Introduction
Keloid is a type of proliferative fibroma that may
form after trauma, and is characterized by abnormal wound healing,
fibroblast proliferation and extracellular matrix deposition.
Keloid is considered as a benign human tumor without malignant
potential, which invades the adjacent normal skin tissue beyond the
initial boundaries of the trauma (1,2).
Fibroblasts are considered to play a key role in the formation and
development of keloids (3). The
hyperproliferation of fibroblasts and deposition of extracellular
matrix results in collagen metabolism disorder, which is the main
cause of keloid fibrosis. However, the pathogenesis of keloids has
not been fully elucidated (4,5).
Researchers have found that keloids are accompanied
by chronic inflammation and immune abnormalities (5), suggesting that proinflammatory
cytokines may be involved in the keloid formation process. Tumor
necrosis factor (TNF)-α is a type of cytokine, which is secreted by
various cells and is involved in the maintenance and homeostasis of
the immune system, inflammation and host defense. However, TNF-α is
also involved in pathological processes such as chronic
inflammation, autoimmunity and malignant diseases (6). In particular, TNF-α is involved in the
pathogenesis of various fibrotic disorders, including liver
fibrosis, pulmonary fibrosis, cardiac fibrosis and scleroderma,
among others (7). The level of
serum TNF-α in African American individuals with keloids was
observed to be higher compared with that in normal subjects without
keloids (8), and the expression of
TNF-α in the keloid was shown to be higher compared with that in
normal tissues (9). Zhu et
al (10) found that stimulation
with TNF-α at a low concentration (50 ng/ml) may lead to the
proliferation of keloid fibroblasts (KFs) in vitro. However,
the specific role and mechanism of action of TNF-α in the
pathogenesis of keloids are unclear. In tumor research, TNF-α was
found to promote cell proliferation through activating the nuclear
factor (NF)-κB, c-Jun N-terminal kinase (JNK) and p38
mitogen-activated protein kinase (MAPK) pathways, which were
regulated by the concentration of TNF-α (11). However, further experimental
research is required to determine whether the function and
mechanism of action of TNF-α in tumors is involved in keloid
pathogenesis.
Based on the keloid characteristics and the role of
TNF-α in fibrotic and neoplastic diseases, it was hypothesized that
TNF-α may be involved in the pathogenesis of keloids. Therefore,
the aim of the present study was to explore the effect and
mechanism of action of TNF-α in keloid formation. First, the
expression of TNF-α and its main receptors, sTNFR1 and sTNFR2, was
detected in the peripheral blood of patients with keloids and
healthy participants, whereas keloid and normal skin tissues and
fibroblast culture supernatants were cultured in vitro.
Subsequently, keloid and normal skin fibroblasts cultured in
vitro were stimulated using recombinant human TNF-α protein,
and the effects of different TNF-α concentrations on the cell
survival of the two types of fibroblasts and the underlying
mechanisms were investigated, in the hope of elucidating the role
of TNF-α in keloid pathogenesis and providing theoretical support
and an experimental basis for the diagnosis and treatment of
keloids.
Materials and methods
Surgical specimens and peripheral
blood samples
Patients with keloids (n=20) and healthy control
patients (n=18) who underwent surgical treatment between February
2016 and March 2018 at the Department of Plastic and Burn Surgery
of West China Hospital, Sichuan University (Chengdu, China) were
enrolled in the present study. All keloids included in the study
shared certain characteristics and were confirmed by pathological
examination. A brief description is as follows (12): Exhibits continuous growth, generally
exceeding 2 years, and does not subside spontaneously. It appears
as a hard, mildly tender, raised tumor with a shiny surface and,
occasionally, with telangiectasia. The surface epithelium becomes
thinner, ranging in color from pink to purple, and may be
accompanied by hyperpigmentation. The boundary is clearly
delineated, but the outline is irregular. The patient may
experience itching and pain in the affected area.
The inclusion criteria patients with keloids were as
follows: i) Keloid diagnosed by a plastic surgeon; ii) no keloid
treatments, such as radiotherapy and injection therapy, during the
previous 5 years; and iii) no ulceration or infection in keloid
tissue or surrounding skin.
The exclusion criteria patients with keloids were as
follows: Patients i) diagnosed with malignant tumors; ii) diagnosed
with psychological diseases; iii) diagnosed with metabolic
diseases; iv) diagnosed with rheumatic immune diseases; and v)
receiving radiotherapy, injection therapy and other keloid
treatments over the previous 5 years.
The inclusion criteria of healthy control patients
were as follows: i) Non-keloid as diagnosed by a plastic surgeon;
ii) the skin had not undergone radiation or chemotherapy; and iii)
no skin disease.
The exclusion criteria of healthy control patients
were as follows: i) Exclusion criteria for patients with keloids as
aforementioned; patients ii) diagnosed with inflammatory or immune
responses; and iii) diagnosed with a skin disorder or skin
abnormality.
The demographics of the patients are summarized in
Table I. There was no statistically
significant difference in age and sex distribution between the two
groups.
 | Table IDemographics of participants included
in the present study. |
Table I
Demographics of participants included
in the present study.
| Characteristics | Keloid group
(n=20) | Control group
(n=18) |
|---|
| Age, years | 28.00±6.58 | 31.47±10.06 |
| Sex, male/female | 9/11 | 6/12 |
The protocol of the present study was reviewed and
approved by the Ethics Committee of West China Hospital of Sichuan
University (approval no. 2014-65) on April 29, 2014 and written
consent was obtained from all the participants.
Keloid samples (n=20) were obtained from the chest,
shoulders or abdomen. The areas were marked with a surgical pen and
collected using a scalpel following local anesthesia with 2%
lidocaine. Normal skin samples (n=18) were obtained from the
abdomen or chest via an incision of a 2-4-mm rim of normal skin
peripheral border without lesions. A tension-free primary closure
was ensured. Following excision and removal of the epidermis,
connective tissue and subcutaneous fat, each sample was divided
into three parts: One part was immediately stored in liquid
nitrogen (-196˚C) for RNA extraction, one was immersed in 4%
formaldehyde at room temperature for 24 h to be used for
immunohistochemistry, and the remaining sample was used for primary
culture of fibroblasts.
Fasting peripheral blood samples (1 ml) were
collected from 10 patients with keloids and 9 healthy individuals,
who were randomly selected from the participants used for skin
sample collection, using heparin as an anticoagulant. Within 30 min
of collection, the samples were centrifuged at 3,000 x g for 15 min
at room temperature, and the supernatants were preserved at
-80˚C.
