Introduction
Acute myocardial infarction (AMI) is associated with
adverse cardiac remodeling and impaired ventricular function, which
is a leading cause of mortality worldwide (1,2).
Myocardial fibrosis following myocardial infarction is a crucial
mechanism of cardiac insufficiency in patients with AMI. A previous
study reported that myocardial fibrosis was characterized by the
abnormal proliferation of fibroblasts and the accumulation of
extracellular matrix (ECM) in the heart, which affected the
diastolic and contractile function of the heart, eventually leading
to heart failure (HF) (3,4). Due to the adverse impact of AMI on the
survival rate of patients, developing novel therapeutic strategies
to improve cardiac performance by reducing the number of myocardial
fibroblasts following AMI are desirable.
Long non-coding (lnc)RNAs are a type of RNA
transcript, longer than 200 nucleotides in length and with no open
reading frame (5). A previous study
suggested that lncRNAs serve important roles in diverse biological
processes, and their vital role in the regulation of fibrosis is
increasingly being elucidated (6).
X-inactive specific transcript (XIST) was highly expressed in
hypertrophic mouse hearts and promoted the progression of cardiac
hypertrophy by targeting microRNA(miR)-101 to upregulate TLR2
expression (7). However, the
biological roles of lncRNA XIST in myocardial injury and fibrosis
have not been widely reported. McKiernan et al (8) reported that XIST was aberrantly
expressed in cystic fibrosis bronchial epithelium in vivo,
which may be associated with inflammation. Furthermore, XIST
promoted bleomycin-induced pulmonary fibrosis by inhibiting miR-139
to enhance the proliferative ability of human/mouse fibroblasts and
ECM protein expression (9).
Notably, XIST-silencing may protect against renal interstitial
fibrosis in diabetic nephropathy by miR-93-5p-dependent CDKN1A
inhibition (10). Therefore, we
hypothesized that XIST may contribute toward myocardial fibroblasts
following AMI.
Angiotensin II (Ang II) is the main active protein
in the rennin-angiotensin-aldosterone system (RAAS), which serves a
crucial role in myocardial interstitial fibrosis and cardiac
remodeling (11,12). Under pathological conditions, Ang II
stimulates the proliferation of cardiac fibroblast cells (CFs) by
autocrine and paracrine signaling, and promotes the synthesis of
collagen (Col) I to repair injury in myocardial tissues (13-15).
However, the continuous abnormal synthesis of Col I, caused by the
excessive proliferation of CFs, may lead toward myocardial
interstitial fibrosis and decreased compliance of the ventricle,
which ultimately affects the diastolic and contractile function of
the heart.
In the present study, human cardiac fibroblast cells
(HCFs) were used and treated with Ang II, while pcDNA3.1 XIST and
short hairpin (sh)RNA-XIST was used to induce XIST overexpression
and silencing in cells, respectively. The present study was
designed to determine whether myocardial fibrosis could be
regulated by XIST and to elucidate the related mechanism. The
results demonstrated that XIST promoted the proliferation of
myocardial fibroblasts and the accumulation of ECM, suggesting that
XIST may be a novel potential target for AMI treatment.
Materials and methods
Cell culture and transfection
Primary HCFs were obtained from the American Type
Culture Collection and were maintained in Dulbecco's modified
Eagle's medium (DMEM, Hyclone; Cytival), supplemented with 10% FBS
(Invitrogen; Thermo Fisher Scientific, Inc.) and 1% penicillin and
1% streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37˚C for
48 h in a humidified incubator with 5% CO2, followed by
Ang II (Sigma-Aldrich; Merck KGaA) treatment (0.01 mM) at 37˚C for
24 h to induce the myocardial fibrosis phenotype.
At 70-80% confluence, HCFs were seeded onto a
corresponding culture plate and starved for 12 h in serum-free DMEM
(Hyclone; Cytival) prior to treatment. The plasmids, including 1
µg/well pcDNA3.1 XIST (forward, 5'-CCAAGCTTTGCACACGGCCTATCTCATC-3'
and reverse, 5'-CCGCTCGAGTGAAAAGAGGTGGGGCATCC-3') inserted into
pcDNA3.1, 100 pmol of short hairpin RNA (shRNA)-XIST (shRNA-XIST-1,
5'-TTCTCCGAACGTGTCACGT-3'; 100 pmol of shRNA-XIST-2,
5'-GCTGCTAGTTTCCCAATGATA-3'), 100 pmol of shRNA-NC
(5'-TTCTCCGAACGTGTCACGT-3'), 20 nM of miR-155-5p mimic (forward,
5'-UUAAUGCUAAUCGUGAUAGGGGU-3' and reverse,
5'-CCCUAUCACGAUUAGCAUUAAUU-3'), 20 nM of mimic-negative control
(NC; forward, 5'-UUCCUCCGAACGUGUCACGUTT-3' and reverse,
5'-ACGUGACACGUUCGGAGAATT-3'), 100 nM of miR-155-5p inhibitor
(5'-ACCCCUAUCACGAUUAGCAUUAA-3') and 100 nM of inhibitor-NC
(5'-CAGUACUUUUGUGUAGUACAA-3') were purchased from Shanghai
GenePharma Co., Ltd., and transfected into the HCFs using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
interaction between XIST and miR-155-5p was predicted using the
Starbase website (http://starbase.sysu.edu.cn/index.php). After 48 h,
the HCFs were used for further experiments and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) was
performed to examine transfection efficiency.
