Introduction
Intracranial aneurysms (IAs) are pathological
dilations of blood vessels in the cerebral region (1). They occur when the wall of an
intracranial vessel becomes weak and forms bulges, which are
susceptible to rupture. An aneurysm ruptures when the wall finally
becomes too weak to resist blood pressure and this causes
subarachnoid hemorrhage (1,2). Surgical treatments such as ligation of
the aneurysm neck are available for IA patients with unruptured
aneurysms (3). However, all
currently available surgical methods are associated with high risks
of rupture, which can potentially result in catastrophic
subarachnoid hemorrhage (3). To
date, details of the underlying mechanism for the development and
progression of IA are poorly understood. Due to the severe impacts
on patients and high risks of the available surgical treatments, a
less invasive medical treatment for IA is required.
Phenotypic modulation is a property of smooth muscle
cells, which refers to the changes in phenotypes that occur in
response to environmental cues, and includes migration,
proliferation and the production of extracellular matrix (4-8).
This property is profound in vascular smooth muscle cells (VSMCs)
as they adapt to changes in blood pressure-induced tension and
damage, and triggers the repair of damage by accelerating cell
migration and proliferation (4-8).
Unfortunately, phenotypic modulation also contributes to the
progression of a number of vascular diseases, since cells are very
susceptible to various signaling cues from the environment
(9). It has been reported that
phenotypic modulation of smooth muscle cells occurs in the early
stage before aneurysm formation (4,5). It
has also been revealed that VSMCs in the cerebral aneurysm wall
switch to a synthetic phenotype, in comparison with the contractile
phenotype in normal cerebral arteries (10). A previous study has suggested that
tumor necrosis factor-α plays a role in such phenotype switching,
by suppressing the expression of contractile proteins and promoting
the pro-inflammatory/matrix remodeling phenotype in cultured VSMCs
(6). However, further investigation
is required regarding the mechanisms for phenotypic modulation in
IA.
MicroRNAs (miRNAs/miRs) serve an important role in
gene regulation, which in turn affects the expression of certain
proteins. They are short, non-coding, single-stranded RNA sequences
that are ~22 nucleotides in length (11-13).
They act as inhibitors for mRNAs by base-pair binding to the
3'-untranslated region (UTR) of the target mRNA, which either
prevents it from being translated or induces degradation. miRNA has
been observed to be involved in numerous cardiac diseases,
including cardiomyocyte hypertrophy, cardiac fibrosis and heart
failure (11-13).
Notably, it also allows the manipulation of gene expression in
vivo, via the use of miRNA mimics or inhibitors (13). A previous study has suggested that
miRNAs may be important in the development of IA, as 157 miRNAs
were identified to be differentially expressed in tissues with
aneurysm (14). Among them, miR-29b
is known to be involved in cell proliferation and apoptosis in
various diseases (15). miR-29b is
a member of the miR-29 miRNA family and is encoded by the precursor
stem sequence pre-miR-29b. Studies have previously demonstrated
miR-29b to be a potential therapeutic agent for cardiac fibrosis
(16,17). Furthermore, it has also been
reported that miR-29b was significantly downregulated in serum
samples from patients with IA (18).
The transforming growth factor β (TGF-β) family is a
group of proteins that are known to regulate cell differentiation,
with TGF-β1 being the most abundant isoform (19-20).
The main function of TGF-β is to bind to its receptor protein, the
TGF-β receptor, which essentially causes the phosphorylation of
Smad3(18). Phosphorylated Smad
(Smad2 and Smad3) proteins eventually form a complex that
translocates into the nucleus and activates a number of
transcription factors responsible for cell differentiation and
migration (20). Notably,
bioinformatics analysis was performed in the present study to
predict potential miR-29b binding targets, where screening
conducted in the present study revealed that TGF-β1 is one of the
potential targets of miR-29b. Therefore, it was speculated that
miR-29b affects cell migration in IA by targeting the TGF-β/Smad
pathway. The present study aimed to investigate the role of miR-29b
in the phenotypic modulation of VSMC in patients with IA and its
potential underlying mechanisms, in order to facilitate the search
for potential therapeutic treatments for patients with IA.
Materials and methods
Cell culture
Human umbilical artery smooth muscle cells (HUASMCs;
cat. no. BH-X005; https://www.biomart.cn/infosupply/67539695.htm?from=search_2)
were purchased from Shanghai Bohu Biotechnology Co., Ltd. and
cultured in Dulbecco's modified Eagle's medium (DMEM; Hyclone; GE
Healthcare Life Sciences), supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.), 50 U/ml penicillin G
and 250 µg/ml streptomycin (Gibco; Thermo Fisher Scientific Inc.)
at 37˚C with 5% CO2 in an incubator. VSMCs (cat. no.
DC740; https://www.biomart.cn/infosupply/48373937.htm?from=search_1)
were obtained from Shanghai Zeye Biotechnology Co., Ltd. and
cultured in DMEM supplemented with 10% FBS at 37˚C with 5%
CO2 in an incubator. When the cell density reached
>80%, cells were washed twice with sterilized PBS. Then, 0.25%
trypsin was used to dissociate cell-to-cell contacts until cells
appeared detached, followed by trypsin inactivation by the addition
of complete medium (DMEM supplemented with 10% FBS). The suspended
cells were gently pipetted into a centrifuge tube and collected by
centrifugation (157 x g, 5 min) at room temperature. Lastly, the
supernatant was removed, and cells were re-suspended in complete
medium with 10% FBS for further experiments.
