Introduction
Hereditary anemias are a group of common but highly
pleiotropic disorders, exhibiting great molecular disparity. As
highly heterogenous hematological disorders (1), the clinical knowledge of hereditary
anemias remains poor. To date, ≥70 genes have been identified to be
involved in anemia (2). Mutations
in these genes occur with low to rare frequency. The mechanisms
underlying the onset of anemias include deficiencies in
regeneration capacity, red blood cell (RBC) deformities, enzymatic
deficiencies and other aspects of hematopoiesis. Precise diagnosis
is of paramount importance, since the therapeutic approaches for
anemias and contraindications are distinctly different (3). Furthermore, verifying genetic defects
in adults is always challenging, largely due to the low penetrance
rate, pleiotropic expression and multifactorial mechanisms.
Therefore, the diagnosis and management of anemia remain
challenging.
Drug-induced liver injury (DILI) is a potentially
serious adverse reaction (4,5).
Current diagnostic scales heavily rely on causality assessment,
such as the Roussel Uclaf Causal Relationship Assessment Method
(RUCAM) scoring system (6),
partially due to the lack of knowledge on its determinants.
Downstream intrahepatic cellular events such as oxidative stress
may aggravate liver injury following the initial insult (7,8). The
phenotype of DILI may mimic almost any other liver disorder. The
clinical presentation may vary significantly among treated
patients, even following treatment with the same substance.
Therefore, determining protective and predisposing factors remains
difficult. The overlap of DILI with a rare form of hereditary
anemia has not been previously reported. The present study reported
on a case of DILI presenting with bland cholestasis in the context
of a chronic rare anemia. The potential interactions between these
two disorders were evaluated and discussed.
Materials and methods
Clinical presentation
An otherwise healthy 21-year-old male was initially
admitted to Ruijin Hospital (Shanghai, China) in March 2020 due to
afebrile painless jaundice for 20 days. The patient was in a normal
state until he underwent laparoscopic cholecystectomy after several
episodes of abdominal pain and cholelithiasis in September 2019. No
transfusion was required during the surgery. Ιn October 2019, the
patient developed generalized pruritus of unclear etiology. The
patient underwent self-treatment with ephedra (9), which slightly improved the symptoms.
In the meantime, the patient also complained of fatigue and
anorexia. The patient noticed yellowish eyes and dark urine, which
prompted a clinical evaluation. In addition, the patient reported
pale stool, which was not observed during hospitalization. The
paternal grandfather of the patient had died of cirrhosis at 36
years of age due to unknown etiology. Apart from the above medical
and surgical history, the patient's medical history was
unremarkable. The patient had no allergies and did not smoke or
drink alcohol. On arrival, the patient's vital signs were stable
and normal and he was conscious. On examination, the patient had
icterus with jaundiced skin, with no visible lesions. The abdomen
was distended and the spleen and liver were non-palpable below the
costal margin. The remainder of the physical examination was
unremarkable. Written informed consent for data usage in scientific
research, including genetic testing and publication of the results
was provided by the patient and his parents.
Next-generation sequencing and Sanger
confirmation
Given the family history of cirrhosis, whole-exome
sequencing of the pedigree, including the proband and his parents
was performed to detect any possible genetic abnormalities. Genomic
DNA was extracted using QIAamp DNA Blood Mini Kit (Qiagen, Inc.)
from peripheral blood and was randomly fragmented using Covaris
S220 Focused-ultrasonicator system (Covaris Inc.) to an average
size of 180-280 bp. Libraries were constructed using paired-end
protocols from Illumina, Inc. The fragmented DNA was captured using
the Agilent SureSelect Human All ExonV6 kit (Agilent Technologies
Inc.) and sequencing was performed on the Illumina Novaseq 6000
platform (Illumina Inc.). Paired-end 150 bp-long reads with a
coverage of ≥10X for 99% of the genome (mean coverage, x100) were
generated. Candidate variants, amplified by PCR, were confirmed by
Sanger sequencing according to Applied Biosystems®
3730/3730xl DNA Analyzers User Guide on the ABI 3730 Prism DNA
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.)
using two sets of self-designed primer pair (first pair:
5'-GGTGATATCTCCCCCTGCTC-3' and 5'-GCTTACTGCTGCGAGAACTG-3'; second
pair: 5'-TTATCCTGCCCAAGTCCCAG-3' and 5'-ATTTGGGGGATCAGGCAAGA-3')
encompassing the region of interest.