Antibodies and reagents
Human TNF-α recombinant protein (cat. no. 300-01A)
was purchased from PeproTech, Inc.; infliximab (Remicade) was from
Janssen Biotech, Inc.; Johnson & Johnson; the Cell Counting
Kit-8 (CCK-8; cat. no. CK04) was from Dojindo Molecular
Technologies, Inc.; human TNF-α (cat. no. ZC-35733), sTNFR1 (cat.
no. ZC-54321) and sTNFR2 (cat. no. ZC-54322) ELISA kits were
purchased from ZCI Bio, China (http://www.zcibio.com/chanpinzhongxin_2073.html); the
cDNA reverse transcription kit (PrimeScript™ RT Reagent Kit; cat.
no. RR037A) was obtained from Takara Biotechnology Co., Ltd. and 2X
SYBR® Green premix was from Bio-Rad Laboratories, Inc.;
propidium iodide (cat. no. ST511) was from Beyotime Institute of
Biotechnology; rabbit monoclonal antibodies against TNF-α (cat. no.
ab6671), TNFR1 (cat. no. ab19139) and TNFR2 (cat. no. ab109322)
were all purchased from Abcam; rabbit polyclonal antibodies against
inhibitor of NF-κB (IκB-α; cat. no. 9242s), phosphorylated
(p)-SAPK/JNK (cat. no. 4668s), p38 MAPK (cat. no. 9212), p-p38 MAPK
(cat. no. 4511s), p-IκB-α (cat. no. 9246s) and SAPK/JNK (cat. no.
9252s) were all obtained from Cell Signaling Technology, Inc.;
mouse polyclonal antibody against β-actin (cat. no. 60008-1-lg) was
purchased from PeproTech, Inc.; horseradish peroxidase
(HRP)-labeled goat anti-mouse IgG (H+L) (cat. no. ZB-2305) and
HRP-labeled goat anti-rabbit IgG (H+L) (cat. no. ZB-2301) were
obtained from OriGene Technologies, Inc.
Primary culture of fibroblasts
On a clean bench, tissue samples were washed with
PBS solution containing 100 U/ml penicillin/streptomycin three
times, cut into small pieces (0.5-1 mm3) with eye
scissors, and then seeded every 1 cm into a T25 culture bottle
(Thermo Fisher Scientific, Inc.). The culture bottle was placed
upside down for 2 h at 37˚C, 5% CO2 and 95% saturated
humidity, after which time the cells were supplemented with DMEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin/streptomycin. Culture was continued in the incubator
until the fibroblasts grew out of the tissue block and covered the
bottom of the culture bottle. Cells used for experiments were
between passages 3 and 5. Keloid and normal skin fibroblasts were
respectively treated with 20, 40, 80 ng/ml TNF-α for 0, 24, 48, 72
and 96 h to detect the cell survival rate using CCK-8 assay. Keloid
fibroblasts were treated with 40 ng/ml TNF-α, 20 µg/ml infliximab
or 20 µg/ml infliximab + 40 ng/ml TNF-α for 0, 24 and 48 h to
detect the cell survival rate using CCK-8 assay and for 48 h to
analyze the cell cycle by flow cytometry, and were also treated
with 40 ng/ml TNF-α for 3 h to detect the phosphorylation of JNK,
p38 and IκBα by western blot.
ELISA
A total of 50 µl diluted sample (100X diluted serum,
or 10X diluted cell culture supernatant) were added into each well
and incubated for 1 h at 37˚C, followed by the addition of 100 µl
biotin antibody to each well and further incubation for 1 h at
37˚C. Each well was aspirated and washed, and the process was
repeated twice for a total of three washes. Subsequently, 100 µl
HRP-avidin was added into each well and incubated for 1 h at 37˚C.
After washing, 100 µl 3,3',5,5'-tetramethylbenzidine substrate
solution was added to each well, followed by an incubation for 30
min at 37˚C. Stop solution (50 µl) was then added, and the optical
density of each well was determined within 15 min, using a
microplate reader (Multiskan™ FC; Thermo Fisher Scientific, Inc.)
set to 450 nm.
Total RNA extraction and quantitative
PCR (qPCR) analysis
The keloid and normal skin dermis stored in liquid
nitrogen were ground in a mortar filled with liquid nitrogen and
then total RNA was extracted according to the manufacturer's
protocol of the RNA extraction kit (Aidlab Biotechnologies Co.,
Ltd.). Total RNA was quantified using a microspectrophotometer
(NanoDrop 2000; NanoDrop Technologies; Thermo Fisher Scientific,
Inc.), and reverse transcribed into cDNA using the aforementioned
reagent (1 h at 42˚C and 10 min at 70˚C). The qPCR primers were
synthesized by Sangon Biotech Co., Ltd., and their sequences are as
follows: β-actin forward, 5'-CGAGGCCCAGAGCAAGAGAG-3' and reverse,
5'-CGGTTGGCCTTAGGGTTCAG-3'; TNF-α forward,
5'-GGCAGTCAGATCATCTTCTCGA-3' and reverse,
5'-CGGTTCAGCCACTGGAGCT-3'; TNFR1 forward,
5'-CAAGTGCCACAAAGGAACCTAC-3' and reverse,
5'-CAGCTGAGGCAGTGTCTGA-3'; TNFR2 forward,
5'-CTATGACCAGACAGCTCAGATG-3' and reverse,
5'-CAGTTCCAGAGCTGGGTGTAT-3'; IL-6 forward,
5'-ATGCAATAACCACCCCTGAC-3' and reverse, 5'-CTGCGCAGAATGAGATGAGT-3'.
The SYBR-Green qPCR amplification conditions were as follows:
Initial denaturation for 1 min at 95˚C; 40 cycles of 10 sec at 95˚C
and 30 sec at 58˚C. Sequence detection software, version 1.2.3
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
analyze the Cq value of each amplification reaction in the qPCR,
and the 2-ΔΔCq method was used for relative quantitative
analysis (13).
Immunohistochemistry
Fixed tissue was embedded in paraffin and cut into
4-µm serial sections. After dewaxing sections to water, the
sections were treated with 3% hydrogen peroxide in methanol for 30
min, then washed with 0.01 M PBS for 5 min and the process was
repeated three times. After blocking with 5% bovine serum albumin
(Biosharp Life Sciences) in PBS for 20 min at 37˚C, the sections
were incubated with diluted primary antibodies against TNF-α
(1:100), TNFR1 (1:50) or TNFR2 (1:50) overnight at 4˚C, then washed
with 0.01M PBS for 5 min and the process was repeated three times.