Cell transfection
The oligonucleotides miR-29b mimic
(5'-UAGCACCAUUUGAAAUCAGUGUU-3'), mimic NC
(5'-GAAUGCUGGUUUUCAUAUGGUAGA-3'), miR-29b-specific inhibitor
(5'-AACACUGAUUUCAAAUG GUGCUA-3') and the corresponding negative
control inhibitor (5'-CAGUACUUUUGUGUAGUACAA-3') were purchased from
Sigma-Aldrich; Merck KGaA. I total, 1 µg miR-29b mimic, miR-29b
inhibitor or the corresponding negative control was transfected
into HUASMCs and VSMCs via Lipofectamine® 2000 (Thermo
Fisher Scientific, Inc.). Following transfection (24 h) with the
miR-29b mimic, treatment with TGF-β1 at a final concentration of 1
µg/ml was performed. After transfection for 6 h at 37˚C with 5%
CO2, cells were collected for further
experimentation.
RNA interference (RNAi) and
transfection
For a further knockdown experiment of TGF-β1 mRNA, a
small interfering RNA (siRNA) targeting TGF-1 (si-TGF-β1: 5'-GCAU
CUCACUCAUGUUGAUGGUCUA-3') was custom synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). A non-specific scrambled siRNA
(si-control: 5'-UUCUCCG AACGUGUCACGUTT-3'; MDbio, Inc.) was used as
a control. Si-TGF-β1 and si-control were each transfected into
VSMCs with the Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.). The final siRNA concentration was 50 nM,
depending on the optimal test. After transfection for 48 h, cells
were collected for the following experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to manufacturer's protocol. Relative expression of
miR-29b in each group was measured by qPCR using a One-Step
SYBR® PrimeScript™ RT-PCR kit II (Takara Bio., Inc.)
according to the manufacturer's protocol 3 days after infection.
The reverse transcription conditions were as follows: 37˚C for 60
min; 85˚C for 5 min and 4˚C for 5 min. The qPCR reaction
thermocycling conditions were as follows: Initial denaturation at
95˚C for 1 min, followed by 40 cycles of 95˚C for 10 sec and 55˚C
for 50 sec. The expression level of miR-29b was normalized by U6,
whilst the level of TGF-β1 was normalized to GAPDH using the
2-ΔΔCq method (21). The
sequences for primers used for RT-qPCR were: miR-29b forward,
5'-UAGCACCAUUUGAAAUC-3' and reverse, 5'-AACGCTTCACGAATTTGCGT-3';
TGF-β1 forward, 5'-CAATTCCTGGCGATACCTCAG-3' and reverse,
5'-GCACAACTCCGGTGACATCAA-3'; U6 forward, 5'-GCTTCGGCAGCACATATAC-3'
and reverse, 5'-AACGCTTCACGAATTTGCGT-3'; GAPDH forward,
5'-GTCAACGGATTTGGTCTGTATT-3' and reverse, 5'-AG
TCTTCTGGGTGGCAGTGAT-3'.
Cell viability
A total of 9,000 cells/well was used for each set,
following which MTT assay was performed using an MTT Cell Growth
assay kit (Sigma-Aldrich; Merck KGaA) by following the
manufacturer's protocol. DMSO was used to dissolve the purple
formazan crystals. The OD490 absorbance values for the
samples were measured using a plate reader.
Transwell migration assay
A Transwell Migration assay ELISA kit (Thermo Fisher
Scientific, Inc.) was used to evaluate cell migration in the
different groups of VSMCs and HUASMCs. The migration assay was
performed by adding 200 µl suspended cell sample (4x108
cells/l) to the upper chamber and 500 µl DMEM with 10% FBS to the
lower chamber, followed by incubation for 48 h at room temperature.
The medium from the upper chamber was then discarded, and cells on
the upper surface of the membrane were cleared away using a swab.
Cells on the lower surface of the membrane were then fixed for 10
min using 4% paraformaldehyde at room temperature and stained with
0.1% crystal violet solution at room temperature for 10 min. The
membranes were subsequently stained and viewed under a light
microscope (magnification, x20) for cell counting, from images
taken from five visual fields per chamber.
Luciferase reporter assay
TargetScan 7.2 (http://www.targetscan.org) used to predict potential
binding targets of miR-29b. The wild-type (WT) or mutant (mut)
3'-UTR of TGF-β1 was cloned into the pGL3 luciferase reporter
vector (Shanghai GeneChem Co., Ltd.). The corresponding mutant
sequence was designed to be identical to that of miR-29b to prevent
sequence-specific binding. The pGL3-TGF-WT or pGL3-TGF-mut plasmid
(1 µg cDNA) was co-transfected with miR-29b mimic (20 pmol RNA)
into cultured VSMCs A firefly-Renilla dual luciferase assay was
performed on samples 48 h after transfection to determine
luciferase activities, using a Dual-Luciferase® Reporter
assay system (Promega Corporation) following the manufacturer's
protocol. Results were detected using a Synergy 2™ Microplate
Reader (BioTek Instruments, Inc.).