Filtering of rare variants and
pedigree analysis
Rare exonic variants with a minor allele frequency
of <0.01 were selected from the 1000 Genomes Project (10), the Exome Sequencing Project (ESP;
esp6500siv2, https://esp.gs.washington.edu/drupal/) and The Genome
Aggregation Database (gnomAD release v3.1, https://gnomad.broadinstitute.org/). Known pathogenic
dominant or recessive variations, homozygous or compound
heterozygous, as well as de novo variants, were all
considered as candidate variations in the pedigree analysis.
Liver biopsy
Ultrasound-guided liver biopsy was performed and the
specimen was stained with hematoxylin and eosin (H&E) and
Masson trichrome (11) using
standard protocols. Liver biopsy sample was fixed in 10% neutral
phosphate buffered formalin at room temperature for 2 h. Xylene and
ethanol gradient method was applied in the dehydration and
rehydration process. Paraffin was used as embedding reagent and the
thickness of slices was 3-5 µm. The cells were stained sequentially
with hematoxylin and eosin at room temperature for 4 and 10 min,
respectively. The slides were observed under x100 and x400
magnification using light microscopy. Slides were evaluated by one
experienced pathologist who was blinded to the clinicopathological
data of the patient.
Treatment and follow-ups
The patient was treated with ursodeoxycholic acid
(UDCA) 250 mg t.i.d. po, starting on day 4 from
hospitalization, for one month. At 3 and 6 months after discharge,
follow-up checks, including abdominal ultrasound, complete blood
count, liver and renal tests, were performed.
Results
Clinical and laboratory findings
Examination of the otherwise healthy young male
patient with jaundice for 20 days and pruritus for 6 months
following cholecystectomy revealed potential ephedra-induced liver
injury with iron overload. In contrast to the obvious cholestasis,
the liver injury was limited compared with other reported cases
(9,12), where patients were also self-treated
with the same medication. CT scans indicated no focal lesions in
the liver. Magnetic resonance cholangiopancreatography revealed a
slightly dilatated post-cholecystectomy common bile duct with no
evidence of choledocholithiasis (Fig.
1), moderate hepatosplenomegaly and intrahepatic lymphatic
stasis. Furthermore, laboratory testing revealed hyperbilirubinemia
(total/direct bilirubin, 253.8/134.9 µmol/l; normal range,
4.7-24/0-6.8 µmol/l), increased bile acid levels (338.6 µmol/l;
normal range, 1.0-10.0 µmol/l), and normal prealbumin, alanine
transaminase, aspartate aminotransferase, gamma-glutamyl
transferase and alkaline phosphatase levels, prothrombin time and
international normalized ratio value. The model for end-stage liver
disease (MELD) score (13) was 17
with an R-factor value of 0.7. Viral and autoimmune hepatitis were
excluded. RUCAM scoring did not exclude DILI (6). Of note, the patient also suffered from
chronic and mild hemolytic anemia with no identifiable cause
(hemoglobin level, 109 g/l; normal range, 131-172 g/l) with an
elevated mean corpuscular volume (91 fl), low haptoglobin level
(<0.1 g/l; normal range, 0.3-2.0 g/l) and moderate elevated
percentages of reticulocytes (2.5%; normal range, 0.5-1.5%). Other
findings included hyperferritinemia (623 ng/ml), increased
transferrin saturation (58.6%) and normal ceruloplasmin levels. RBC
size variations may be readily observed under a microscope. During
hospitalization, the RBC distribution width (15.3-17.8%; normal
range, 11.6-14.0%) and the percentage of basophils (1.6-2%; normal
range; <1.0%) were consistently elevated. The levels of lactate
dehydrogenase (LDH) were marginally increased, while those of
ammonia were normal. These results indicated chronic mild to
moderate macrocytic anemia. However, the Coombs tests (IgG and
C3d), Isopropanol test, Ham's test, Glucose-6-phosphate
dehydrogenase (G6PD) deficiency test and pyruvate kinase enzymatic
activity were negative for autoimmune hemolytic anemia. Finally,
high-performance liquid chromatography indicated normal levels of
hemoglobin α (HbA), HbA2 and fetal hemoglobin, whilst no visible
inclusion body was observed inside RBCs.