Next, the sections were incubated with secondary antibodies (1:200;
cat. no. ZB-2301; OriGene Technologies, Inc.) labelled with HRP for
1 h at 37˚C. After staining with 3,3'-diaminobenzidine for 10 min
and hematoxylin solution for 20 sec at room temperature, the
expression and distribution of the target proteins were observed
under an inverted phase contrast microscope (magnification, x400;
Olympus Corporation). The results were analyzed using Image-Pro
Plus software 6.0 (Media Cybernetics, Inc.) and scored according to
Crambert et al (14).
CCK-8 assay
Cells were seeded in a 96-well plate at a density of
5x103 cells/well. When observed at exponential growth,
cells were treated as aforementioned in the ‘Primary culture of
fibroblasts’ section and incubated for 24, 48, 72 and 96 h. Next,
10 µl CCK-8 solution was added to each well and incubated for 4 h
at 37˚C. Subsequently, the absorbance was measured on a microplate
reader (Thermo Fisher Scientific, Inc.) at 450 nm. Cell viability
was calculated as follows: Cell viability (%)=[treated (OD)-blank
(OD)/control (OD)-treated (OD)] x100%.
Flow cytometry
The cells were harvested, fixed in 70% cold ethanol
for 16 h overnight at 4˚C, and centrifuged at 300 x g for 5 min at
4˚C. The supernatants were discarded. Then, the cell pellet was
stained with 0.4 ml propidium iodide staining solution in a bath at
37˚C for 30 min in the dark. Cell cycle distribution was analyzed
using FACSCalibur flow cytometer (BD Biosciences), and the
percentage of cells in each cell cycle phase was evaluated
(FACSCalibur software version 1.3; BD Biosciences).
Western blot analysis
The cells were collected and lysed in RIPA buffer
(Beyotime Institute of Biotechnology), then total cell proteins
were harvested, and protein concentration was determined using a
BCA Protein Assay kit (Thermo Fisher Scientific, Inc.). A total of
15 µg protein per lane was loaded on 10% sodium dodecyl sulfate
polyacrylamide gel (Bio-Rad Laboratories, Inc.), protein
electrophoresis was performed (120 V, 1 h), followed by a transfer
to a PVDF membrane (90 V, 90 min), which was then blocked with 5%
non-fat milk for 2 h at 4˚C. The membrane was then incubated with
the aforementioned primary antibodies against IκB-α (1:1,000),
p-IκB-α (1:1,000), p38 MAPK (1:1,000), p-p38 MAPK (1:1,000),
SAPK/JNK (1:1,000), p-SAPK/JNK (1:1,000) and β-actin (1:5,000)
overnight at 4˚C. Subsequently, the PVDF membranes were washed
three times in Tris-buffered saline with 0.1% Tween-20 (TBST), and
incubated with the aforementioned HRP-conjugated secondary
antibodies (1:5,000) for 2 h at 37˚C. After washing with TBST and
incubation with an ECL solution (Bio-Rad Laboratories, Inc.), the
membranes were exposed and images were captured. Quantity One
version 4.6.6 software (Bio-Rad Laboratories, Inc.) was used to
analyze the expression of target proteins and internal reference
protein β-actin.
Statistical analysis
The data are presented as the mean ± standard
deviation and statistical analysis was performed using SPSS 16.0
(SPSS Inc.). For normally distributed data, independent samples
t-test was applied to analyze differences between two groups, while
one-way ANOVA followed by Tukey's post hoc was used to determine
the statistical significance of the differences among multiple
groups, and Welch's test followed by Games-Howell post hoc test was
applied to data with unequal variances. Non-parametric Mann-Whitney
test was applied to data with non-normal distribution. P<0.05
was considered to indicate statistically significant
differences.
Results
