2011.PubMed/NCBI View Article : Google Scholar
|