Western blotting
VSMCs and HUASMCs were lysed using RIPA buffer
(Beyotime Institute of Biotechnology). Protein quantification was
measured by performing bicinchoninic acid assay (Beyotime Institute
of Biotechnology). Proteins were extracted (30 µg) followed by
separation using 10% SDS-PAGE and then transferred onto a
polyvinylidene difluoride membranes. The membrane was then blocked
with 5% fat-free dry milk for 2 h at room temperature, followed by
overnight incubation with mouse anti-TGF-β1 (cat. no. sc-130348),
phosphorylated (p)-Smad3 (cat. no. sc-517575), Smad3 (cat. no.
sc-101154) or GAPDH (cat. no. sc-32233) primary antibodies (1:500;
Santa Cruz Biotechnology, Inc.) at 4˚C. Next, the membrane was
incubated with horseradish peroxidase-conjugated goat anti-mouse
IgG secondary antibody (1:6,000; cat. no. 7076; Cell Signaling
Technology, Inc.) for 2 h at room temperature. Protein expression
was then detected by enhanced chemiluminescence (ECL) using
Immobilon ECL Ultra Western HRP substrate (EMD Millipore) and
quantified using Quantity One software 4.6 (Bio-Rad Laboratories,
Inc.).
Statistical analysis
SPSS version 16.0 statistics software (SPSS, Inc.)
was used for statistical analysis throughout this study. All
statistical results are presented as the mean ± standard deviation.
The data analysis between multiple groups was based on one-way
analysis of variance, followed by the Student-Newman-Keuls post hoc
tests (three groups) or Bonferroni post hoc tests (>3 groups).
Two-way ANOVA with Bonferroni post hoc test were performed when
comparing the MTT results. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-29b affects cell proliferation and
migration
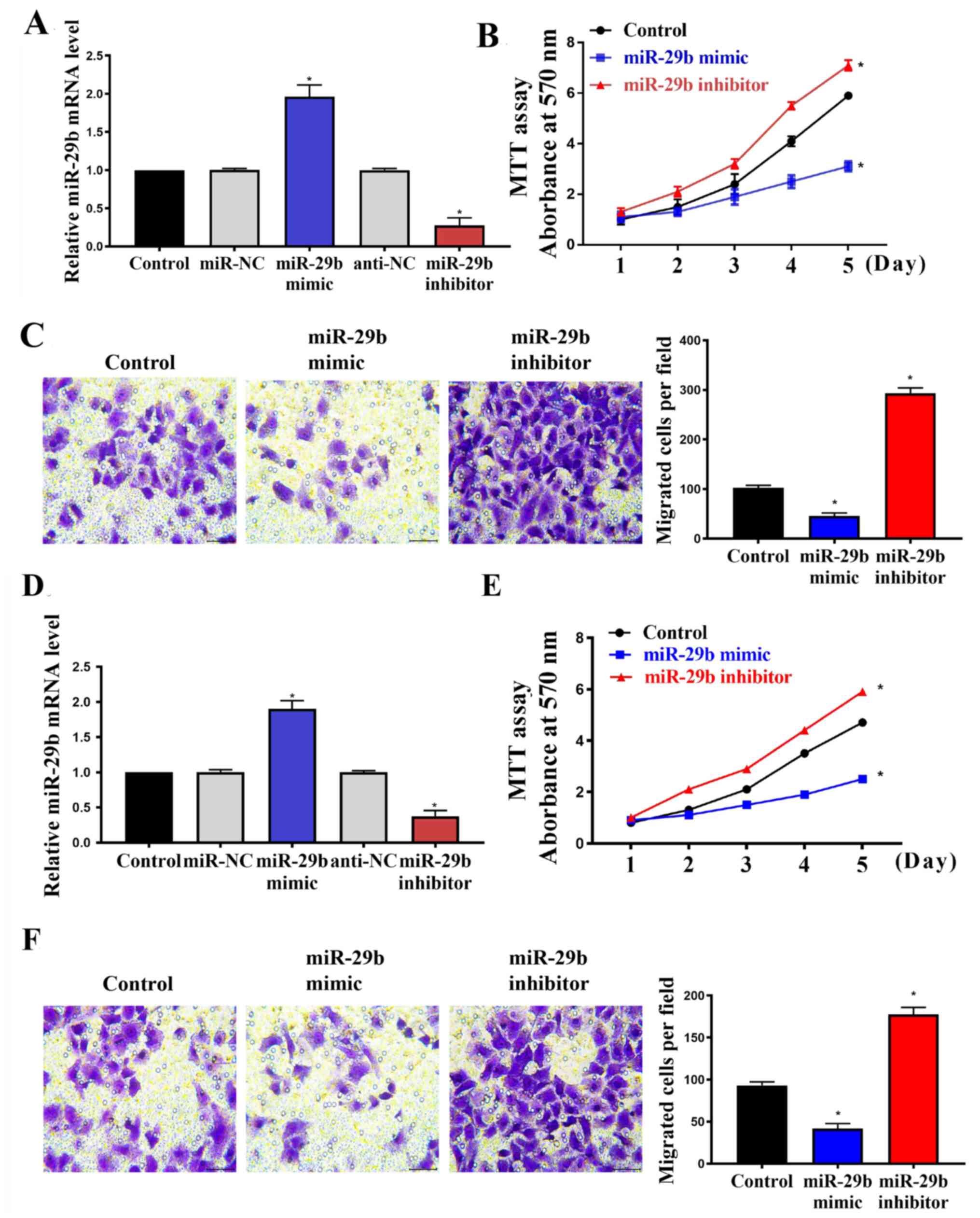
To investigate the effect of miR-29b on cell
proliferation and migration, HUASMCs and VSMCs were infected with
miR-29b mimic, miR-29b inhibitor and their respective controls. The
transfection efficiency was evaluated by RT-qPCR and the results
confirmed that miR-29b was upregulated by miR-29b mimic and
downregulated by miR-29b inhibitor significantly in both HUASMCs
(Fig. 1A; P<0.05) and VSMCs
(Fig. 1D; P<0.05) compared with
the respective control. Regarding the role of miR-29b, MTT
(Fig. 1B and E; P<0.05) and Transwell migration
assays (Fig. 1C and 1F; P<0.05) indicated that the
upregulation of miR-29b via the incorporation of miR-29b mimic
significantly suppressed cell proliferation and migration compared
with the control, whereas downregulation of the miRNA using the
miR-29b inhibitor significantly increased these properties.