RT-qPCR
Total RNA was extracted from the HCFs using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. First strand cDNA was
synthesized at 42˚C for 60 min and 95˚C for 5 min, using a
PrimeScript RT reagent kit. qPCR was performed using a
SYBR® Green Real-Time PCR Master Mix (Takara Bio, Inc.).
qPCR amplification parameters were: Annealing at 25˚C for 10 min,
extension at 42˚C for 30 min; inactivation at 85˚C for 10 min,
after extension of the detection of fluorescence signals, a total of
40 cycles. The 2-ΔΔCq method (16) was used to quantify the relative mRNA
expression of the target and housekeeping genes. The following
primers were used: XIST forward, 5'-ACGCTGCATGTGTCCTTAG-3 and
reverse, 5'-GAGCCTCTTATAAGCTGTTTG-3'; miR-155-5p forward,
5'-GAGGGTTAATGCTAATCGTGATAGG-3' and reverse,
5'-GCACAGAATCAACACGACTCACTAT-3'; GAPDH forward,
5'-CTGGGCTACACTGAGCACC-3' and reverse, 5'-AAGTGGTCGTTGAGGGCAATG-3';
and U6 forward, 5'-GAGGGTTAATGCTAATCGTGATAGG-3' and reverse,
5'-GCACAGAATCAACACGACTCACTAT-3'.
Cell proliferation
After the HCFs (4x103/well) from
different experimental groups, were plated onto 96-well plates and
cultured for 24 h, the Cell Counting Kit-8 (CCK-8; Dojindo
Molecular Technologies, Inc.) was added (10 µl/well) and incubated
with the HCFs for another 2 h according to manufacturer's protocol.
A microplate reader was used to detect the absorbance at 450 nm.
The experiment was repeated independently three times.
Flow cytometry
The Annexin V-FITC/PI apoptosis detection kit
(Beyotime Institute of Biotechnology) was used according to the
manufacturer's protocols. The harvested cells (1x105
cells/ml) were resuspended in 500 µl of 1X binding buffer (Beyotime
Institute of Biotechnology). Next, the cells were incubated with 5
µl Annexin V-FITC followed by 10 µl PI staining solution. Following
incubation in the dark, at room temperature for 20 min, the
fluorescence was detected using a flow cytometer (FACScan™; BD
Biosciences) and ModFit software (version 3.2; Verity Software
House, Inc.) to analyze the apoptotic rate of HCFs.
Western blot analysis
The HCFs in each group were lysed with RIPA lysis
buffer (Beyotime Institute of Biotechnology). Total protein was
determined using a bicinchoninic acid assay (Thermo Fisher
Scientific, Inc.). Next, the protein samples (25 µg) were separated
using 10% SDS-PAGE, transferred to polyvinylidene difluoride
membranes, and blocked with 5% skimmed milk at room temperature for
1 h. The membranes were incubated overnight at 4˚C with the primary
antibodies against Bcl-2 (1:1,000; cat. no. 2875; Cell Signaling
Technology, Inc.), Bax (1:1,000; cat. no. 2772, Cell Signaling
Technology, Inc.), cleaved-caspase-9 (1:500; cat. no. 9509; Cell
Signaling Technology, Inc.), cleaved caspase-3 (1:500; cat. no.
9654; Cell Signaling Technology, Inc.), caspase-3 (1:1,000; cat.
no. 9668; Cell Signaling Technology, Inc.), caspase-9 (1:1,000;
cat. no. 9502; Cell Signaling Technology, Inc.), Col I (1:1,000;
ab34710; Abcam), Col III (1:1,000; ab6310; Abcam), α-smooth muscle
actin (α-SMA; 1:1,000; cat. no. 68463; Cell Signaling Technology,
Inc.), cytochrome C (1:1,000; cat. no. 12959; Cell Signaling
Technology, Inc.), cleaved PARP (1:500; cat. no. 9185; Cell
Signaling Technology, Inc.), and PARP (1:1,000; cat. no. 9542; Cell
Signaling Technology, Inc.). Next, the membranes were incubated
with horseradish peroxidase-conjugated horseradish
peroxidase-conjugated anti-rabbit IgG secondary antibodies
(1:1,000; cat. no. 7074; Cell Signaling Technology, Inc.) at room
temperature for 2 h and visualized using a Tanon-5200
chemiluminescence imager (Tanon Science and Technology Co., Ltd)
using an enhanced chemiluminescence kit and the intensity of the
bands was normalized to GAPDH expression.
Luciferase reporter assay
The HCFs (1x105) were seeded onto 24-well
plates and cultured for 24 h. The luciferase reporter vector for
the wild-type (WT) or mutant (MUT) type 3'-untranslated region
(UTR) of XIST, which contained the binding sites between miR-155-5p
was cloned into the pMIR-reporter luciferase system (Thermo Fisher
Scientific, Inc.). The MUT 3'-UTR of XIST was constructed using the
QuikChang Site-Directed Mutagenesis kit (Agilent Technologies).
miR-155-5p mimic or mimic-NC combined with the vector expressing
firefly luciferase reporter fused with WT or MUT 3'-UTR of XIST was
co-transfected into the HCFs using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Following incubation
for 24 h, the HCFs were collected and the luciferase activities
were determined using the dual-luciferase reporter assay system
(Promega Corporation) and the firefly luciferase activity was
normalized to Renilla luciferase activity.