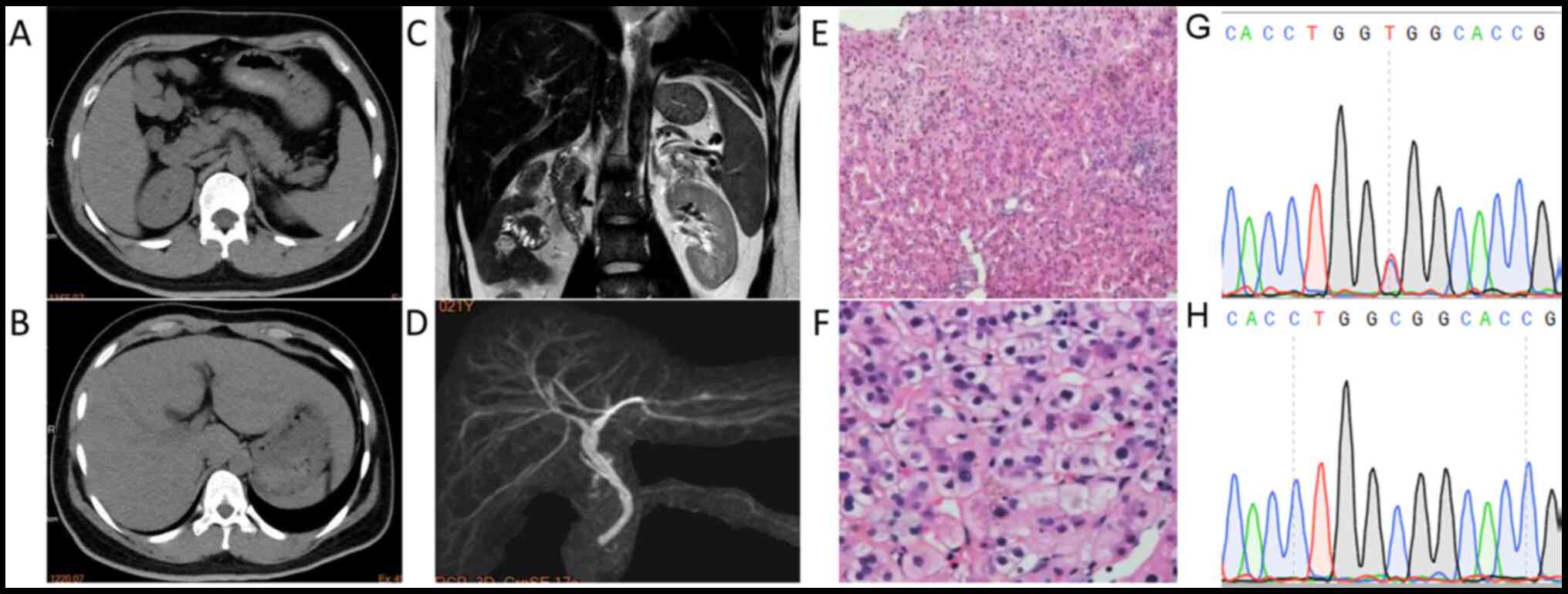 | Figure 1CT/MRCP and liver biopsy indicated
characteristics of cholestasis with mild inflammatory events. CT
scan revealed a slightly enlarged (A) spleen and (B) liver. (C)
MRCP and (D) 3D reconstruction indicated slightly dilated
intrahepatic bile ducts with no evidence of lithiasis (sequence
parameters: T2-weighted; echo time, 120 msec; repetition time,
1,100 msec). (E) H&E staining of the liver biopsy specimen
revealing central lobular focal necrosis with mild portal
inflammation (magnification, x100). (F) H&E staining indicating
cholestasis with dilated canaliculi, bile plug, apoptotic bodies
and mild microvesicular steatosis (magnification, x400). (G) The C
to T nucleotide substitution in the CDAN1 gene, causing
R1067H, was verified by Sanger sequencing. (H) Sanger sequencing of
the wild-type CDAN1 gene from the mother. CT, computed
tomography; MRCP, magnetic resonance cholangiopancreatography;
H&E, hematoxylin and eosin; CDAN1, codanin 1. |
Liver biopsy and pathology
Staining of liver specimens with H&E revealed
that the lobular structure remained intact, while intrahepatocyte
cholestasis and bile plug were observed in the dilated canaliculus.
