Introduction
Severe hypertriglyceridemia (HTG) is the third major
cause of acute pancreatitis (AP) after alcohol abuse and
cholelithiasis worldwide (1). The
severity and complication rates of HTG-induced pancreatitis (HTGP)
are higher than those of AP that originate from other causes
(2). The mechanism by which high
triglyceride (TG) levels trigger AP is unknown (3), but may be due to the toxicity of free
fatty acids (FFAs) to the pancreas. High concentrations of FFAs are
generated from TG hydrolysis by lipases in pancreatic tissues and
may trigger the self-digestion of the pancreas by damaging
pancreatic acinar cells (PACs) and vascular endothelial cells,
activating trypsinogen and protein kinase C, and releasing
intracellular calcium (3,4).
Pancreatic ductal epithelial cells (PDECs) are
considered the most important component of the pancreatic duct
mucosal barrier, and serve a critical role in preventing the reflux
of bile and pancreatic enzymes (5). Previous studies have reported that
barrier disfunction in PDECs is a major contributor to the
occurrence of pancreatitis (6-9).
Our previous study revealed that the synthetic cholecystokinin
analogue caerulein (CAE) induced AP in the human pancreatic ductal
epithelial cell line HPDE6-C7(10). In addition, high-fat diet-derived
FFAs can impair intestinal epithelial cell permeability (11-13);
nonetheless, the effect of HTG on PDECs in AP is incompletely
understood.
The cytoskeleton is a complex and dynamic system
composed primarily of actin filaments, which is essential for key
cellular functions, including adhesion, spreading, migration and
interaction with the environment (14). Previous studies have reported that
the maintenance of the endothelial barrier function and cell-cell
junctions is critically dependent on actin filament dynamics
(15-18),
which are regulated by actin-binding proteins (ABPs) (19). Moreover, FFAs can modulate actin
reorganization and have toxic effects on PACs by increasing
cytosolic calcium concentrations (20-22),
potentially leading to HTGP. However, to the best of our knowledge,
the underlying mechanism of actin dynamics in PDECs in HTGP has not
been explored.
Gelsolin (GSN) is a calcium-regulated ABP that
controls actin dynamics by nucleating, capping and severing actin
filaments, and participates in cell morphology, motility,
metabolism, apoptosis and phagocytosis (23). Moreover, GSN has been reported to
be involved in cell-cell junctions and can regulate cell adhesion
strength by remodeling actin (24,25).
Our unpublished data revealed that GSN is expressed in PDECs
(HPDE6-C7) (Yang et al, unpublished data); however, the role
of GSN in PDECs in HTGP is not fully understood.
The present study hypothesized that GSN may impair
barrier function in PDECs by regulating actin filaments in HTGP. To
test this hypothesis, GSN gene expression was knocked down and HTGP
was induced using CAE (13) and TG
in HPDE6-C7 cells, in order to assess the effect of GSN-regulated
actin filaments on barrier function in PDECs in HTGP in
vitro.
Materials and methods
Cell culture
The human PDEC line HPDE6-C7 (Guangzhou Jenniobio
Biotechnology) was cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum
(Shanghai Shuangru Biotechnology Co., Ltd.), 1% L-glutamine
(Beijing Solarbio Science & Technology Co., Ltd.) and 1%
penicillin-streptomycin mixture (Beijing Solarbio Science &
Technology Co., Ltd.) at 37˚C in a humidified atmosphere containing
5% CO2. Cells were grown on coverslips (24x24 mm) in
6-well plates or 25-cm2 flasks.
RNA interference-mediated GSN gene
silencing
Three pairs of short hairpin RNA (shRNA) sequences
were designed by BLOCK-iT™ RNAi Designer (Thermo Fisher Scientific
Inc.) according to GSN coding DNA sequences and were synthesized by
Sangon Biotech Co., Ltd. (Table
I). The third generation lentiviral packaging system was used
to inhibit GSN expression analysis. The shRNAs were cloned into the
pcDNA6.2-GW/EmGFP-miR plasmid vector (Shanghai R&S
Biotechnology Co., Ltd.). The cloned DNA fragments were amplified
by PCR and subcloned into the pLenti6.3/V5-DEST vector (Shanghai
R&S Biotechnology Co., Ltd.). A total of 60 µg lentiviral
plasmids and ViraPower™ lentiviral packaging mix (Invitrogen;
Thermo Fisher Scientific Inc.), mixed with POLOdeliverer™ 3000
Transfection Reagent (Shanghai R&S Biotechnology Co., Ltd.),
were co-transfected into 293T cells (Shanghai R&S Biotechnology
Co., Ltd.) in a 1:1 ratio. The transfected cells were incubated at
37˚C for 48 and 72 h, respectively, and the lentivirus supernatant
was collected and concentrated. HPDE6-C7 cells were infected with
lentivirus for 72 h, with a multiplicity of infection of 8. The
lentivirus-infected cells derived from the third pairs of shRNAs
were selected for subsequent experiments by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
screening and validation. The stable cell lines were created using
1.0 µg/ml blasticidin for selection and 0.5 µg/ml for maintenance.