Overexpression of miR-29b suppresses
the TGF-β/Smad3 signaling pathway in HUASMCs and VSMCs
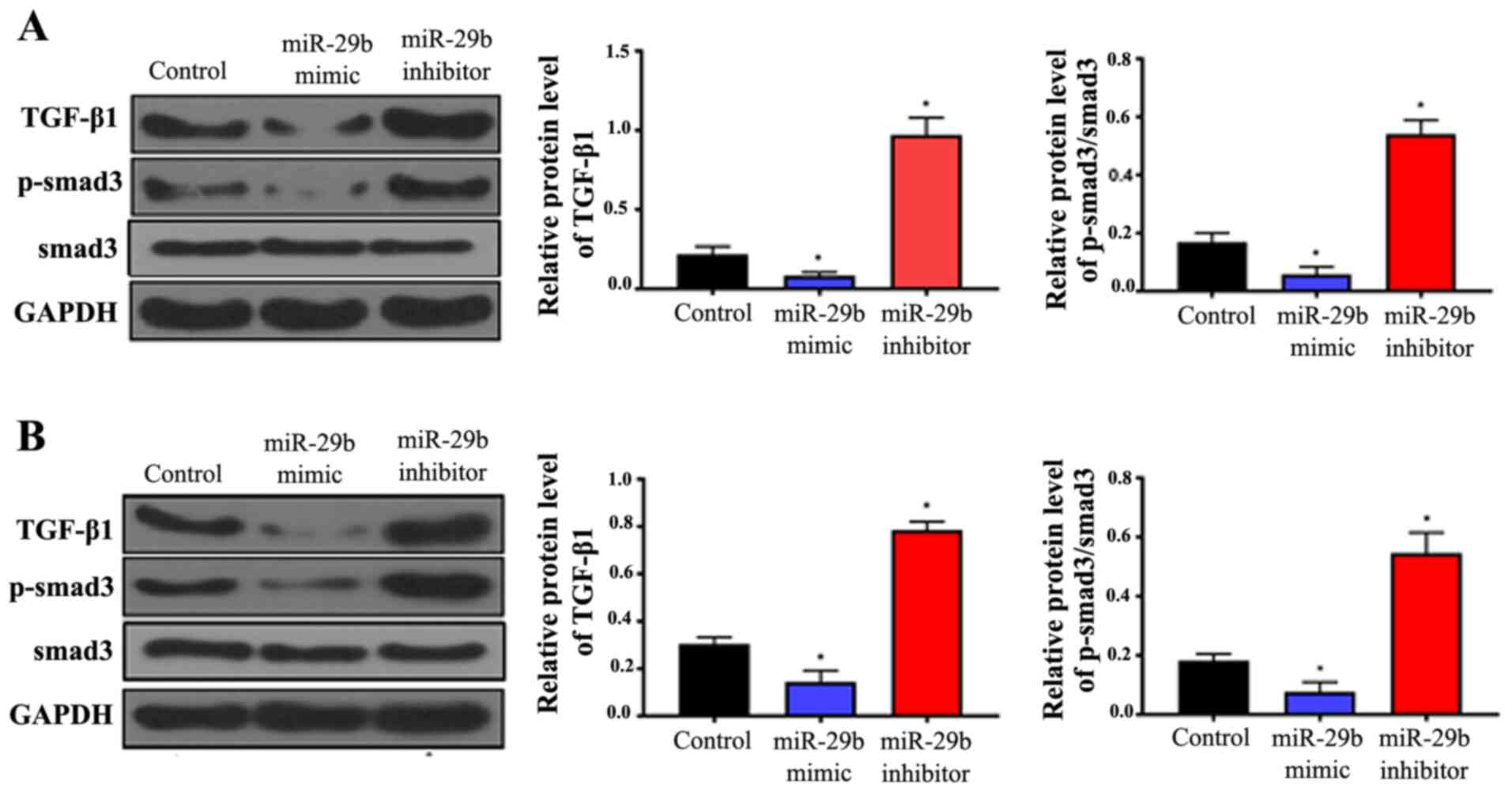
The expression levels of TGF-β1 and Smad3 were
analyzed in all sample groups by western blotting. The results
indicated that overexpression of miR-29b significantly suppressed
the expression of TGF-β1 and decreased the phosphorylation of
Smad3, whereas inhibition of miR-29b significantly increased TGF-β1
expression and Smad3 phosphorylation compared with the controls
(Fig. 2; P<0.05). These effects
were observed in HUASMCs (Fig. 2A)
and VSMCs (Fig. 2B). This suggests
that miR-29b functions by inhibiting the TGF-β/Smad3 pathway.