In addition, a small number of foci of intralobular necrosis,
apoptotic bodies, microvesicular steatosis and minimal portal
inflammation were observed. The Masson trichrome staining was
normal. The liver tissues were negative for hepatitis B surface and
core antigens and for copper. Staining for cytokeratin 19 revealed
slight ductular proliferation. The pathological data suggested that
the patient suffered from acute cholestatic hepatitis, most likely
caused by DILI (Fig. 1).
Disease-causing variation screening
results
In addition to common homozygous polymorphisms in
cholestasis-associated genes, including ATP binding cassette
subfamily B member 11 (ABCB11) and tight junction protein 2
(TJP2), the pedigree genetic study revealed compound
heterozygous variations in the congenital anemia-causing gene
codanin 1 (CDAN1), including a novel rare mutation of
paternal origin, namely p.R1067H (ClinVar accession no.
SCV001448675). Apart from anemia, defects in the CDAN1 gene
have also been associated with jaundice, iron overload,
splenomegaly (14) and gallstones
(Table I) (15). All of the aforementioned
abnormalities were present in the patient of the present study.
 | Table IHomozygous gene polymorphisms
associated with cholestasis and congenital anemia causing
mutations. |
Table I
Homozygous gene polymorphisms
associated with cholestasis and congenital anemia causing
mutations.
| A, Common
cholestasis-associated polymorphisms |
|---|
| | Minor allele
frequency | |
|---|
| Gene | AA Change | GnomAD | 1000g_Chinese | Disease (ID: OMIM or
HGMD) | Inheritance
pattern | Note |
|---|
| ABCB11 | p.V444A | 0.568838 | 0.664452 | Cholestasis, benign
recurrent intrahepatic, 2 (605479); cholestasis, drug-induced,
association with (CM071525) | Autosomal