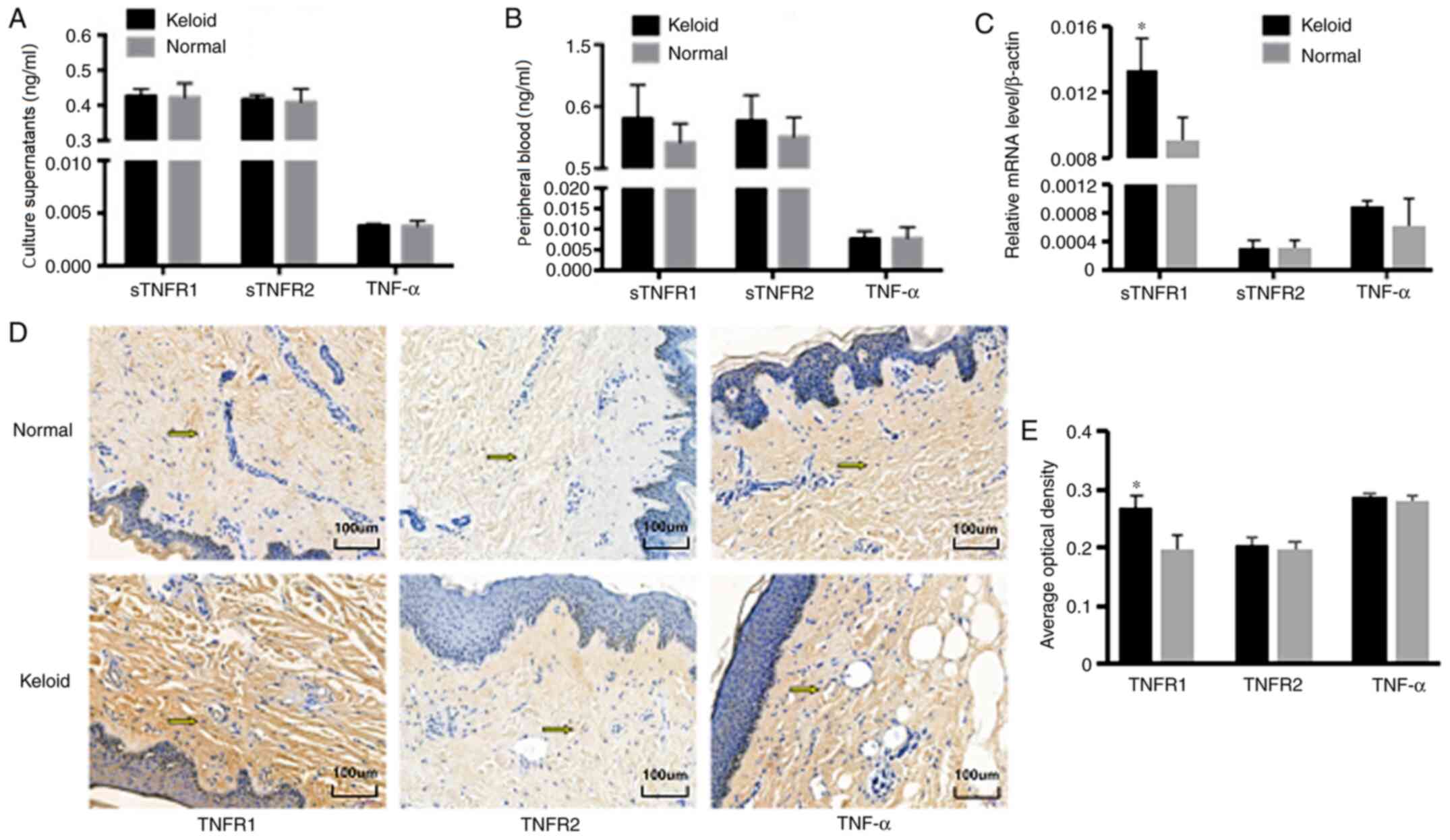
Expression of TNF-α, sTNFR1 and sTNFR2
in keloid and normal skin
Fibroblasts from keloid and normal skin were
isolated, and the content of TNF-α and its receptors, sTNFR1 and
sTNFR2, was detected in the fibroblast culture supernatants by
ELISA. There was no significant difference in the content of TNF-α,
sTNFR1 or sTNFR2 in the culture supernatants between keloid and
normal skin fibroblasts (P>0.05; Fig. 1A). To further verify these results,
peripheral blood samples were obtained from patients with keloids
and healthy participants, and the content of TNF-α, sTNFR1 and
sTNFR2 in the plasma was detected by ELISA. As in culture
supernatants, there was no statistical difference in the content of
TNF-α, sTNFR1 or sTNFR2 in the plasma between keloid and normal
skin (P>0.05; Fig. 1B). Surgical
specimens of keloids and normal skin were used to study the
expression of the aforementioned molecules at the mRNA and protein
level, by qPCR analysis of fibroblasts cultured in vitro and
by immunohistochemistry and microscopic examination of tissue
samples, respectively. The results demonstrated that the mRNA
levels of TNF-α and sTNFR2 in fibroblasts did not differ
significantly between the two groups (P>0.05), while the sTNFR1
mRNA level in KFs was significantly higher compared with that in
normal skin (P<0.05; Fig. 1C).
On immunohistochemical analysis, the TNF-α and sTNFR2 protein
levels exhibited no significant difference between the two groups
(P>0.05), while the sTNFR1 protein level in keloid tissue was
significantly higher compared with that in normal skin (P<0.05;
Fig. 1D and E). These data suggested that there was no
difference in the expression of TNF-α and sTNFR2 in the peripheral
blood, skin tissues and fibroblasts cultured in vitro
between patients with keloids and healthy individuals; however, the
expression of sTNFR1 in keloid tissue was found to be significantly
higher compared with that in normal skin.
TNF-α regulates the proliferation of
KFs
In order to verify this hypothesis, different
concentrations (0, 10, 20, 40 and 80 ng/ml) of TNF-α recombinant
protein were applied to treat KFs and normal skin fibroblasts, and
the CCK-8 assay was used to evaluate the effect of the treatment on
cell proliferation. The results revealed that TNF-α promoted the
proliferation of KFs at 48, 72 and 96 h after stimulation (Fig. 2A), whereas it had little effect on
normal skin fibroblasts (Fig. 2B).
To further study the proliferation-promoting effect of TNF-α on
KFs, cells were pretreated with infliximab, a specific antagonist
of TNF-α. The CCK-8 assay results revealed that the
proliferation-promoting effect of TNF-α on KFs was reversed by this
treatment (Fig. 2C). The cell cycle
analysis by flow cytometry supported the results mentioned above.
Treatment with TNF-α increased the proportion of KFs in the S and
G2 phases of the cell cycle, and reduced the proportion
of G1 phase cells compared with the NT group, while
pretreatment with infliximab eliminated this increased proportion
of S and G2 phase cells (Fig. 2D and E). These results indicated that TNF-α may
promote the proliferation of KFs.
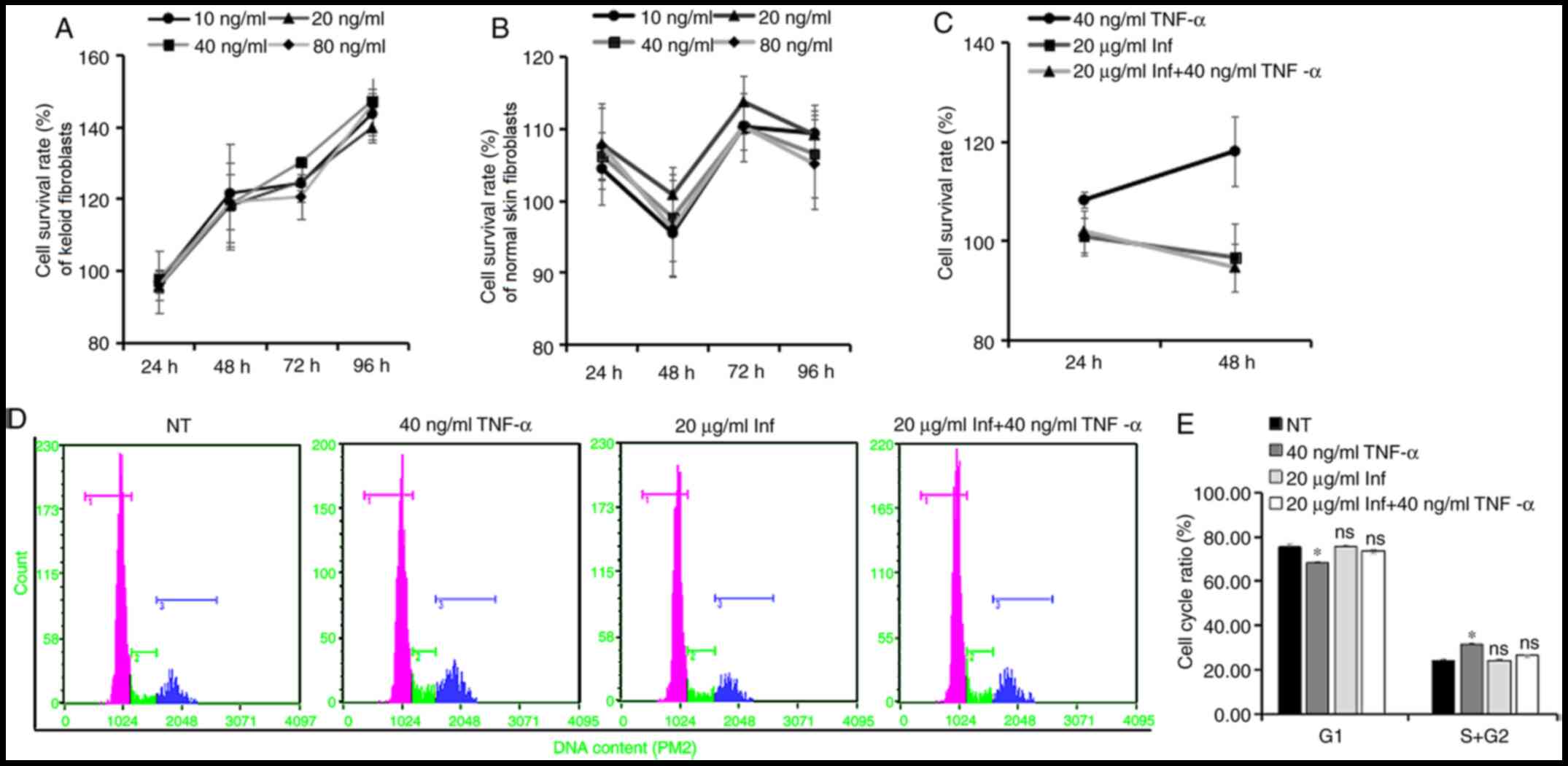 | Figure 2TNF-α regulates the proliferation of
keloid fibroblasts. (A) Keloid or (B) normal skin fibroblasts were
stimulated with different concentrations (0, 10, 20, 40 and 80
ng/ml) of TNF-α recombinant protein, and the CCK-8 assay results
were used to evaluate the effect of the treatment on cell
proliferation (n=6). The untreated group at each time point was set