Statistical analysis
The data are presented as the mean ± standard error
of the mean and analyzed using GraphPad Prism v5.0 (GraphPad
Software, Inc.). The difference between two groups was determined
using a two-tailed Student's t-test, while a one-way analysis of
variance, followed by Bonferroni's post hoc test, was used for
multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
XIST overexpression promotes
proliferation and the protein expression level of fibrosis-related
proteins in HCFs
To investigate the association between XIST and
myocardial fibrosis, RT-qPCR was performed to detect XIST mRNA
expression following treatment of the HCFs with Ang II. As shown in
Fig. 1A, Ang II treatment
significantly increased the relative mRNA expression of XIST,
compared with that in the control group. Subsequently, the
pcDNA3.1-XIST plasmid was used to induce XIST overexpression, which
was confirmed by the RT-qPCR results (Fig. 1B). The CCK-8 assay was performed to
analyze the cell viability of the HCFs. As shown in Fig. 1C, the HCFs in the pcDNA3.1-XIST
group had a higher rate of viability, compared with that in the
control and pcDNA3.1 groups. Furthermore, cell apoptosis was
detected using flow cytometry and the protein expression level of
the apoptosis-related proteins were quantified using western blot
analysis. As shown in Fig. 1D and
E, the apoptotic rate was
significantly decreased in the pcDNA3.1-XIST group, compared with
that in the control group. In addition, XIST-overexpression induced
upregulation of Bcl-2, while Bax, cleaved caspase-3 and -9,
cytochrome c and cleaved PARP were downregulated (Fig. 1F and G). Finally, the protein expression levels
of Col I, Col III and α-SMA were also determined using western blot
analysis and the results revealed that the expression of these
proteins was significantly increased by XIST overexpression
(Fig. 1H). These results suggested
that XIST-overexpression promoted the proliferation of HCFs and the
expression of the fibrosis-related proteins in vitro.
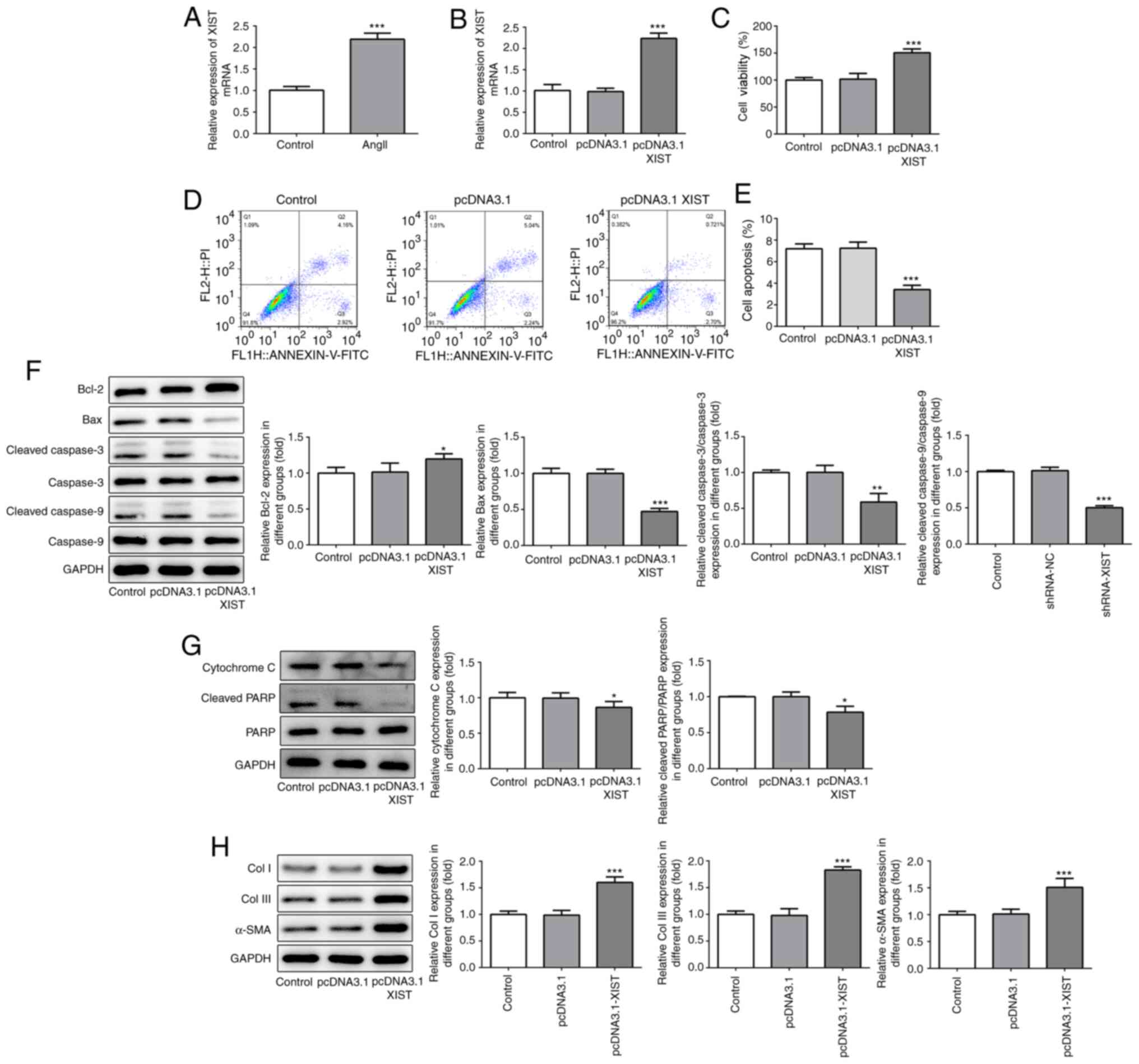 | Figure 1XIST-overexpression promoted cell
proliferation and increased the expression of fibrosis-related
proteins in the HCFs. (A and B) The relative mRNA expression level
of XIST was determined using reverse transcription-quantitative
polymerase chain reaction. (C) The viability of the HCFs was
analyzed using a Cell Counting Kit-8 assay. (D and E) The apoptotic
rates of the HCFs transfected with or without pcDNA3.1 XIST were
determined using flow cytometry. The protein expression of (F)
Bcl-2, Bax, cleaved cacpase-3, cacpase-3, cleaved cacpase-9,
cacpase-9; (G) cytochrome c, cleaved PARP and PARP; and (H)
Col I, Col III and α-SMA were detected using western blot analysis.