miR-29b may inhibit the TGF-β/Smad3
signaling pathway by directly targeting the TGF-β1 gene
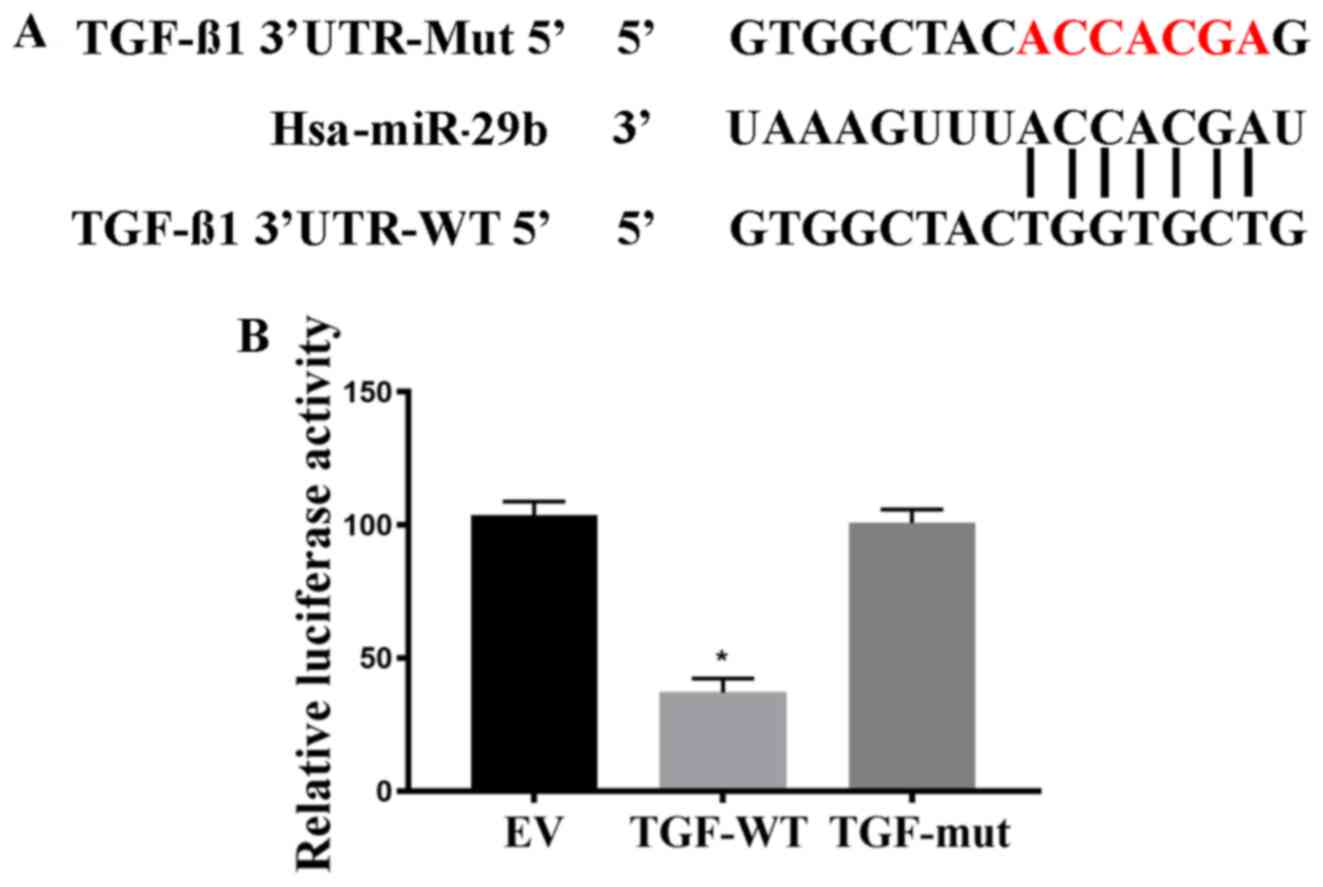
TargetScan analysis found that miR-29b had a
sequence complementary to the 3'UTR of TGF-β1 mRNA. To investigate
whether TGF-β1 is the direct target of miR-29b, a Dual-luciferase
assay was performed using the putative binding site of miR-29b in
the 3'-UTR of TGF-β1 as presented in Fig. 3A. The results indicated that
luciferase activity was significantly lower for the WT 3'-UTR of
TGF-β1 compared with the EV control (Fig. 3B; P<0.05), whereas the mutant
version showed no change in luciferase response. This provides
evidence that miR-29b targets TGF-β1 by directly binding to its
3'-UTR.