The lentivirus obtained from empty pLenti6.3/V5-DEST was used as an
empty vector and HPDE6-C7 cells transduced with an empty vector
were used as the negative control (NC).
 | Table IShort hairpin RNA sequences used for
GSN silencing. |
Table I
Short hairpin RNA sequences used for
GSN silencing.
| Name | Oligonucleotide
sequence (5'-3') |
|---|
| GSN-1 | F:
TGCTGAAGAAGTCTCCATAAAGGTTGGTTTTGGCCACTGACTGACCAACCTTTGGAGACTTCTT |
| | R:
CCTGAAGAAGTCTCCAAAGGTTGGTCAGTCAGTGGCCAAAACCAACCTTTATGGAGACTTCTTC |
| GSN-2 | F:
TGCTGAGAACTGTCCATATGTGGCAGGTTTTGGCCACTGACTGACCTGCCACATGGACAGTTCT |
| | R:
CCTGAGAACTGTCCATGTGGCAGGTCAGTCAGTGGCCAAAACCTGCCACATATGGACAGTTCTC |
| GSN-3 | F:
TGCTGTACAGAATGATGTAGCTGTCGGTTTTGGCCACTGACTGACCGACAGCTATCATTCTGTA |
| | R:
CCTGTACAGAATGATAGCTGTCGGTCAGTCAGTGGCCAAAACCGACAGCTACATCATTCTGTAC |
Cell treatment and experimental
grouping
Cells with or without GSN silencing were treated
with 100 nM CAE (MilliporeSigma), 2.5 mM TG (cat. no. T9420;
Beijing Solarbio Science & Technology Co., Ltd.), or CAE + TG
for 24 h at 37˚C. CAE was resuspended in phosphate-buffered saline
(PBS), and TG was resuspended in PBS containing 0.1 mg/ml bovine
serum albumin (cat. no. A8020; Beijing Solarbio Science &
Technology Co., Ltd.). AP was induced in vitro using CAE,
the effects of HTG on PDECs were assessed using TG, and HTGP was
induced by CAE + TG. Cells were divided into 12 groups, as follows:
Blank control (BC) group, cells without lentiviral transduction;
CAE-treated BC group (CAE group); TG-treated BC group (TG group);
CAE + TG-treated BC group (CAE + TG group); NC group, cells
transduced with an empty vector; CAE-treated NC group (NC + CAE
group); TG-treated NC group (NC + TG group); CAE + TG-treated NC
group (NC + CAE + TG group); knockdown (KD) group, cells with GSN
KD; CAE-treated KD group (KD + CAE group); TG-treated KD group (KD
+ TG group); and CAE + TG-treated KD group (KD + CAE + TG
group).
Inverted biological microscopy
analysis of cell morphology
Cells at a density of 4x105 cells/well
were seeded into a six-well plate and cultured at 37˚C in a
humidified atmosphere containing 5% CO2 until they
reached 75-80% confluence. Then the cells were treated with
purified trilaurin (TG; cat. no. T9420; Beijing Solarbio Science
& Technology Co., Ltd.) at concentrations of 0.5, 1.0, 2.5, 5
and 10 mM for 24 h at 37˚C. Cell morphology was analyzed by an
inverted biological microscopy (CKX53; Olympus Corporation) at x100
magnification.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was determined using CCK-8 (cat. no.
CA1210; Beijing Solarbio Science & Technology Co., Ltd.)
according to the manufacturer's protocols. HPDE6-C7 cells at a
density of 1x104 cells/well were seeded into a 96-well
plate with 100 µl growth medium/well and cultured for 12 h.
Subsequently, the cells were treated with TG (10 µl/well) at
concentrations of 0.5, 1.0, 2.5, 5.0 and 10 mM for 24 h at 37˚C and
10 µl CCK-8 reagent was added to each well for 1 h at 37˚C. The
absorbance was measured at 450 nm using a microplate reader
(Varioskan LUX; Thermo Fisher Scientific Inc.).
DAPI staining
The cells were seeded into a 6-well plate at a
density of 1x105 cells/well and cultured for 24 h, then
treated with 100 nM CAE for 6, 12, 24 and 48 h or TG at
concentrations of 0.5, 1.0, 2.5, 5.0 and 10 mM for 24 h at 37˚C.
Subsequently, the cells were fixed in 4% paraformaldehyde (cat. no.
P1110; Beijing Solarbio Science & Technology Co., Ltd.) for 30
min and stained with 100 µl DAPI solution (10 µg/ml; cat. no.
C0065; Beijing Solarbio Science & Technology Co., Ltd.) in the
dark for 5 min (all steps at room temperature). The cell nuclei
were imaged at 460-495 nm under a fluorescence microscope. The
cells stained light blue represented the DAPI positively stained
cells.