recessive | Co-existed with
another heterozygous cholestasis-related polymorphism,
p.A1028A |
| TJP2 | p.T909T p.A913A | 0.265188
0.264951 | 0.460133
0.458472 | Cholestasis,
progressive familial intrahepatic 4 (615878); hypercholanemia,
familial (607748) | Autosomal
recessive | - |
| B, Congenital
anemia-associated mutations |
| | Minor allele
frequency | |
| Gene | AA Change | GnomAD | 1000g_Chinese | Disease (ID: OMIM or
HGMD) | Inheritance
pattern | Note |
| CDAN1 | p.R1067H | / | Novel | Dyserythropoietic
anemia, congenital, type Ia (224120) | Autosomal
recessive | Paternal origin
co-existed with another two maternal heterozygous variations with
unknown significancea |
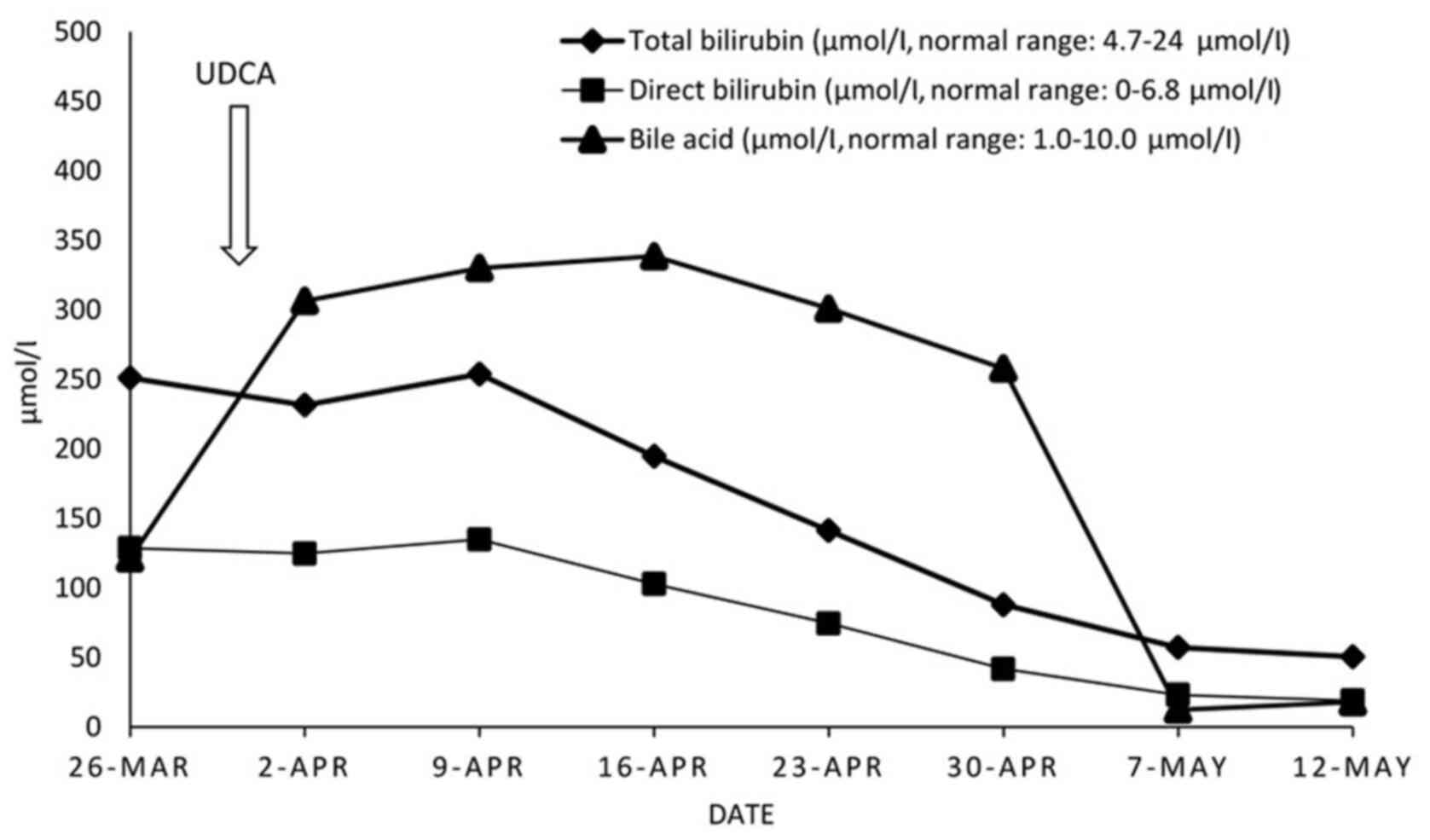
Treatment and outcome
The patient was diagnosed with drug-induced bland
cholestasis, with iron overload in the context of a rare form of
congenital anemia. Since anemia was mild, the patient was treated
only with UDCA, and cholestasis and pruritus swiftly subsided 2
weeks later (Fig. 2). Three and six
months after discharge, follow-up examinations revealed that the
patient remained in a good overall condition.
Discussion
The present study reported on the case of an
otherwise healthy young male patient with an episode of
cholestasis. According to the LiverTox.nih.gov website (16) (https://www.ncbi.nlm.nih.gov/books/NBK548711/),
ephedra is known to promote liver injury. However, all reported
cases, including hepatocellular and biliary injury (9), with or without co-treatment with
steroids (12), were more severe
compared to the present case. A comparison of the present and
previously reported cases is provided in Table II. The previously reported cases
were young adults, which is consistent with the current case.