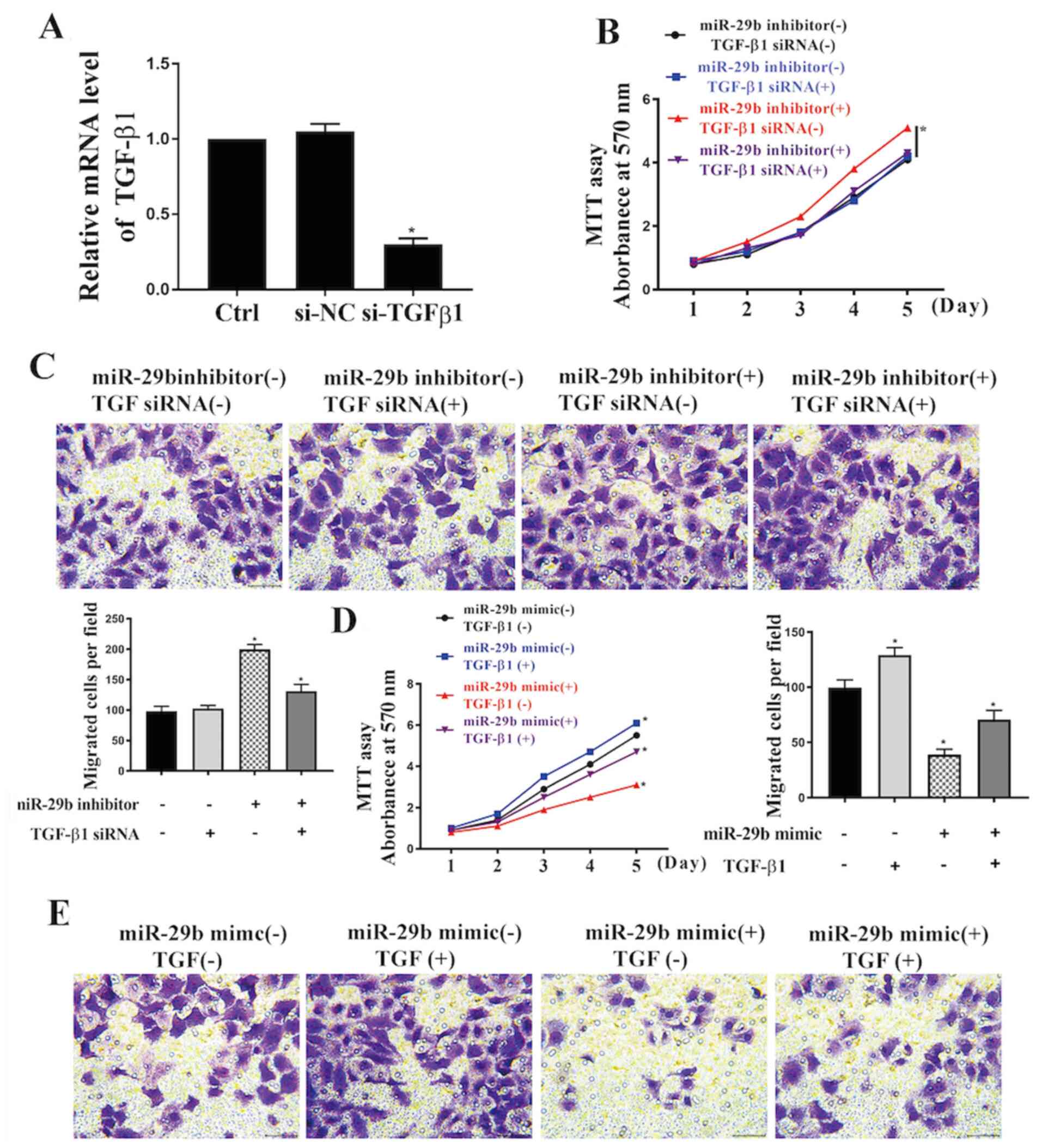
Knockdown of TGF-β1 reverses miR-29b
inhibition-induced increases in cell viability and migration
To further examine the role of the TGF-β/Smad3
pathway in the miR-29b-induced reduction of cell viability, miR-19b
inhibitor was transfected into VSMCs with or without TGF-β1 siRNA.
Reduction of the expression level of TGF-β1 following transfection
with TGF-β1 siRNA was confirmed by RT-qPCR (Fig. 4A). As shown in Fig. 4B and C, TGF-β1 siRNA significantly suppressed
miR-29b inhibitor-induced increases in cell viability and
migration. Furthermore, miR-29b mimic was transfected into VSMCs
with or without TGF-β1 treatment (Fig.
4D). The MTT and migration assay results in Fig. 4D and E show that miR-29b mimic significantly
suppressed cell viability and migration, and the TGF-β1 treatment
significantly reversed this effect.