Measurement of intracellular
calcium
HPDE6-C7 cells were grown on 60-mm culture dishes
until they reached 60-65% confluence. The cells were then washed
three times with Hanks' balanced salt solution without calcium,
magnesium and phenol red (cat. no. H1046; Beijing Solarbio Science
& Technology Co., Ltd.) and were stained with Calcium Crimson
(5 µM; 1.0 ml/dish; cat. no. C3018; Invitrogen; Thermo Fisher
Scientific, Inc.) at 37˚C for 30 min in the dark. Subsequently,
cells were washed with D-HBSS three times and stained with Hoechst
33258 (30 µg/ml; 1.0 ml/dish; cat. no. H3569; Invitrogen; Thermo
Fisher Scientific, Inc.) at 37˚C for 20 min in the dark.
Fluorescence in the cytosol and nucleus were measured at 530-550
and 460-495 nm, respectively, under an inverted fluorescence
microscope (IX71; Olympus Corporation). Images were analyzed using
ImageJ software (version 1.8.0; National Institutes of Health).
Transmission electron microscopy
(TEM)
Control and GSN-silenced HPDE6-C7 cells were
cultured in 25-cm2 flasks until they reached 70-80%
confluence. Cells were harvested using a cell scraper, centrifuged
at 1,500 x g for 10 min at room temperature, fixed in 3%
glutaraldehyde for 2.5 h at 4˚C and washed three times with PBS (10
mM; 10 min/wash). Subsequently, the samples were fixed in 1% osmium
for 2 h at 4˚C, washed three times with PBS (10 min/wash),
dehydrated through a graded ethanol series, embedded in epoxy resin
at 37˚C overnight. Epoxy resin samples were heated at 35˚C for 12
h, 45˚C for 15 h and 60˚C for 24 h, successively, cut into 70-nm
sections and stained with 3% uranium acetate-lead citrate.
Ultrastructural changes in tight junctions (TJs) were analyzed
using TEM (Hitachi, Ltd.).
Western blotting
Proteins were lysed from HPDE6-C7 cells using RIPA
buffer (Beijing Solarbio Science & Technology Co., Ltd.)
containing 1% phenylmethyl sulfonyl fluoride (Beijing Solarbio
Science & Technology Co., Ltd.) on ice for 30 min, and were
centrifuged at 12,000 x g for 20 min at 4˚C. Protein concentration
was determined using a bicinchoninic acid protein assay kit (cat.
no. P0012; Beyotime Institute of Biotechnology). Proteins samples
(50 µg/well) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and electrotransferred
to polyvinylidene fluoride membranes. Membranes were then blocked
with 5% non-fat milk for 1 h at room temperature and incubated with
primary antibodies against GSN (1:1,000; cat. no. ab74420; Abcam),
ZO-1 (1:1,000; cat. no. 21173-1-AP; Wuhan Sanying Biotechnology),
nectin-2 (1:1,000; cat. no. ab135246; Abcam), E-cadherin (1:1,000;
cat. no. 20874-1-AP; Wuhan Sanying Biotechnology), occludin
(1:1,000; cat. no. 27260-1-AP; Wuhan Sanying Biotechnology) and
GAPDH (1:10,000; cat. no. ab181602; Abcam) overnight at 4˚C.
Subsequently, membranes were incubated with DyLight 680-conjugated
secondary anti-rabbit antibody (1:10,000; cat. no. 5366; Cell
Signaling Technology, Inc.) for 1 h at room temperature in the
dark. Immunoreactive bands were imaged using the Odyssey infrared
imaging system (LI-COR Biosciences) and fluorescence intensity was
semi-quantified using ImageJ software (version 1.8.0; National
Institutes of Health).
RT-qPCR
Total RNA was isolated from HPDE6-C7 cells using
RNAiso Plus (cat. no. 9108; Takara Bio, Inc.) and converted to cDNA
by RT using PrimeScript RT reagent kit with gDNA Eraser (cat. no.
RR047B; Takara Bio, Inc.) according to manufacturer's protocol.
qPCR was performed using TB Green Premix Ex Taq II (cat. no.
RR820B; Takara Bio, Inc.) and corresponding primers (Table II). The thermocycling conditions
were as follows: Pre-denaturation at 95˚C for 30 sec, followed by
40 cycles of denaturation at 95˚C for 5 sec, annealing and
extension at 60˚C for 34 sec. GAPDH was used as internal reference
gene and the relative expression levels of target genes were
measured using the 2-ΔΔCq method (26).