The data are expressed as the mean ± standard error of the mean,
from three independent experiments. *P<0.05,
**P<0.01, ***P<0.001 vs. control. Col,
collagen; α-SMA, α smooth muscle actin; HCFs, human cardiac
fibroblast cells. |
XIST-silencing inhibits proliferation
and decreases the expression of fibrosis-related proteins in
HCFs
To confirm the role of XIST in myocardial fibrosis,
shRNA-XIST-1 and shRNA-XIST-2 were used to induce XIST-silencing.
As shown in Fig. 2A, the RT-qPCR
results demonstrated that a lower mRNA expression level of XIST was
observed in the HCFs in the shRNA-XIST-1 group, compared with that
in the shRNA-XIST-2 group; therefore, shRNA-XIST-1 was selected in
the following experiments. The CCK-8 assay results demonstrated
that XIST-silencing significantly suppressed cell viability of the
HCFs (Fig. 2B). The flow cytometry
results revealed that XIST-silencing significantly enhanced the
apoptotic rate of the HCFs (Fig. 2C
and D). In addition, XIST-silencing
increased the protein expression level of Bax, cleaved caspase-3
and -9, cytochrome c and cleaved PARP, while the protein
expression level of Bcl-2, Col I, Col III and α-SMA was decreased
(Fig. 2E-G). These results
indicated that XIST-silencing inhibited proliferation of the HCFs
and the protein expression level of fibrosis-related proteins in
vitro.
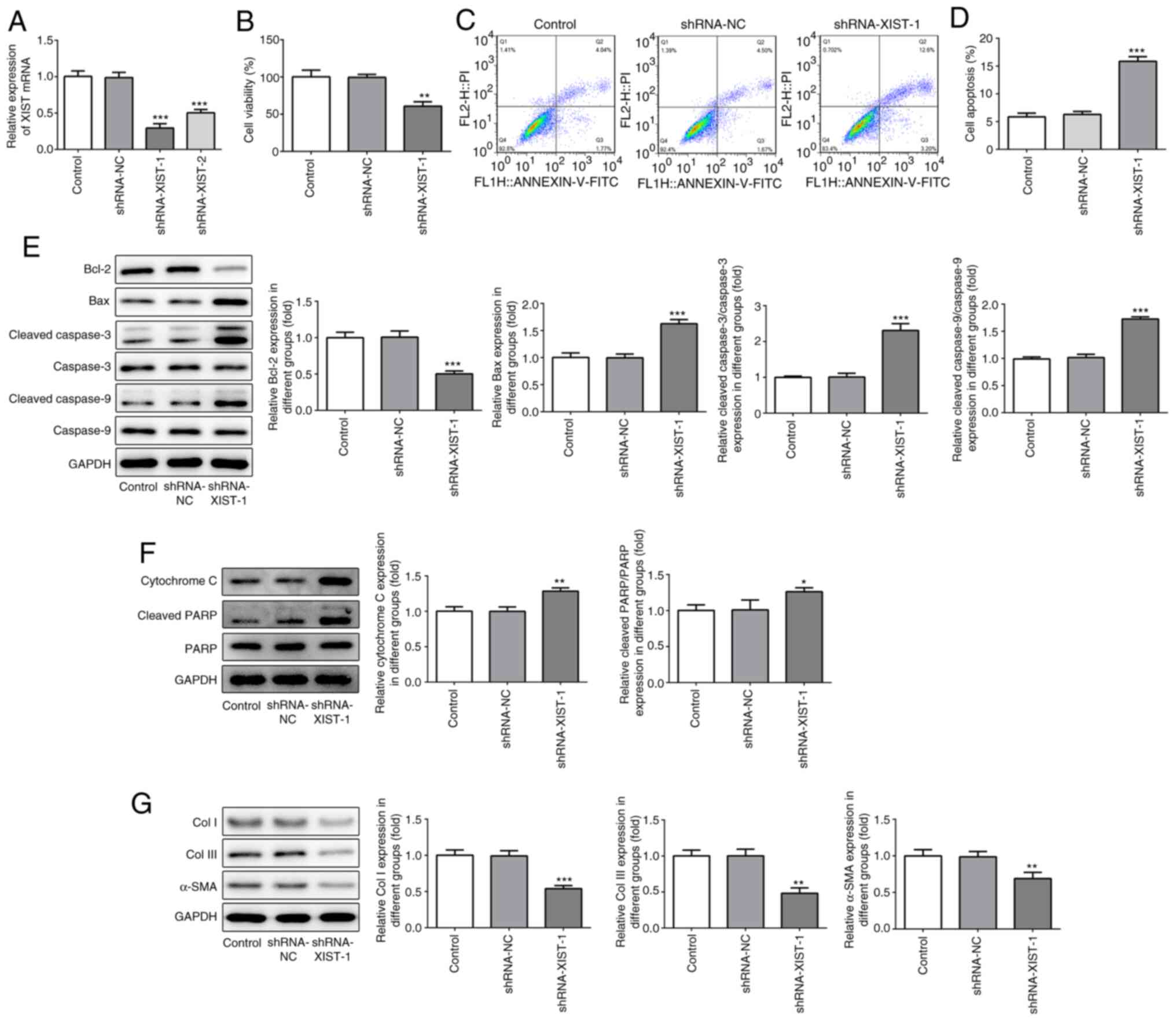 | Figure 2XIST-silencing inhibited the
proliferation and expression of fibrosis-related proteins in the
HCFs. (A) The relative mRNA expression level of XIST was determined
using reverse transcription-quantitative polymerase chain reaction.