Discussion
The role of miR-29b has been investigated
extensively in the past decade, due to the fact that its expression
is known to be altered in many cancer and diseased cells compared
with normal cells (16,19,22,23).
These studies suggest that its major effects include the
suppression of cell proliferation and migration, which serve an
important role in cancer-associated studies. The present study
sought to identify potential targets for miR-29b using TargetScan.
TGF-β1 was identified as one of the targets of miR-29b, and is
known to induce VSMC proliferation and synthesis of extracellular
matrix through the Smad3 signaling pathway (24). TGF-β1 is activated by binding to
transmembrane kinase receptors, which phosphorylate Smad3 for
downstream signaling (25,26). One possible signaling pathway of
Smad3 is the upregulation of monocyte chemoattractant protein-1,
which is known to serve an important role in angiogenesis and tumor
progression by stimulating VSMC migration, and is overexpressed in
tumor cells (27,28). In addition, miR-29b has been
identified to have a protective role in cardiac fibrosis, whereby
thickening of heart valves occurs due to the proliferation of
vascular muscle cells, by targeting the TGF-β/Smad3 signaling
pathway (16,29,30).
The present study investigated the involvement of miR-29b in the
TGF-β/Smad3 signaling pathway in VSMCs and HUASMCs, as well as the
underlying mechanism.
Results from migration assays in the present study
clearly demonstrated that the overexpression of miR-29b suppressed
the viability and migration of VSMCs and HUASMCs, while suppression
of its expression exhibited the reverse effect. This is consistent
with the previous finding that miR-29b is significantly
downregulated in the VSMCs of patients with IA (19). Furthermore, the present study
demonstrated that miR-29b overexpression reduces the expression of
TGF-β1 and phosphorylation of Smad3, while miR-29b downregulation
has the opposite effect in VSMCs and HUASMCs. The luciferase assay
suggested that miR-29b targets the 3'-UTR of TGF-β1, which in turn
reduces the expression of TGF-β1 and suppresses the TGF-β/Smad3
signaling pathway, thus reducing cell viability and migration. For
further validation, changes in cell migration were monitored while
the expression of TGF-β1 was manipulated. This was achieved by
either providing a TGF-β1 treatment to increase its level, or
transfecting with an siRNA that interferes with the translation of
TGF-β1. For the cells with increased viability due to miR-29b
inhibition and hence overexpressed TGF-β, the TGF-β1 siRNA
partially reversed the effect. Furthermore, with miR-29b mimic,
where miR-29b was upregulated, TGF-β expression was reduced and
viability was reduced, the addition of TGF-β1 treatment increased
proliferation. These findings are consistent with the proposed
mechanism.
Overall, the results indicated the potential role of
miR-29b in IA. Inhibition of miR-29b in the VSMCs is responsible
for the overexpression of TGF-β, which may promote cell
proliferation and migration via the TGF-β/Smad3 signaling pathway.
Increased rates of cell proliferation and migration are important
aspects in the switch to the synthetic phenotype (31). In response to a changing
environment, such as vascular injury, this phenotype would promote
cell growth and the synthesis of extracellular matrix, which helps
in vascular repair. Unfortunately, this property is also the main
contributor in the progression of IA (32,33).
The present study has shown that these effects can be suppressed
in vitro by transfecting cells with miR-29b mimic to
upregulate miR-29b, or by the suppression of TGF-β1 by transfecting
cells with siRNA to interfere with its translation. These results
provide a possible approach for a less-invasive treatment for
patients with IA. However, further investigation of the expression
of miR-29b in VSMCs from patients with IA is required in order to
explore the upstream gene regulation for miR-29b.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LL designed the study and wrote the manuscript. LL,
SR and XH performed the experiments. ZZ, LJ and HJ collected and
analyzed data. All authors have read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Frösen J, Tulamo R, Paetau A, Laaksamo E,
Korja M, Laakso A, Niemelä M and Hernesniemi J: Saccular
intracranial aneurysm: Pathology and mechanisms. Acta Neuropathol.
123:773–786. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bor AS, Rinkel GJ, Adami J, Koffijberg H,
Ekbom A, Buskens E, Blomqvist P and Granath F: Risk of subarachnoid
haemorrhage according to number of affected relatives: A population
based case-control study. Brain. 131:2662–2665. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Louw DF, Asfora WT and Sutherland GR: A
brief history of aneurysm clips. Neurosurg Focus.
11(E4)2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ailawadi G, Moehle CW, Pei H, Walton SP,
Yang Z, Kron IL, Lau CL and Owens GK: Smooth muscle phenotypic
modulation is an early event in aortic aneurysms. J Thorac
Cardiovasc Surg. 138:1392–1399. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nakajima N, Nagahiro S, Sano T, Satomi J
and Satoh K: Phenotypic modulation of smooth muscle cells in human
cerebral aneurysmal walls. Acta Neuropathol. 100:475–480.
2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ali MS, Starke RM, Jabbour PM, Tjoumakaris
SI, Gonzalez LF, Rosenwasser RH, Owens GK, Koch WJ, Greig NH and
Dumont AS: TNF-α induces phenotypic modulation in cerebral vascular
smooth muscle cells: Implications for cerebral aneurysm pathology.