| Table IITarget gene primers used in reverse
transcription-quantitative polymerase chain reaction. |
Table II
Target gene primers used in reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer
(5'-3') | Reverse primer
(5'-3') |
|---|
| GAPDH |
ACATCGCTCAGACACCA |
GTAGTTGAGGTCAATGAAGGG |
| GSN |
AGCTGGCCAAGCTCTACAAG |
TGTTTGCCTGCTTGCCTTTC |
| E-cadherin |
AGGATGACACCCGGGACAAC |
TGCAGCTGGCTCAAGTCAAAG |
| Nectin-2 |
ACCTGCAAAGTGGAGCATGAGA |
CCGGAGATGGACACTTCAGGA |
| ZO-1 |
ATAAAGTGCTGGCTTGGTCTGTTTG |
GCACTGCCCACCCATCTGTA |
| Occludin |
AGTGCCACTTTGGCATTATGAGA |
CTTGTGGCAGCAATTGGAAAC |
Immunofluorescence staining of actin
filaments
Cells were plated onto coverslips (24x24 mm) in
6-well plates until they reached 55-60% confluence. The cells were
fixed in 4% paraformaldehyde for 15 min at room temperature,
permeabilized in 0.1% Triton X-100 for 5 min at room temperature,
blocked in PBS containing 2% bovine serum albumin for 20 min at
room temperature and stained with tetramethyl rhodamine
isothiocyanate-phalloidin (100 nM; 50 µl/well; Beijing Solarbio
Science & Technology Co., Ltd.) for 30 min at room temperature
in the dark. The samples were washed with PBS three times at 5-min
intervals between each step and mounted with anti-fading medium
containing DAPI (cat. no. S2100; Beijing Solarbio Science &
Technology Co., Ltd.) for 3 min at room temperature in the dark.
Actin filaments were observed at 530-550 nm and cell nuclei were
observed at 460-495 nm under an upright fluorescence microscope
(BX53; Olympus Corporation).
Fluorescein isothiocyanate
(FITC)-dextran fluorescence
HPDE6-C7 cell suspensions (2x105
cells/well) were cultured on microporous polycarbonate membranes
(pore size, 0.4 µm) until they reached 100% confluence in the upper
compartment of 12-well Transwell plates (cat. no. 3401; Corning,
Inc.). The culture medium in the upper compartment was removed and
adhered cells were washed with PBS twice. Subsequently,
FITC-dextran (4 kDa; 0.5 mg/ml; 500 µl/well; MilliporeSigma) was