(B) The viability of the HCFs was analyzed using Cell Counting
Kit-8 assay. (C) The apoptotic rate of the HCFs transfected with or
without shRNA-XIST-1 was determined using flow cytometry and was
(D) subsequently quantified. The protein expression of (E) Bcl-2,
Bax, cleaved cacpase-3, cacpase-3, cleaved cacpase-9, cacpase-9;
(F) cytochrome c, cleaved PARP and PARP; and (G) Col I, Col
III and α-SMA was detected using western blot analysis. The data
are presented as the mean ± standard error of the mean, from three
independent experiments. *P<0.05,
**P<0.01, ***P<0.001 vs. control. Col,
collagen; α-SMA, α smooth muscle actin; sh, short hairpin; NC,
negative control; HCFs, human cardiac fibroblast cells. |
XIST directly targets miR-155-5p and
downregulates its mRNA expression level
To investigate the specific mechanism of XIST in
myocardial fibrosis, RT-qPCR was performed to detect miR-155-5p
mRNA expression, following treatment with Ang II. As shown in
Fig. 3A, Ang II treatment
significantly decreased the relative mRNA expression of miR-155-5p.
In addition, the RT-qPCR results demonstrated that the mRNA
expression of miR-155-5p could be increased by XIST-silencing
(Fig. 3B). Subsequently, miR-155-5p
mimic was used to induce miR-155-5p-overexpression. The RT-qPCR
results demonstrated that miR-155-5p mimic was transfected into the
cells successfully and upregulated miR-155-5p mRNA expression
(Fig. 3C). Notably, XIST expression
was significantly decreased by miR-155-5p-overexpression (Fig. 3D). The interaction between XIST and
miR-155-5p was predicted using the Starbase website (http://starbase.sysu.edu.cn/index.php),
as presented in Fig. 3E.
Subsequently, the luciferase reporter assay was performed to
confirm whether miR-155-5p was a direct target of XIST. As shown in
Fig. 3F, the relative luciferase
activity was significantly lower in the HCFs co-transfected with
WT-XIST and miR-155-5p mimic than that in the control group. These
results indicated that XIST specifically bound to the 3'-UTR of
miR-155-5p to decrease its expression.
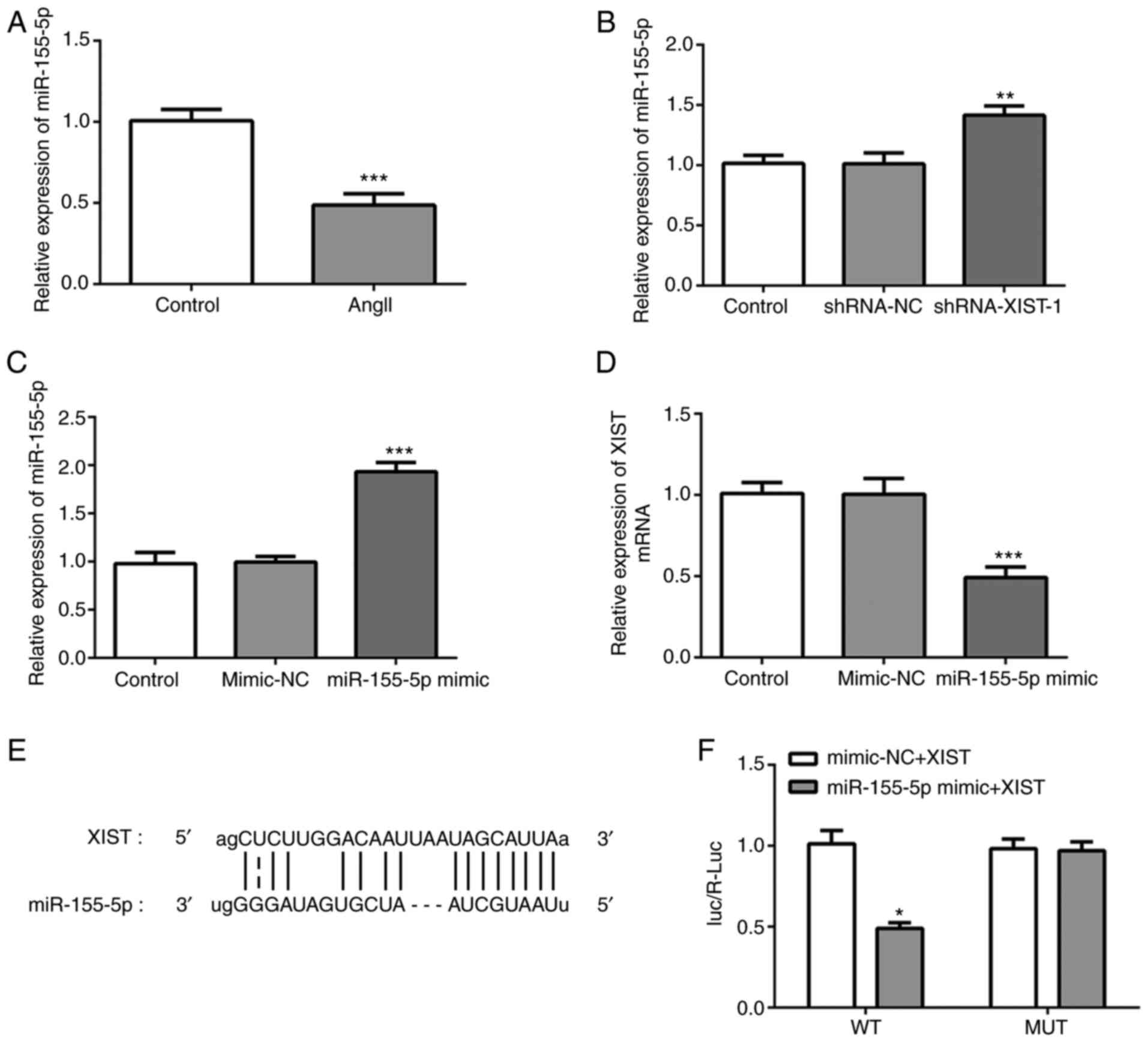 | Figure 3XIST directly targets miR-155-5p and
downregulates its expression. (A-C) Relative mRNA expression of
miR-155-5p was determined using RT-qPCR. (D) Relative mRNA
expression of XIST was determined using RT-qPCR. (E) The binding
sites between XIST and miR-155-5p. (F) The relative luciferase
activities in the human cardiac fibroblast cells were evaluated
using a luciferase reporter assay. The data are presented as the
mean ± standard error of the mean, from three independent
experiments. *P<0.05, **P<0.01,
***P<0.001 vs. control. miR, microRNA; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; WT,
wild-type; MUT, mutant; NC, negative control; Ang II, angiotensin
II; sh, short hairpin. |
miR-155-5p-downregulation abolishes
the effect of XIST-silencing on cell viability and the protein
expression level of the fibrosis-related proteins in the HCFs
To further investigate the molecular mechanism of
XIST in myocardial fibrosis, the HCFs were transfected with an
miR-155-5p inhibitor. As shown in Fig.