to 100% and compared with the cell survival rates of the
corresponding treated groups. Keloid fibroblasts were pretreated
with Inf, and were subsequently stimulated with or without TNF-α
recombinant protein (40 ng/ml). (C) CCK-8 assay results were used
to evaluate the effect of the treatment on cell proliferation
(n=6). (D) Cell cycle distribution was analyzed after 48 h treated
with TNF-α and infliximab by flow cytometry (rose color indicates
the G1 cell count; green indicates the S phase cell
count; and blue indicates the G2 phase cell count), and
(E) cell cycle ratio was plotted as a histogram to compare the
number of cells in different phases of the cell cycle (n=3).
*P<0.05 vs. NT. Data are presented as the mean ± SD.
TNF, tumor necrosis factor; CCK-8, Cell Counting Kit-8; Inf,
infliximab; ns, not significant; NT, non-treated. |
TNF-α regulates the NF-κB, JNK and p38
MAPK pathways in KFs
The aforementioned results demonstrated that TNF-α
promotes KF proliferation. Subsequently, the expression of IL-6,
which is an important effector of the NF-κB, JNK and p38 MAPK
signaling pathways (11), was first
detected. The IL-6 mRNA level in KFs significantly increased after
TNF-α treatment (P<0.05; Fig.
3A), which suggested that TNF-α stimulation may lead to the
activation of one or more of those pathways. To identify the exact
pathway activated, differences in the activation levels of the
NF-κB, JNK and p38 MAPK pathways were detected in KFs treated with
or without TNF-α. The western blotting results revealed that,
following KF stimulation with TNF-α for 30 min, the relative level
of the p-JNK protein increased significantly compared with that in
the untreated group (P<0.05; Fig.
3B), which indicated activation of the JNK pathway. The IκB-α
protein level decreased significantly (P<0.01), accompanied by a
significant increase in p-IκB-α (P<0.05; Fig. 3C), which indicated activation of the
NF-κB pathway. The relative level of the p-p38 protein also
increased significantly after treatment with TNF-α (P<0.05;
Fig. 3D), which indicated
activation of the p38 MAPK pathway. These data confirmed that TNF-α
simultaneously activated the NF-κB, JNK and p38 MAPK pathways in
KFs.
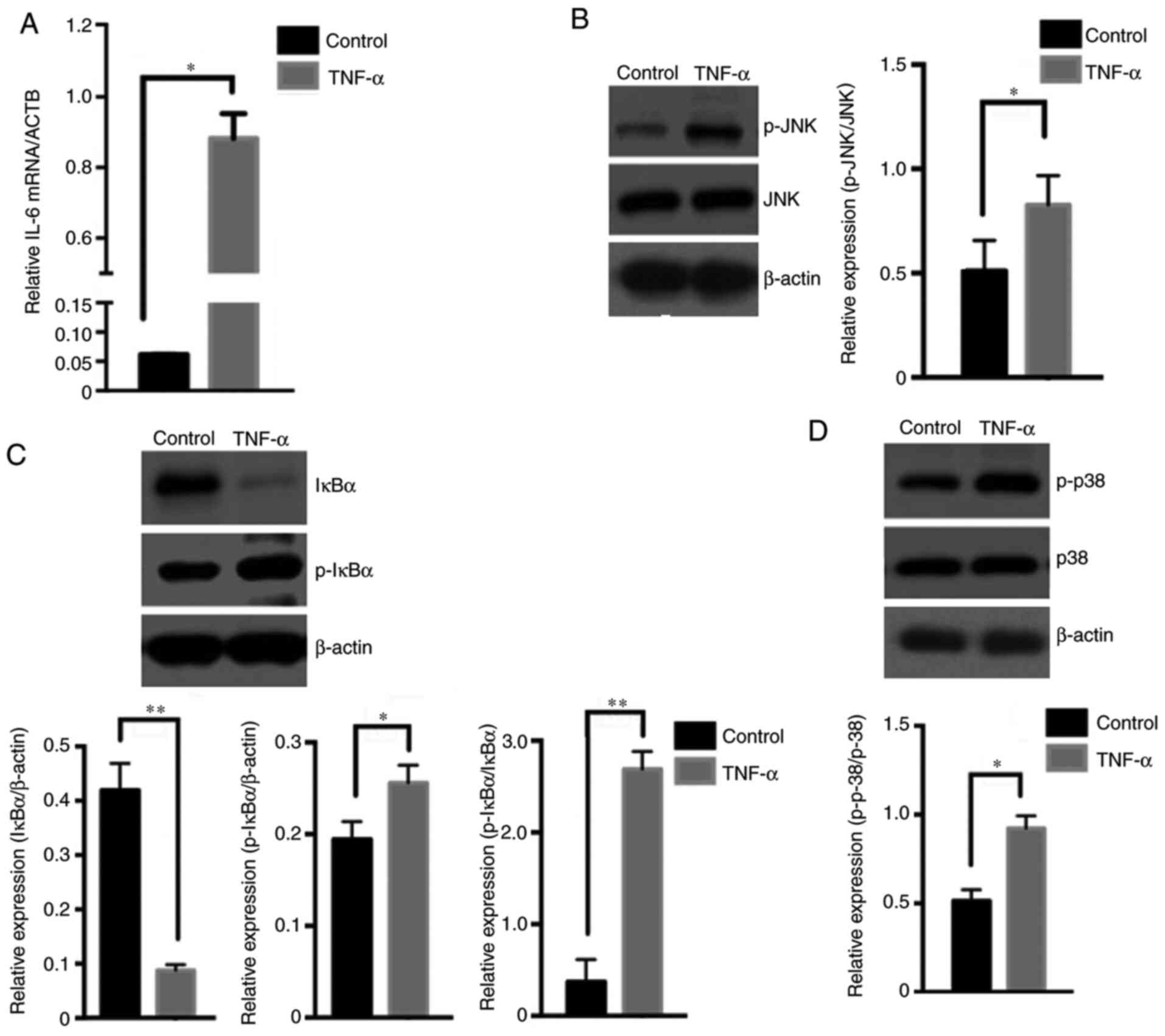 | Figure 3TNF-α regulates NF-κB, JNK and p38
MAPK pathway activation in keloid fibroblasts. (A) Keloid
fibroblasts were stimulated with or without 40 ng/ml TNF-α
recombinant protein for 24 h, and the IL-6 mRNA level in keloid
fibroblasts was detected by quantitative PCR analysis, with ACTB
serving as the reference gene (n=6). Keloid fibroblasts were
stimulated with or without 40 ng/ml TNF-α recombinant protein for
30 min, and the protein levels of (B) p-JNK, JNK and β-actin, (C)
IκB-α, p-IκB-α and β-actin, and (D) p-p38, p38 and β-actin were
detected by western blotting (n=3). *P<0.05 and
**P<0.01 vs. control. Data are presented as the mean
± SD. TNF, tumor necrosis factor; NF-κB, nuclear factor-κB; JNK,
c-Jun N-terminal kinase; MAPK, p38 mitogen-activated protein
kinase; IL, interleukin; IκB-α, inhibitor of NF-κB; ACTB, β-actin;
p, phosphorylated. |
Discussion
The present study revealed that TNF-α may play an
important role in the hyperproliferation of KFs. A previous gene
chip study confirmed that the expression of pro-inflammatory
factors, such as IL-1α, IL-1β, TNF-α and IL-6, was upregulated in
KFs. McCauley et al (8)
found that the TNF-α protein level in the serum of African American
patients with keloids was increased compared with that in normal
subjects without keloids, and Messadi et al (9) observed that the TNF-α mRNA and protein