Furthermore, not only was the liver injury much milder in the
present case, but a large proportion of the young male patients'
liver functions were also well preserved, despite the fact that
accumulated bile salts should have aggravated the hepatobiliary
injury due to their inherent detergent activities (16). All of the above evidence indicated
the presence of an intrahepatic protective molecule shielding the
liver from further injury.
| Table IIComparison among ephedra-administered
cases. |
Table II
Comparison among ephedra-administered
cases.
| Item | Case by Nadir et
al (9) | Case by Singh et
al (12) | Present case |
|---|
| Age (years) | 33 | 24 | 21 |
| Sex | Female | Male | Male |
| Medication | Ephedra | Ephedra with
steroids | Ephedra |
| Pattern | Hepatocellular | Bland
cholestatic | Bland
cholestatic |
| ALT (U/l) | 1586 | 237 | 37 |
| ALP (U/l) | 175 | 129 | 104 |
| Bilirubin
(µmol/l) | 136.80 | 906.3 | 253.8 |
| Prothrombin time
(sec) | 13.2 | NA | 12 |
| Jaundice | Yes | Yes | Yes |
| Recovery time
(months) | 4 | 3 | 1 |
Of note, the clinical examination suggested a rare
and mild type of chronic hemolytic anemia. These rare disorders may
be caused by any genetic defect affecting RBCs, including
Diamond-Blackfan anemia, congenital dyserythropoietic anemias,
thalassemia, sickle cell disease, enzyme deficiency and red cell
membrane disorders. These disorders are characterized by a variable
degree of anemia, chronic extravascular hemolysis, reduced
erythrocyte lifespan, splenomegaly, jaundice, biliary lithiasis and
iron overload (17). Each type may
require contradictory therapeutic approaches (3); therefore, the precise genetic
diagnosis is of great importance. In the current case, a rare
genetic variant was identified in the CDAN1 gene. This
mutation is involved in the onset of rare congenital
dyserythropoietic anemia type I (CDA-I), which is caused by
bi-allelic mutations in the CDAN1 gene, usually accompanied
by iron overload. In CDA-I, liver tests are commonly normal unless
severe iron overload is present. However, unconjugated bilirubin
and LDH levels are usually elevated due to hemolysis and
ineffective erythropoiesis. It has been reported that abdominal
ultrasound in patients with CDA-I reveals universal splenomegaly
(14), while 50-60% of adult
patients have gallstones (15). In
the current case, all the aforementioned clinical features were
present and the patient had splenomegaly and a history of
gallstones. Furthermore, the dysregulation of erythropoiesis was
evidenced by the marginally elevated percentage of reticulocytes
relative to the degree of anemia, and constantly by the increased
number of basophils in the peripheral blood of the patient. In this
case, genetic testing is recommended, since it may significantly
reduce the time required for accurate molecular diagnosis and
prevent invasive bone marrow aspiration. To the best of our
knowledge, the novel p.R1067H mutation identified in the present
study, located at the disease-related hotspot exon 24(18), has not been previously reported and
is adjacent to the well-established disease-related p.D1043V
mutation. Computational prediction tools, including SIFT
(sift.jcvi.org), PolyPhen-2 (genetics.bwh.harvard.edu/pp2), mutation taster
(www.mutationtaster.org), PROVEAN
(provean.jcvi.org/index.php), SNPs
& GO (snps-and-go.biocomp.unibo.it), PANTHER (pantherdb.org), PhD-SNP (snps.biofold.org/phd-snp/phd-snp.html), I-Mutant
(folding.biofold.org/cgi-bin/i-mutant2.0.cgi) and
SNAP (www.rostlab.org/services/SNAP), unanimously support
the deleterious effect of mutations of this gene (19-21).