added to the upper compartment, and PBS (1 ml/well) was added to
the lower compartment. The plates were incubated in the dark for 60
min at 37˚C. Fluorescence in the lower compartment was measured
using a microplate reader (excitation, 495 nm; emission, 520 nm;
Thermo Fisher Scientific, Inc.) and quantified using a calibration
curve of FITC-dextran.
Statistical analysis
Data are presented as the mean ± standard deviation
and the experiments were repeated at least three times. Statistical
analysis was performed by one-way analysis of variance and Tukey's
post hoc test using SPSS Statistics version 22.0 (IBM Corp.),
GraphPad Prism version 6.0 (GraphPad Software, Inc.) and ImageJ
(version 1.8.0; National Institutes of Health). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of TG on cell viability and
cell-cell junctions of PDECs
Cell viability and cell apoptosis of PDECs treated
with TG (0.5, 1.0, 2.5, 5.0 and 10 mM) for 24 h were assessed by
light microscope, CCK-8 assay and DAPI staining. The optimal TG
intervention concentration was selected and its effects on
cell-cell junctions was analyzed by TEM. The results showed that
high levels of TG (≥2.5 mM) significantly inhibited cell viability
(Fig. S1A and B) and increased cell apoptosis of PDECs
(Fig. S1C). Therefore, 2.5 mM of
TG was selected for subsequent experiments. Moreover, the analysis
of cell-cell junctions showed that CAE and CAE + TG disrupted
cell-cell junctions, particularly TJs, compared with the control.
In addition, the disruptions were more pronounced with CAE + TG,
but TG had no effect on cell-cell junctions compared with the
control (Fig. S2).
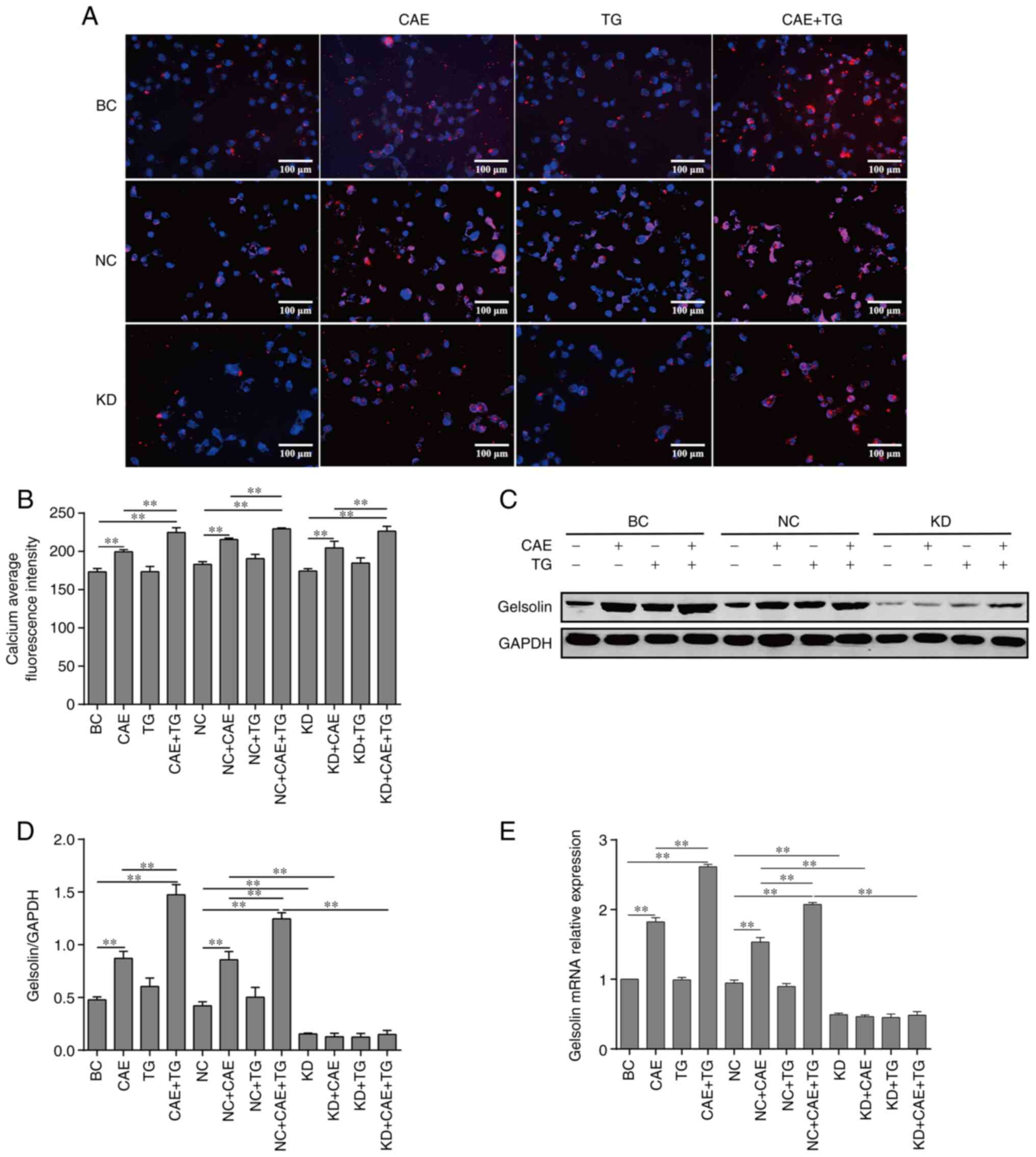
Effect of intracellular calcium levels
on GSN expression in HPDE6-C7 cells treated with CAE and TG
Intracellular calcium was semi-quantified by
fluorescence microscopy, and changes in GSN gene and protein
expression levels in HPDE6-C7 cells treated with CAE and TG were