J Cereb Blood Flow Metab. 33:1564–1573. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kawai-Kowase K and Owens GK: Multiple
repressor pathways contribute to phenotypic switching of vascular
smooth muscle cells. Am J Physiol Cell Physiol. 292:C59–C69.
2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801.
2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lacolley P, Regnault V, Segers P and
Laurent S: Vascular smooth muscle cells and arterial stiffening:
Relevance in development, aging, and disease. Physiol Rev.
97:1555–1617. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mao N, Gu T, Shi E, Zhang G, Yu L and Wang
C: Phenotypic switching of vascular smooth muscle cells in animal
model of rat thoracic aortic aneurysm. Interact Cardiovasc Thorac
Surg. 21:62–70. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
van Rooij E, Marshall WS and Olson EN:
Toward microRNA-based therapeutics for heart disease: The sense in
antisense. Circ Res. 103:919–928. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342.
2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
van Rooij E and Olson EN: MicroRNA
therapeutics for cardiovascular disease: Opportunities and
obstacles. Nat Rev Drug Discov. 11:860–872. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Liu D, Han L, Wu X, Yang X, Zhang Q and
Jiang F: Genome-wide microRNA changes in human intracranial
aneurysms. BMC Neurol. 14(188)2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yan B, Guo Q, Fu FJ, Wang Z, Yin Z, Wei YB
and Yang JR: The role of miR-29b in cancer: Regulation, function,
and signaling. Onco Targets Ther. 8:539–548. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM
and Lan HY: miR-29b as a therapeutic agent for angiotensin
II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol
Ther. 22:974–985. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
McMullen JR and Bernardo BC: Inhibition of
miR-29 protects against cardiac hypertrophy and fibrosis: New
insight for the role of miR-29 in the heart. Non-coding RNA
Investig. 2(14)2018.
|
|
18
|
Wang G, Matsuura I, He D and Liu F:
Transforming growth factor-{beta}-inducible phosphorylation of
Smad3. J Biol Chem. 284:9663–9673. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sun L, Zhao M, Zhang J, Lv M, Li Y, Yang
X, Liu A and Wu Z: MirR-29B downregulation induces phenotypic
modulation of vascular smooth muscle cells: Implication for
intracranial aneurysm formation and progression to rupture. Cell
Physiol Biochem. 41:510–518. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fang J, Xu H, Yang C, Morsalin S,
Kayarthodi S, Rungsrisuriyachai K, Gunnal U, Mckenzie B, Rao VN and
Reddy ES: Ets related gene and Smad3 proteins collaborate to
activate transforming growth factor-beta mediated signaling pathway
in ETS related gene-positive prostate cancer cells. J Pharm Sci
Pharmacol. 1:175–181. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kole AJ, Swahari V, Hammond SM and
Deshmukh M: miR-29b is activated during neuronal maturation and
targets BH3-only genes to restrict apoptosis. Genes Dev.
25:125–130. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Eyholzer M, Schmid S, Wilkens L, Mueller
BU and Pabst T: The tumour-suppressive miR-29a/b1 cluster is
regulated by CEBPA and blocked in human AML. Br J Cancer.
103:275–284. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tsai S, Hollenbeck ST, Ryer EJ, Edlin R,
Yamanouchi D, Kundi R, Wang C, Liu B and Kent KC: TGF-beta through
Smad3 signaling stimulates vascular smooth muscle cell
proliferation and neointimal formation. Am J Physiol Heart Circ
Physiol. 297:H540–H549. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Flanders KC, Sullivan CD, Fujii M, Sowers
A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB,
et al: Mice lacking Smad3 are protected against cutaneous injury
induced by ionizing radiation. Am J Pathol. 160:1057–1068.
2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ma J, Wang Q, Fei T, Han JD and Chen YG:
MCP-1 mediates TGF-beta-induced angiogenesis by stimulating
vascular smooth muscle cell migration. Blood. 109:987–994.
2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Salcedo R, Ponce ML, Young HA, Wasserman
K, Ward JM, Kleinman HK, Oppenheim JJ and Murphy WJ: Human
endothelial cells express CCR2 and respond to MCP-1: Direct role of
MCP-1 in angiogenesis and tumor progression. Blood. 96:34–40.
2000.PubMed/NCBI
|
|
29
|
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H,
Qi Y and Du J: Macrophage-stimulated cardiac fibroblast production
of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis
induced by angiotensin II. PLoS One. 7(e35144)2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Davis-Dusenbery BN, Wu C and Hata A:
Micromanaging vascular smooth muscle cell differentiation and
phenotypic modulation. Arterioscler Thromb Vasc Biol. 31:2370–2377.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
ten Dijke P and Arthur HM: Extracellular
control of TGFbeta signalling in vascular development and disease.
Nat Rev Mol Cell Biol. 8:857–869. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Petsophonsakul P, Furmanik M, Forsythe R,
Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees
B and Schurgers L: Role of vascular smooth muscle cell phenotypic
switching and calcification in aortic aneurysm formation.
Arterioscler Thromb Vasc Biol. 39:1351–1368. 2019.PubMed/NCBI View Article : Google Scholar
|