levels in keloid tissue were higher compared with those in
surrounding normal skin using specific cDNA microarrays, western
blot analysis and immunohistochemistry. However, in the present
study, the expression of TNF-α in the plasma, tissue specimens and
culture supernatants of skin fibroblasts did not differ
significantly between patients with keloids and healthy
individuals, which is inconsistent with the results of previous
studies. It may be hypothesized that the inconsistency for plasma
test results may be due to the differences among different
ethnicities and geographic regions, since McCauley et al
(8) collected blood samples from
African American patients, while the samples collected in the
present study were collected from Asian patients. Furthermore,
McCauley et al (8) detected
the change in TNF-α level in peripheral blood mononuclear cells,
which is different from the direct detection of serum TNF-α level
in the present study. The inconsistency for tissue test results may
be associated with the processing of surgical specimens. In the
present study, the epidermis and connective tissue were removed
from the skin samples, and only the dermis was retained. Thus, the
expression level of TNF-α was only detected in fibroblasts, which
helped to more accurately study its function in these cells.
There are two main receptors of TNF-α: TNFR1 (also
referred to as p55 or CD120a) and TNFR2 (also referred to as p75 or
CD120b). Both receptors are classed as type I transmembrane
proteins. TNFR1 is expressed in almost all tissues and cells, and
may be activated by two forms (soluble and membrane bound) of
TNF-α; TNFR2 is mainly expressed in immune and endothelial cells,
and mainly interacts with membrane bound TNF-α. TNFR1 mediates
almost all known biological effects of TNF-α. The activation of
TNFR1 can further activate a variety of signal transduction
pathways by recruiting a series of intracellular adaptor proteins
(7,11). Since sTNFR1, the main receptor of
TNF-α, has been found to be more highly expressed in keloid tissue
compared with normal skin, it was hypothesized that KFs exhibit
increased sensitivity to TNF-α compared that in normal skin
fibroblasts. The expression of the TNF-α receptors sTNFR1 and
sTNFR2, was also detected in the current study. There was no
significant difference in the expression of TNFR2 between keloid
and normal skin, but the expression of TNFR1 in keloid tissues and
fibroblasts cultured in vitro was higher compared with that
in normal skin. It was previously reported that the expression of
TNFR1 in normal skin was significantly higher compared with that in
keloid tissue (15), but this was
contradictory to the hypothesis that the upregulation of TNFR1
inhibits apoptosis and promotes the proliferation of fibroblasts.
In addition, Peruccio et al (16) observed that the TNFR1 mRNA level in
hypertrophic scars after burn injury was lower compared with that
in keloids. This conclusion was different from the findings of the
present study, as hypertrophic scars after burn injury were not
examined in the present study, and it may be hypothesized that this
is due to the different scar types and testing sites of surgical
specimens, as previous studies either focused on hypertrophic scars
or did not explicitly mention the testing sites of specimens
(15,16). Based on the results of the present
study, as TNFR1 is the main receptor of TNF-α mediating its
biological effects, its higher expression may lead to more potent
sensitization of KFs to the stimulatory action of TNF-α.