4A, miR-155-5p-downregulation was achieved via miR-155-5p
inhibitor transfection. The CCK-8 assay results demonstrated that
miR-155-5p inhibitor abolished the suppressive effects of
XIST-silencing on cell viability (Fig.
4B). The flow cytometry results demonstrated that
XIST-silencing promoted apoptosis in the HCFs, which was partly
reversed by miR-155-5p downregulation (Fig. 4C and D). The western blot result showed that
miR-155-5p downregulation abolished the regulatory effect of XIST
on the protein expression level of the apoptosis-related proteins
(Fig. 4E and F), as well as the fibrosis-related
proteins (Fig. 4G). These results
suggested that XIST promoted the proliferation of the HCFs and the
expression level of the fibrosis-related proteins by sponging
miR-155-5p.
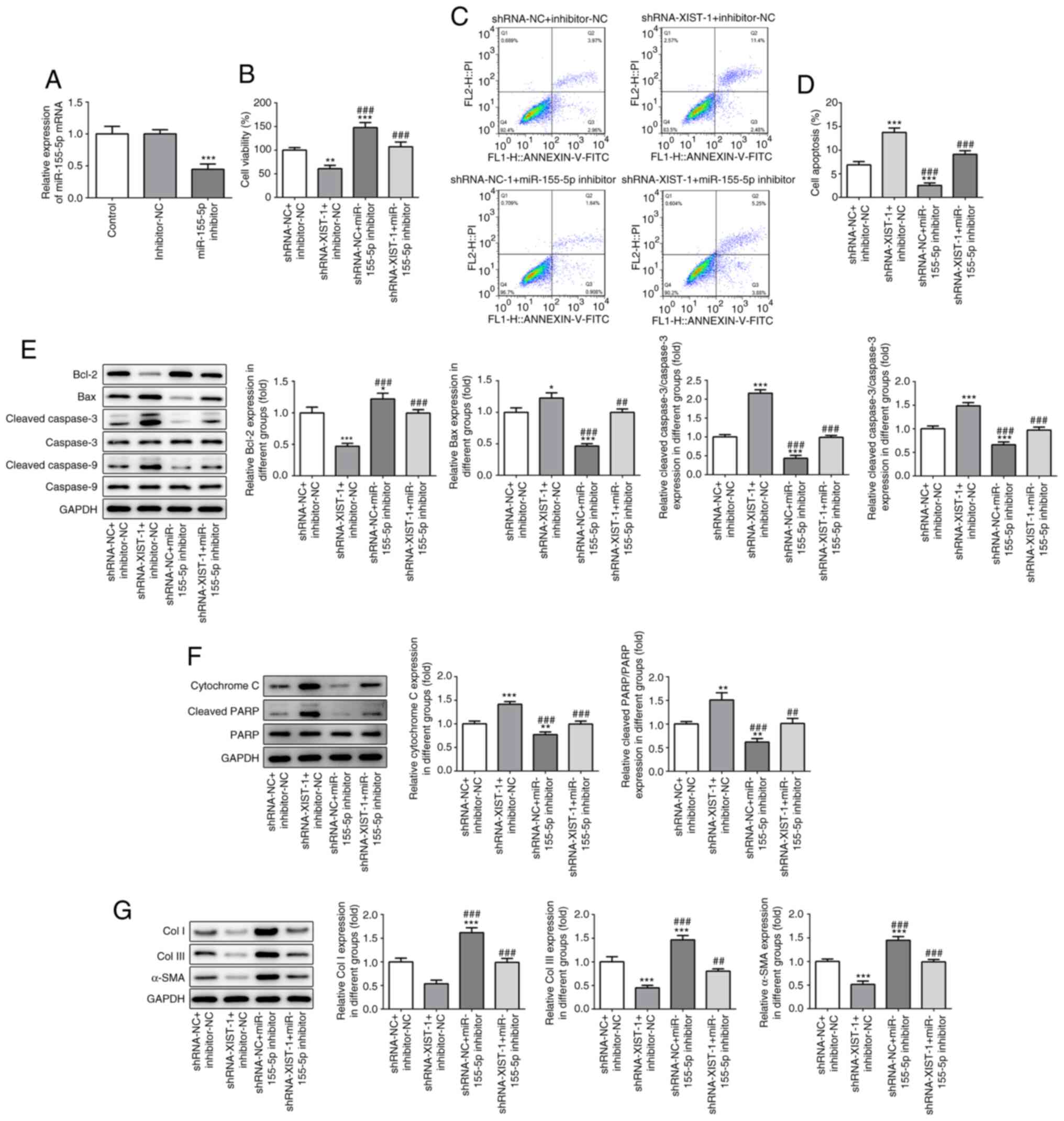 | Figure 4miR-155-5p-downregulation abolished
the effect of XIST-silencing on cell viability and the expression
of fibrosis-related proteins in the HCFs. (A) The relative mRNA of
miR-155-5p was determined by reverse transcription-quantitative
polymerase chain reaction. (B) The viability of the HCFs was
analyzed using a Cell Counting Kit-8 assay. (C) The apoptotic rate
of the HCFs in each group was determined using flow cytometry and
was (D) subsequently quantified. The protein expression of (E)
Bcl-2, Bax, cleaved caspase-3, caspase-3, cleaved-caspase-9,
caspase-9; (F) cytochrome c, cleaved PARP and PARP; and (G)
Col I, Col III and α-SMA was detected using western blot analysis.