analyzed by RT-qPCR and western blotting. The results revealed that
CAE and CAE + TG, as treatments of the BC and NC groups, increased
intracellular calcium levels (Fig.
1A and B) and the mRNA and
protein expression levels of GSN relative to baseline (Fig. 1C-E), and the increase was more
pronounced with CAE + TG. Notably, as treatment of the BC and NC
groups, TG had no detectable effect on intracellular calcium levels
(Fig. 1A and B) and GSN expression (Fig. 1C-E). Moreover, GSN silencing in the
KD + CAE and KD + CAE + TG groups did not affect intracellular
calcium levels compared with the NC + CAE and NC + CAE + TG groups
(Fig. 1A and B), and lentiviral-mediated RNA
interference in the KD group significantly reduced the mRNA and
protein expression of GSN in PDECs compared with the NC group
(Fig. 1C-E).
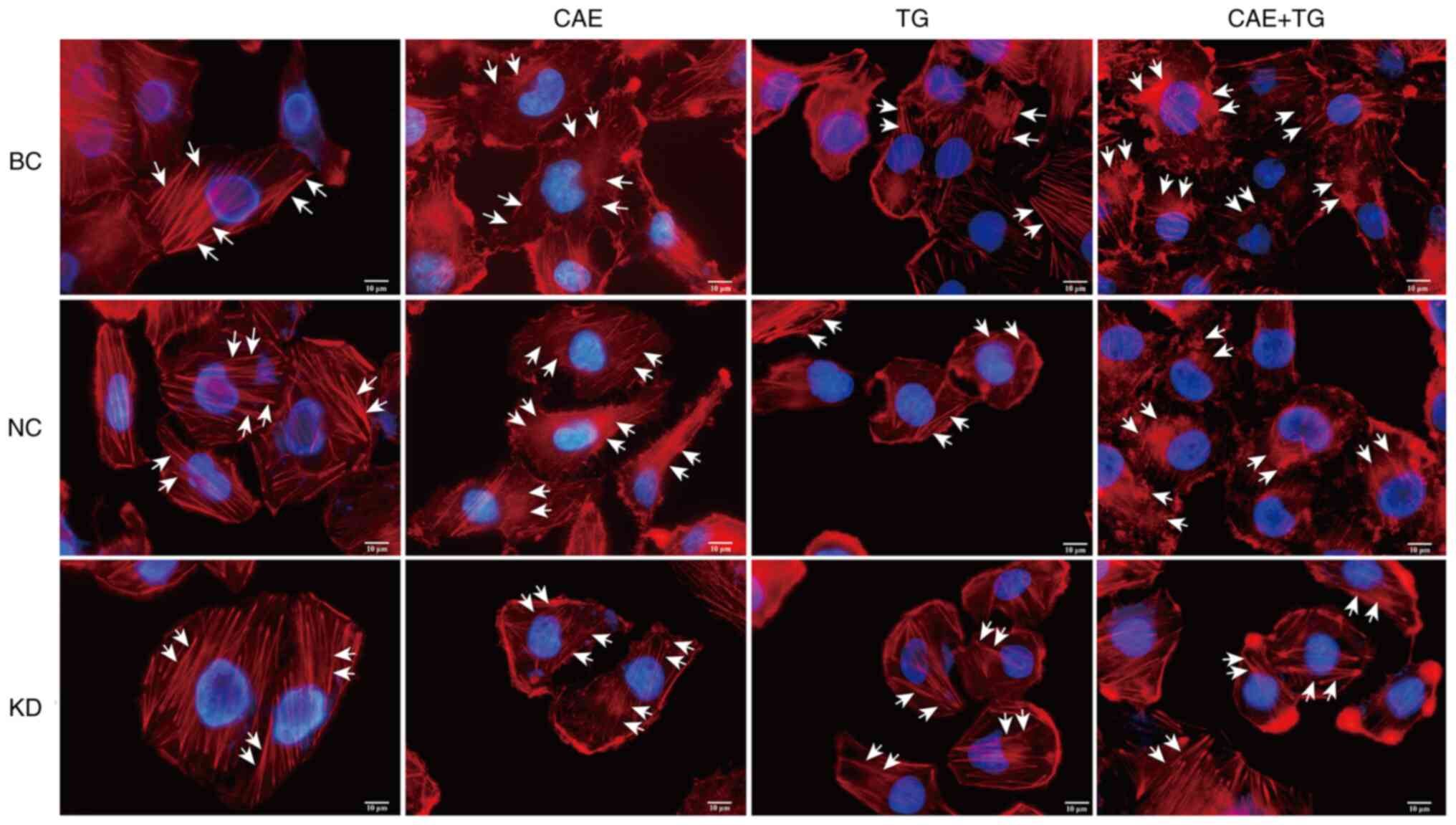
Effect of GSN on actin filament
dynamics in HPDE6-C7 cells treated with CAE and TG
Changes in actin filament dynamics by GSN silencing
were analyzed by immunofluorescence. The results demonstrated that
CAE and CAE + TG, as treatments of the BC and NC groups, disrupted
the actin filament network, and the effect was stronger with CAE +
TG, whereas GSN silencing reduced this effect in the comparisons of
the NC + CAE and KD + CAE groups or the NC + CAE + TG and KD + CAE
+ TG groups. Notably, TG, as treatment of the BC and NC groups, did
not cause actin depolymerization (Fig.
2).
Effect of GSN on the major components
of cell-cell junctions in HPDE6-C7 cells treated with CAE and
TG
The expression levels of major components of
cell-cell junctions, including E-cadherin, nectin-2, ZO-1 and
occludin, were analyzed by RT-qPCR and western blotting. The
results revealed that CAE and CAE + TG, as treatments of the BC and
NC groups, reduced the mRNA (Fig.
3A-D) and protein (Fig. 3E-I)
expression levels of these markers relative to baseline.
Conversely, GSN silencing reduced the effects of CAE and CAE + TG
treatment in the comparisons of the NC + CAE and KD + CAE groups or