While the hemolytic process may predispose the
patient's liver to cholestatic injury, most likely through iron
overload, lithiasis and splenomegaly, it may also guarantee a
steady flow of slightly elevated bilirubin in the blood
(hyperbilirubinemia). Bilirubin, a pigment with potent
anti-oxidative properties (22), is
known to protect against inflammation-mediated diseases (23,24)
all-cause mortality (25) and even
possesses therapeutic value (23,24,26).
However, the intrahepatic protective role of mildly-elevated
unconjugated bilirubin has not been reported in the literature, to
the best of our knowledge. Therefore, the present case provided
hint that the hemolysis-mediated mild hyperbilirubinemia may
prepare and protect the liver from further injury.
In conclusion, the present study reported on a case
of DILI, in the context of a mild form of rare congenital anemia
resulting in bland cholestasis. Hemolysis-induced mildly-elevated
unconjugated bilirubin may contribute to the milder DILI observed
in the current case compared to the previously reported cases.
Therefore, the present study highlighted the importance of genetic
pedigree analysis, particularly when a given condition is
manifested in additional family members. In addition, a novel rare
mutation in the CDAN1 gene was identified in the present
case, enhancing the spectrum of mutations known to be associated
with such pathologies.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Science Foundation of China (grant no. 82002126), the
Shanghai Jiao Tong University (SJTU) medical X engineering project
(grant no. YG2017MS63) and the Excellent Youth B Project (grant no.
GCQN-2017-B20).
Availability of data and materials
The next-generation sequencing datasets generated
and analyzed during the current study are available in the National
Center for Bioinformatics Information (NCBI) Sequence Read Archive
repository (accession no. PRJNA690696; https://www.ncbi.nlm.nih.gov/sra/PRJNA690696).
Authors' contributions
YH drafted the manuscript and performed the
laboratory tests. YZ, WT, LC and YC performed the follow-up checks
and analyzed the clinical data. The experiments were designed by QG
and XZ. QG and XZ checked and approved the authenticity of the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Ruijin Hospital (Shanghai, China; ID: 2016-17).
Written informed consent was provided by the patient.
Patient consent for publication
The patient and his parents provided written
informed consent for publication of the results.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gambale A, Iolascon A, Andolfo I and Russo
R: Diagnosis and management of congenital dyserythropoietic
anemias. Expert Rev Hematol. 9:283–296. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Russo R, Andolfo I, Manna F, Gambale A,
Marra R, Rosato BE, Caforio P, Pinto V, Pignataro P, Radhakrishnan
K, et al: Multi-gene panel testing improves diagnosis and
management of patients with hereditary anemias. Am J Hematol.
93:672–682. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bolton-Maggs PH, Stevens RF, Dodd NJ,
Lamont G, Tittensor P and King MJ: General Haematology Task Force
of the British Committee for Standards in Haematology. Guidelines
for the diagnosis and management of hereditary spherocytosis. Br J
Haematol. 126:455–474. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
European Association for the Study of the
Liver. Electronic address: simpleeasloffice@easloffice.eu;
Clinical Practice Guideline Panel: Chair; Panel members and EASL
Governing Board representative. EASL clinical practice guidelines:
Drug-induced liver injury. J Hepatol. 70:1222–1261. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lucena MI, Sanabria J, García-Cortes M,
Stephens C and Andrade RJ: Drug-induced liver injury in older
people. Lancet Gastroenterol Hepatol. 5:862–874. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Danan G and Teschke R: RUCAM in drug and
herb induced liver injury: The update. Int J Mol Sci.
14(17)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Russmann S, Jetter A and Kullak-Ublick GA:
Pharmacogenetics of drug-induced liver injury. Hepatology.
52:748–761. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Garcia-Cortes M, Robles-Diaz M, Stephens
C, Ortega-Alonso A, Lucena MI and Andrade RJ: Drug induced liver
injury: An update. Arch Toxicol. 94:3381–3407. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nadir A, Agrawal S, King PD and Marshall
JB: Acute hepatitis associated with the use of a Chinese herbal
product, ma-huang. Am J Gastroenterol. 91:1436–1438.