The data are presented as the mean ± standard error of the mean,
from three independent experiments. *P<0.05,
**P<0.01, ***P<0.001 vs.
shRNA-NC+inhibitor-NC. ##P<0.05,
###P<0.001 vs. shRNA-XIST-1+inhibitor-NC. miR,
microRNA; HCFs, human cardiac fibroblast cells; sh, short hairpin;
NC, negative control; Col, collagen; α-SMA, α smooth muscle
actin. |
Discussion
AMI is one of the diseases with the highest
morbidity and mortality rates in the world (17), and it may induce cardiac remodeling
and HF (18). Myocardial fibrosis
has been reported to be associated with sparseness of cardiac
microvessels, and found to contribute toward HF, which is caused by
myocardial fibroblast accumulation and imbalance of synthesis,
metabolism and degradation of collagen in ECM deposition (19). CFCs are the primary cells that
secrete the ECM of the myocardium. In the present study, HCFs were
treated with Ang II to induce the myocardial fibrosis phenotype.
Notably, it was observed that XIST was highly expressed in the HCFs
treated with Ang II, suggesting that XIST was associated with the
onset and development of myocardial fibrosis.
A recent study demonstrated that XIST was
upregulated in ethanol-induced human hepatic stellate cells and
contributed toward liver fibrogenesis (20), which was consistent with the results
of the present study. Furthermore, the results of the present study
suggested that XIST-overexpression promoted proliferation and
enhanced the protein expression of fibrosis-related proteins in
HCFs, while XIST-silencing had the opposite effect. These data
indicated that XIST may promote the development of myocardial
fibrosis.
To investigate the specific mechanisms underlying
the role of XIST in myocardial fibrosis, the interaction between
XIST and miR-155-5p was predicted using the Starbase website. It
was found that miR-155-5p was one of the potential targets of XIST.
In the present study, Ang II treatment led to a decrease in
miR-155-5p mRNA expression, whereas XIST downregulation
significantly increased miR-155-5p mRNA expression. These data
suggested that XIST may directly target miR-155-5p and downregulate
its mRNA expression level. A recent study has demonstrated that
miR-155-5p suppressed the proliferation, migration and invasion of
vascular smooth muscle cells (VSMCs) and human umbilical vein
endothelial cells in atherosclerosis by regulating Akt (21). Chen et al (21) reported that miR-155-5p inhibition
prevented the development of abdominal aortic aneurysms in mice by
regulating macrophage-mediated inflammation (22). It was reported that
miR-155-overexpression decreased cellular proliferation in VSMCs,
and the expression of miR-155 was decreased and negatively
correlated with calcification in the aorta of chronic kidney
disease rats (23). Additionally, a
previous study reported that miR-155-5p mediated the regulatory
network for myocardial infarction (24), suggesting that miR-155-5p may be a
putative therapeutic target for AMI. In addition,
miR-155-5p-upregulation decreased vascular thickening and fibrosis
in a vascular calcification model (25). In the present study,
miR-155-5p-downregulation promoted the proliferation of the HFCs
and suppressed cell apoptosis. It is well-known that collagen, an
insoluble fibrous protein, is the main component of the ECM
(26,27). miR-155-5p downregulation abolished
the inhibitory effect of XIST-silencing on the protein expression
level of Col I, Col III and α-SMA in the HCFs, suggesting that XIST
promoted the proliferation of the HCFs and the protein expression
of fibrosis-related proteins by sponging miR-155-5p, as shown in
Fig. S1.
In summary, XIST-silencing inhibited the
proliferation of the HCFs and collagen expression, which was
partially reversed by miR-155-5p-downregulation; therefore, XIST
promoted cell proliferation and the protein expression of the
fibrosis-related proteins by sponging miR-155-5p. Therefore, XIST
may be a novel effective target for AMI treatment.
Supplementary Material
Schematic illustration of the proposed
model for the role of XIST in myocardial fibrosis. lncRNA-XIST
promotes the proliferation of cardiac fibroblasts and the
accumulation of extracellular matrix by sponging miR-155-5p.
lncRNA, long non-coding RNA; miRNA/miR, microRNA; Col, collagen;
α-SMA, α smooth muscle actin.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ designed the study. HZ and JM were responsible
for the data collection and analysis. HZ and FL collaborated to
perform the data analysis. All authors collaborated to interpret
results and develop the manuscript. All authors read and approved
the final version of the manuscript.
Ethics approval and consent for
participation
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kurose H and Mangmool S: Myofibroblasts
and inflammatory cells as players of cardiac fibrosis. Arch Pharm
Res. 39:1100–1113. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cho N, Razipour SE and McCain ML: Featured
Article: TGF-β1 1 dominates extracellular matrix rigidity for
inducing differentiation of human cardiac fibroblasts to
myofibroblasts. Exp Biol Med (Maywood). 243:601–612.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yuan X, Pan J, Wen L, Gong B, Li J, Gao H,
Tan W, Liang S, Zhang H and Wang X: MiR-144-3p enhances cardiac
fibrosis after myocardial infarction by targeting PTEN. Front Cell
Dev Biol. 7(249)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Puvvula PK: LncRNAs regulatory networks in
cellular senescence. Int J Mol Sci. 20(2615)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yarani R, Mirza AH, Kaur S and Pociot F:
The emerging role of lncRNAs in inflammatory bowel disease. Exp Mol
Med. 50:1–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xiao L, Gu Y, Sun Y, Chen J, Wang X, Zhang
Y, Gao L and Li L: The long noncoding RNA XIST regulates cardiac
hypertrophy by targeting miR-101. J Cell Physiol. 234:13680–13692.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
McKiernan PJ, Molloy K, Cryan SA,
McElvaney NG and Greene CM: Long noncoding RNA are aberrantly
expressed in vivo in the cystic fibrosis bronchial epithelium. Int
J Biochem Cell Biol. 52:184–191. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang Y, Liang Y, Luo J, Nie J, Yin H, Chen
Q, Dong J, Zhu J, Xia J and Shuai W: XIST/miR-139 axis regulates
bleomycin (BLM)-induced extracellular matrix (ECM) and pulmonary
fibrosis through β-catenin. Oncotarget. 8:65359–65369.