the NC + CAE + TG and KD + CAE + TG groups (Fig. 3).
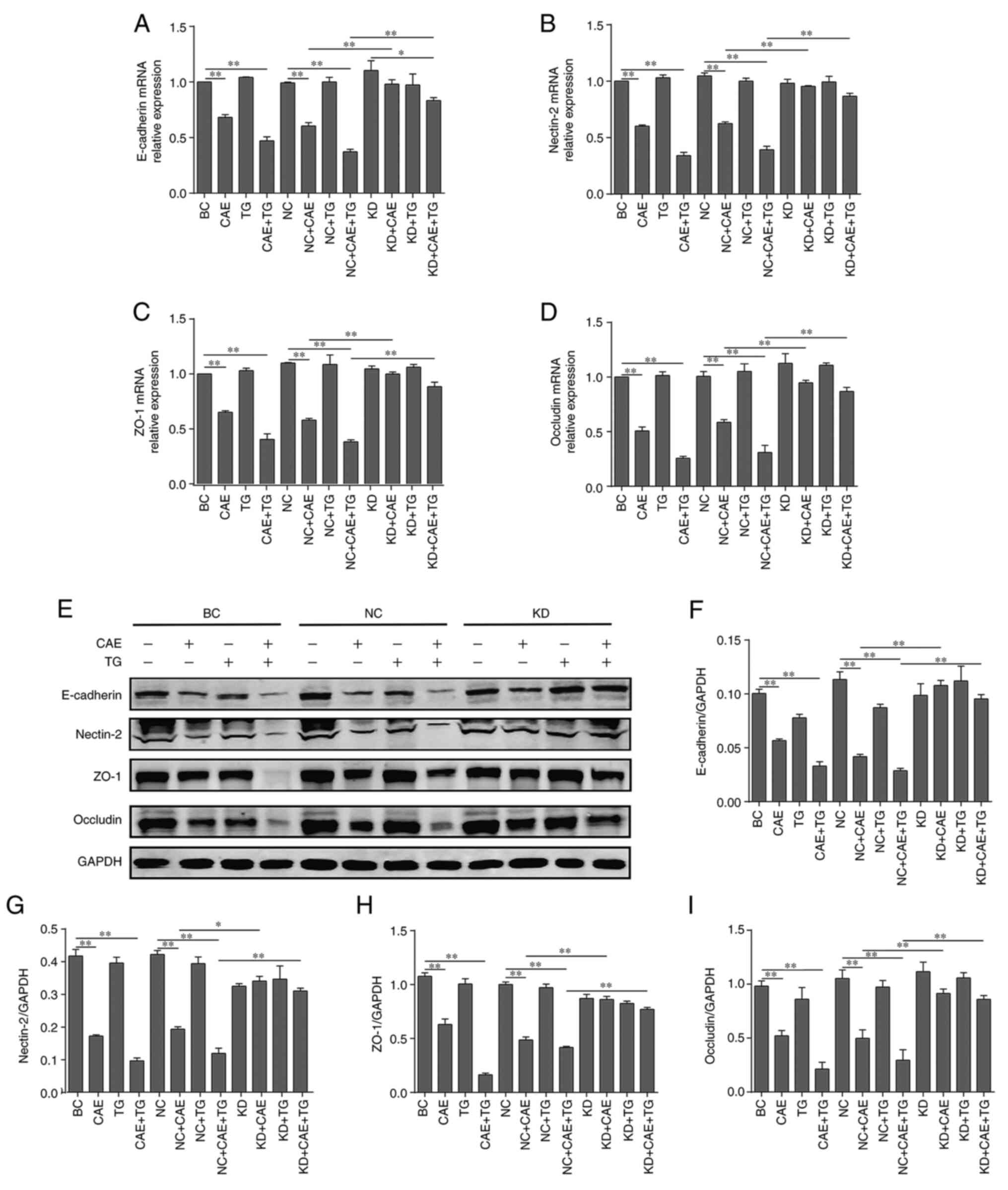 | Figure 3Effect of gelsolin on the major
components of cell-cell junctions in HPDE6-C7 cells treated with
CAE and TG. Relative mRNA expression levels of (A) E-cadherin, (B)
nectin-2, (C) ZO-1 and (D) occludin, as determined by reverse
transcription-quantitative polymerase chain reaction. (E) Western
blot analysis and semi-quantification of the protein expression
levels of (F) E-cadherin, (G) nectin-2, (H) ZO-1 and (I) occludin.
The experiments were repeated at least three times. Data are
presented as the mean ± standard deviation. *P<0.05,
**P<0.01. BC, blank control; CAE, caerulein; KD,
knockdown; NC, negative control; TG, triglycerides. |
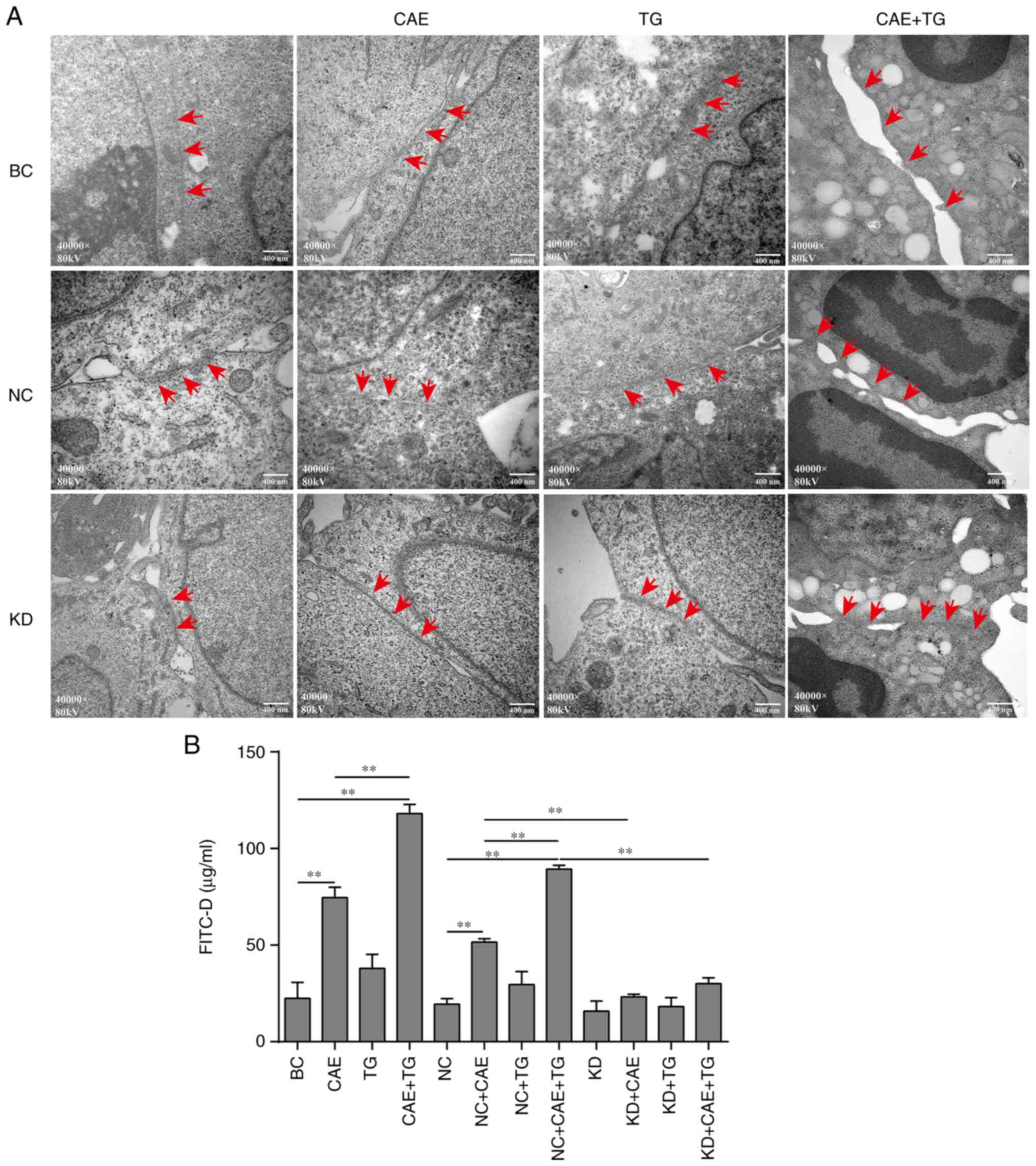
Effect of GSN on TJ ultrastructure and
permeability of HPDE6-C7 cells treated with CAE and TG
The ultrastructure of TJs in HPDE6-C7 cells was
evaluated by TEM. The present study revealed that treatment with
CAE and CAE + TG, as treatments of the BC and NC groups, decreased
the number of TJ strands and disrupted the organization of
cell-cell junctions; some cells in the CAE + TG and NC + CAE + TG
groups had no TJs. GSN silencing mitigated the effects of CAE and
CAE + TG treatment in the comparisons of the NC + CAE and KD + CAE
groups or the NC + CAE + TG and KD + CAE + TG groups (Fig. 4A). In addition, CAE + TG, as
treatment of the BC and NC groups, increased cell permeability more
strongly than CAE, and GSN silencing attenuated this effect in the
comparisons of the NC + CAE and KD + CAE groups or the NC + CAE +
TG and KD + CAE + TG groups, whereas TG, as treatment of the BC and
NC groups, alone did not affect cell permeability (Fig. 4B).
Time-dependent effects of CAE on
apoptosis, GSN protein expression and actin filament dynamics in
HPDE6-C7 cells
HPDE6-C7 cells were treated with CAE for 6, 12, 24
and 48 h. Apoptosis was evaluated using DAPI staining under a