1996.PubMed/NCBI
|
|
10
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Iezzoni JC: Diagnostic histochemistry in
hepatic pathology. Semin Diagn Pathol. 35:381–389. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Singh C, Bishop P and Willson R: Extreme
hyperbilirubinemia associated with the use of anabolic steroids,
health/nutritional supplements, and ethanol: Response to
ursodeoxycholic acid treatment. Am J Gastroenterol. 91:783–785.
1996.PubMed/NCBI
|
|
13
|
Brown RS Jr, Kumar KS, Russo MW,
Kinkhabwala M, Rudow DL, Harren P, Lobritto S and Emond JC: Model
for end-stage liver disease and Child-Turcotte-Pugh score as
predictors of pretransplantation disease severity,
posttransplantation outcome, and resource utilization in United
Network for Organ Sharing status 2A patients. Liver Transpl.
8:278–284. 2002.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Heimpel H, Schwarz K, Ebnöther M, Goede
JS, Heydrich D, Kamp T, Plaumann L, Rath B, Roessler J,
Schildknecht O, et al: Congenital dyserythropoietic anemia type I
(CDA I): Molecular genetics, clinical appearance, and prognosis
based on long-term observation. Blood. 107:334–340. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shalev H, Kapleushnik Y, Haeskelzon L,
Degani O, Kransnov T, Sphilberg O, Moser A, Yaniv I and Tamary H:
Clinical and laboratory manifestations of congenital
dyserythropoietic anemia type I in young adults. Eur J Haematol.
68:170–174. 2002.PubMed/NCBI View Article : Google Scholar
|
|
16
|
LiverTox: Clinical and Research
Information on Drug-Induced Liver Injury. [Internet]. National
Institute of Diabetes and Digestive and Kidney Diseases, Bethesda
(MD), 2012.
|
|
17
|
Zaninoni A, Fermo E, Vercellati C,
Marcello AP, Barcellini W and Bianchi P: Congenital hemolytic
anemias: Is there a role for the immune system? Front Immunol.
11(1309)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Roy NBA and Babbs C: The pathogenesis,
diagnosis and management of congenital dyserythropoietic anaemia
type I. Br J Haematol. 185:436–449. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Borgio JF, Al-Madan MS and AbdulAzeez S:
Mutation near the binding interfaces at alpha-hemoglobin
stabilizing protein is highly pathogenic. Am J Transl Res.
8:4224–4232. 2016.PubMed/NCBI
|
|
20
|
AbdulAzeez S and Borgio JF: In-silico
computing of the most deleterious nsSNPs in HBA1 Gene. PLoS One.
11(e0147702)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Abdulazeez S, Sultana S, Almandil NB,
Almohazey D, Bency BJ and Borgio JF: The rs61742690 (S783N) single
nucleotide polymorphism is a suitable target for disrupting
BCL11A-mediated foetal-to-adult globin switching. PLoS One.
14(e0212492)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fujiwara R, Haag M, Schaeffeler E, Nies
AT, Zanger UM and Schwab M: Systemic regulation of bilirubin
homeostasis: Potential benefits of hyperbilirubinemia. Hepatology.
67:1609–1619. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vitek L, Bellarosa C and Tiribelli C:
Induction of mild hyperbilirubinemia: Hype or real therapeutic
opportunity? Clin Pharmacol Ther. 106:568–575. 2019.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Boon AC, Hawkins CL, Coombes JS, Wagner KH
and Bulmer AC: Bilirubin scavenges chloramines and inhibits
myeloperoxidase-induced protein/lipid oxidation in physiologically
relevant hyperbilirubinemic serum. Free Radic Biol Med. 86:259–268.
2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bulmer AC, Bakrania B, Du Toit EF, Boon
AC, Clark PJ, Powell LW, Wagner KH and Headrick JP: Bilirubin acts
as a multipotent guardian of cardiovascular integrity: More than
just a radical idea. Am J Physiol Heart Circ Physiol.
315:H429–H447. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vitek L and Tiribelli C: Bilirubin,
intestinal integrity, the microbiome, and inflammation. N Engl J
Med. 383:684–686. 2020.PubMed/NCBI View Article : Google Scholar
|