Hyperproliferation of fibroblasts is the main cause
underlying keloid formation and progression (3). Previous studies have demonstrated that
TNF-α is involved in various fibrotic diseases. Miyazaki et
al (17) found that transgenic
mice expressing high levels of TNF-α in the lung were more
susceptible to fibroalveolitis compared with those with low levels
of TNF-α. Guo et al (18)
reported that the renal interstitial volume of
TNFR1-/-/TNFR2-/- double knockout mice was
significantly lower compared with that of the control group.
Gurevitch et al (19) found
that cardiac fibroblasts were able to proliferate and transform
into myofibroblasts under TNF-α stimulation. Weiner et al
(20) demonstrated that TNF-α
directly promoted the proliferation of Ito cells and fibroblasts in
the liver, and Elias et al (21) reported that TNF-α promoted the
proliferation of lung fibroblasts. All these previous findings
indicate that TNF-α can promote fibroblast proliferation. In the
present study, the regulatory effect of TNF-α on the proliferation
of KFs was investigated. The results demonstrated that 40 ng/ml
TNF-α could promote the proliferation of KFs, but not that of
normal fibroblasts. As an increased expression of TNFR1 potentially
increases the sensitivity of KFs to TNF-α, it may be hypothesized
that, during the process of keloid formation, KFs with a higher
sensitivity to TNF-α are under constant stimulation. Consequently,
the intracellular signaling pathways are repeatedly activated,
eventually leading to sustained and excessive fibroblast
proliferation.
The present study further investigated the mechanism
underlying the role of TNF-α in the regulation of KF proliferation.
After binding to TNFR1, TNF-α may activate NF-κB, JNK, p38 MAPK, or
other intracellular signaling pathways. Since the activation of
these signaling pathways may differ among different cell types, the
specific pathways activated in KFs have not yet been fully
elucidated. NF-κB is the most important signaling molecule for
TNF-α to exert its promoting effect on cell proliferation and
inflammation (10,22), whereas the JNK and p38 MAPK pathways
may also promote cell survival and proliferation (11,22,23).
In the present study, it was observed that TNF-α simultaneously
activated the NF-κB, JNK and p38 MAPK pathways in KFs, which
suggested that the proliferation of KFs induced by TNF-α
stimulation was closely associated with the activation of these
pathways. This was consistent with previous findings reporting the
promotion of cell proliferation by these pathways (7,11,22,23).
Therefore, it was inferred that the persistent activation of the
NF-κB, JNK and p38 pathways may be the mechanism underlying
TNF-α-induced KF proliferation (Fig.
4).
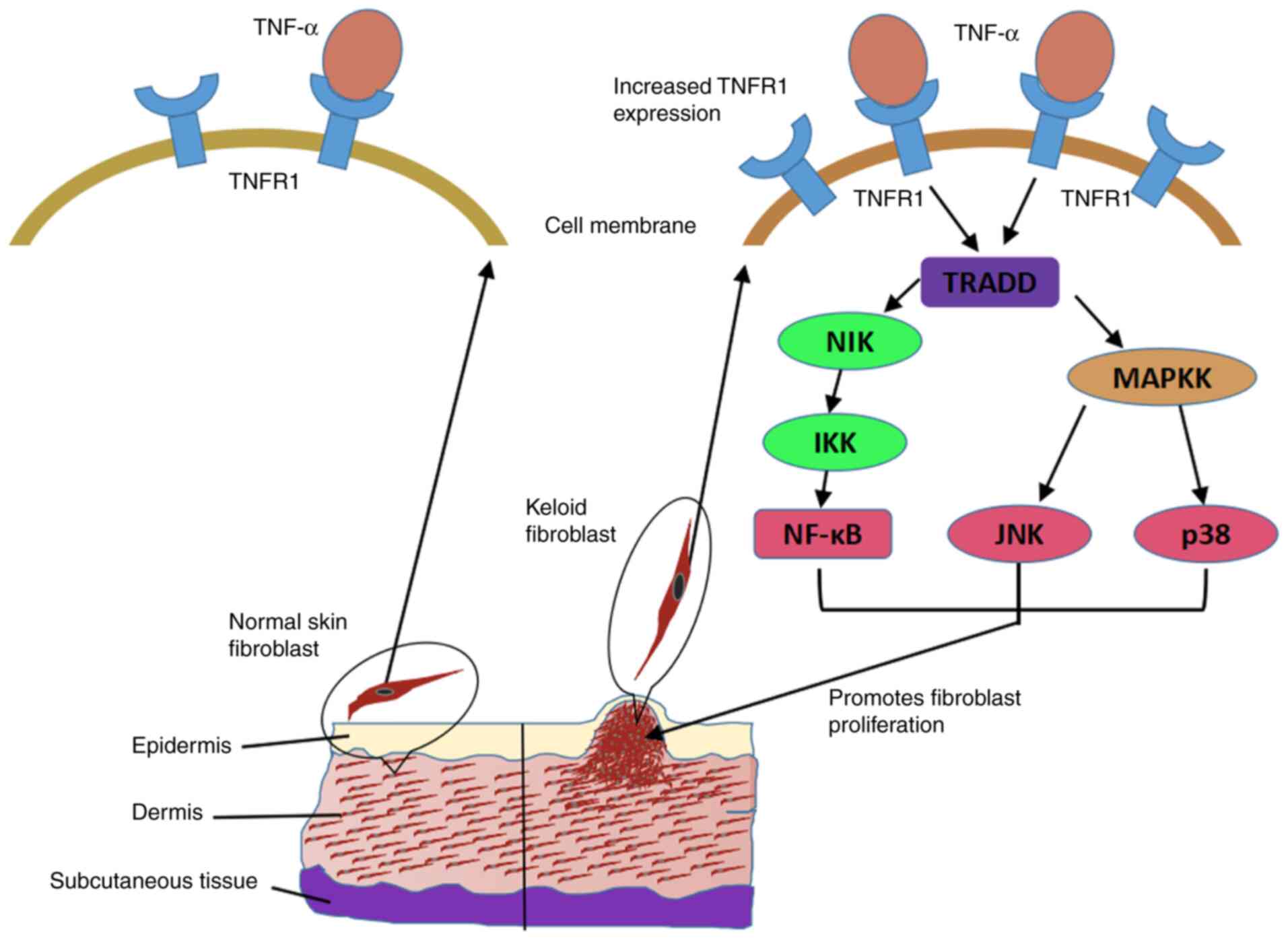 | Figure 4TNF-α promotes KF hyperproliferation
by activating the NF-κB, JNK and p38 MAPK pathways. Increased
expression of TNFR1 in KFs compared with that in normal fibroblasts
may contribute to their enhanced sensitivity to TNF-α, and then the
persistent stimulation by a low concentration of TNF-α may lead to
repeated activation of the NF-κB, JNK and p38 pathways, thereby
promoting the sustained and excessive proliferation of KFs. TNF,
tumor necrosis factor; TNFR, TNF receptor; NF-κB, nuclear
factor-κB; JNK, c-Jun N-terminal kinase; MAPK, p38
mitogen-activated protein kinase; KF, keloid fibroblast; NIK, NF-κB
inducing kinase; TRADD, tumor necrosis factor receptor type
1-associated DEATH domain protein. |
In conclusion, compared with normal fibroblasts,
increased expression of TNFR1 in KFs may confer a unique
sensitivity to TNF-α, and persistent stimulation by a low
concentration of TNF-α may lead to repeated activation of the
NF-κB, JNK and p38 pathways, thereby promoting the sustained and
excessive proliferation of KFs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Sichuan Basic Applied
Research Project (grant no. 2017FZ0055), the Post-Doctor Research
Project, West China Hospital, Sichuan University (grant no.