2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang J, Shen Y, Yang X, Long Y, Chen S,
Lin X, Dong R and Yuan J: Silencing of long noncoding RNA XIST
protects against renal interstitial fibrosis in diabetic
nephropathy via microRNA-93-5p-mediated inhibition of CDKN1A. Am J
Physiol Renal Physiol. 317:F1350–F1358. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu Y, Gao L, Guo S, Zhao X, Li R, Yan X,
Li Y, Wang S, Niu X, Yao L, et al: Kaempferol alleviates
angiotensin II-induced cardiac dysfunction and interstitial
fibrosis in mice. Cell Physiol Biochem. 43:2253–2263.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang ZF, Wang NP, Harmouche S, Philip T,
Pang XF, Bai F and Zhao ZQ: Postconditioning attenuates coronary
perivascular and interstitial fibrosis through modulating
angiotensin II receptors and Angiotensin-converting enzyme 2 after
myocardial infarction. J Surg Res. 211:178–190. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lin C, Wei D, Xin D, Pan J and Huang M:
Ellagic acid inhibits proliferation and migration of cardiac
fibroblasts by Down-regulating expression of HDAC1. J Toxicol Sci.
44:425–433. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tang CM, Zhang M, Huang L, Hu ZQ, Zhu JN,
Xiao Z, Zhang Z, Lin QX, Zheng XL, Yang M, et al: CircRNA_000203
enhances the expression of fibrosis-associated genes by
derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac
fibroblasts. Sci Rep. 7(40342)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhao N, Yu H, Sun M, Zhang Y, Xu M and Gao
W: MiRNA-711-SP1-collagen-I pathway is involved in the
anti-fibrotic effect of pioglitazone in myocardial infarction. Sci
China Life Sci. 56:431–439. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Maruyama T, Nishihara K, Umikawa M,
Arasaki A, Nakasone T, Nimura F, Matayoshi A, Takei K, Nakachi S,
Kariya KI, et al: MicroRNA-196a-5p is a potential prognostic marker
of delayed lymph node metastasis in early-stage tongue squamous
cell carcinoma. Oncol Lett. 15:2349–2363. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Castro-Dominguez Y, Dharmarajan K and
McNamara RL: Predicting death after acute myocardial infarction.
Trends Cardiovasc Med. 28:102–109. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yin Y, Zhang Q, Zhao Q, Ding G, Wei C,
Chang L, Li H, Bei H, Wang H, Liang J, et al: Tongxinluo attenuates
myocardiac fibrosis after acute myocardial infarction in rats via
inhibition of endothelial-to-mesenchymal transition. Biomed Res
Int. 2019(6595437)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
van Putten S, Shafieyan Y and Hinz B:
Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol.
93:133–142. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xie ZY, Wang FF, Xiao ZH, Liu SF, Lai YL
and Tang SL: Long noncoding RNA XIST enhances ethanol-induced
hepatic stellate cells autophagy and activation via miR-29b/HMGB1
axis. IUBMB Life. 71:1962–1972. 2019.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Chen L, Zheng SY, Yang CQ, Ma BM and Jiang
D: MiR-155-5p inhibits the proliferation and migration of VSMCs and
HUVECs in atherosclerosis by targeting AKT1. Eur Rev Med Pharmacol
Sci. 23:2223–2233. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang Z, Liang K, Zou G, Chen X, Shi S,
Wang G, Zhang K, Li K and Zhai S: Inhibition of miR-155 attenuates
abdominal aortic aneurysm in mice by regulating macrophage-mediated
inflammation. Biosci Rep. 38(BSR20171432)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen NX, Kiattisunthorn K, O'Neill KD,
Chen X, Moorthi RN, Gattone VH II, Allen MR and Moe SM: Decreased
microRNA is involved in the vascular remodeling abnormalities in
chronic kidney disease (CKD). PLoS One. 8(e64558)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang G, Shi H, Wang L, Zhou M, Wang Z,
Liu X, Cheng L, Li W and Li X: MicroRNA and transcription factor
mediated regulatory network analysis reveals critical regulators
and regulatory modules in myocardial infarction. PLoS One.
10(e0135339)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhao F, Wu Y, Yang W, Wu D, Wang C and
Zhang F: Inhibition of vascular calcification by microRNA-155-5p is
accompanied by the inactivation of TGF-β1/Smad2/3 signaling
pathway. Acta Histochem. 122(151551)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Halper J and Kjaer M: Basic components of
connective tissues and extracellular matrix: Elastin, fibrillin,
fibulins, fibrinogen, fibronectin, laminin, tenascins and
thrombospondins. Adv Exp Med Biol. 802:31–47. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jabłońska-Trypuć A, Matejczyk M and
Rosochacki S: Matrix metalloproteinases (MMPs), the main
extracellular matrix (ECM) enzymes in collagen degradation, as a
target for anticancer drugs. J Enzyme Inhib Med Chem. 31 (Suppl
1):S177–S183. 2016.PubMed/NCBI View Article : Google Scholar
|