2018HXBH082) and the China Postdoctoral Science Foundation (grant
no. 2019M653413).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL and FC participated in the design of the
experiments, optimization of experimental conditions, conduction of
the experiments and writing of the manuscript. KZ, LF and JW
participated in the recruitment of participants and the collection
of clinical specimens. QX and YC participated in the experimental
design and data analysis. JC and YQ participated in designing the
present study and revising the manuscript and confirmed the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The protocol of the present study was reviewed and
approved by the Ethics Committee of West China Hospital of Sichuan
University written consent was obtained from all the
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ikeda K, Torigoe T, Matsumoto Y, Fujita T,
Sato N and Yotsuyanagi T: Resveratrol inhibits fibrogenesis and
induces apoptosis in keloid fibroblasts. Wound Repair Regen.
21:616–623. 2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hunasgi S, Koneru A, Vanishree M and
Shamala R: Keloid: A case report and review of pathophysiology and
differences between keloid and hypertrophic scars. J Oral
Maxillofac Pathol. 17:116–120. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bettinger DA, Yager DR, Diegelmann RF and
Cohen IK: The effect of TGF-beta on keloid fibroblast proliferation
and collagen synthesis. Plastic Reconst Surg. 98:827–833.
1996.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Harty M, Neff AW, King MW and Mescher AL:
Regeneration or scarring: An immunologic perspective. Dev Dyn.
226:268–279. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dong XL, Mao SL and Wen H: Upregulation of
proinflammatory genes in skin lesions may be the cause of keloid
formation (Review). Biomed Rep. 1:833–836. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Frances B: TNF-alpha in promotion and
progression of cancer. Cancer Metastasis Rev. 25:409–416.
2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bahcecioglu IH, Koca SS, Poyrazoglu OK,
Yalniz M, Ozercan IH, Ustundag B, Sahin K, Dagli AF and Isik A:
Hepatoprotective effect of infliximab, an anti-TNF-α agent, on
carbon tetrachloride-induced hepatic fibrosis. Inflammation.
31:215–221. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
McCauley RL, Chopra V, Li YY, Herndon DN
and Robson MC: Altered cytokine production in black patients with
keloids. J Clin Immunol. 12:300–308. 1992.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Messadi DV, Doung HS, Zhang Q, Kelly AP,
Tuan TL, Reichenberger E and Le AD: Activation of NFkappaB signal
pathways in keloid fibroblasts. Arch Dermatol Res. 296:125–133.
2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhu GY, Cai JL, Zhang J, Zhao YR and Xu B:
Abnormal nuclear factor (NF)-kappaB signal pathway and aspirin
inhibits tumor necrosis factor α-induced NF-kappaB activation in
keloid fibroblasts. Dermatol Surg. 33:697–708. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65.
2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gauglitz GG, Korting HC, Pavicic T,
Ruzicka T and Jeschke MG: Hypertrophic scarring and keloids:
Pathomechanisms and current and emerging treatment strategies. Mol
Med. 17:113–125. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Crambert G, Li C, Claeys D and Geering K:
FXYD3 (Mat-8), a new regulator of Na, K-ATPase. Mol Biol Cell.
16:2363–2371. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu LP, Chen FC, Guo LL and Chen YT:
Expression and significance of apoptosis-related TNFR1 and Bcl-2 in
fibroblasts derived from pathologic scar. Chin J Prac Aesthetic and
Plastic Surg. 15:272–274. 2004.
|
|
16
|
Peruccio D, Castagnoli C, Stella M,
D'Alfonso S, Momigliano PR, Magliacani G and Alasia ST: Altered
biosynthesis of tumour necrosis factor (TNF) alpha is involved in
postburn hypertrophic scars. Burns. 20:118–121. 1994.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Miyazaki Y, Araki K, Vesin C, Garcia I,
Kapanci Y, Whitsett JA, Piguet PF and Vassalli P: Expression of a
tumor necrosis factor-alpha transgene in murine lung causes
lymphocytic and fibrosing alveolitis. A mouse model of progressive
pulmonary fibrosis. J Clin Invest. 96:250–259. 1995.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo G, Morrissey J, McCracken R, Tolley T
and Klahr S: Role of TNFR1 and TNFR2 receptors in
tubulointerstitial fibrosis of obstructive nephropathy. Am J
Physiol. 277:F766–F772. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gurevitch J, Frolkis I, Yuhas Y,
Lifschitz-Mercer B, Berger E, Paz Y, Matsa M, Kramer A and Mohr R:
Anti-tumor necrosis factor-alpha improves myocardial recovery after
ischemia and reperfusion. J Am Coll Cardiol. 30:1554–1561.
1997.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Weiner FR, Giambrone MA, Czaja MJ, Shah A,
Annoni G, Takahashi S, Eghbali M and Zern MA: Ito-cell gene
expression and collagen regulation. Hepatology. 11:111–117.
1990.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Elias JA, Freundlich B, Kern JA and
Rosenbloom J: Cytokine networks in the regulation of inflammation
and fibrosis in the lung. Chest. 97:1439–1445. 1990.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lamb JA, Ventura JJ, Hess P, Flavell RA
and Davis RJ: JunD mediates survival signaling by the JNK signal
transduction pathway. Mol Cell. 11:1479–1489. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ventura JJ, Hübner A, Zhang C, Flavell RA,
Shokat KM and Davis RJ: Chemical genetic analysis of the time
course of signal transduction by JNK. Mol Cell. 21:701–710.
2006.PubMed/NCBI View Article : Google Scholar
|