fluorescence microscope, the protein expression of GSN was
semi-quantified using western blotting, and actin filaments were
analyzed by immunofluorescence. The results showed that CAE
increased apoptosis (Fig. S3A),
GSN protein expression (Fig. S3B
and C) and actin filament
depolymerization (Fig. S3D) in a
time-dependent manner in HPDE6-C7 cells compared with the
control.
Discussion
Previous studies have shown that the disruption of
TJs and adherens junctions, which maintain the integrity and
function of the endothelial barrier in PDECs, serves an important
role in the pathogenesis of AP (27,28).
Our data showed that CAE + TG disrupted cell-cell junctions of
PDECs more compared with CAE, suggesting that the impairment of
barrier structure of PDECs was more pronounced in HTGP compared
with in AP. In addition, actin filament dynamics are associated
with the maintenance of barrier integrity in PDECs (29). Furthermore, GSN has a pivotal role
in controlling the actin filament network (30). The results of the present study
showed that the protein expression of GSN increased, actin
filaments depolymerized and the barrier function of PDECs disrupted
in an in vitro model of HTGP. Moreover, GSN silencing
reduced actin filament depolymerization and barrier disruption in
these cells. These data confirmed that GSN may impair barrier
function in PDECs in vitro by causing the depolymerization
of actin filaments in HTGP.
Calcium overload in PACs is a significant
contributor to AP and also occurs in HTGP (31). GSN is a calcium-sensitive modulator
of actin filament length (23);
however, to the best of our knowledge, the occurrence of calcium
overload and its potential association with GSN in PDECs in HTGP
has not been determined. In the present study, CAE + TG increased
intracellular calcium levels and GSN protein expression more
strongly than CAE alone, indicating that calcium overload and GSN
expression in PDECs were more pronounced in HTGP than in AP.
Conversely, GSN gene silencing did not affect calcium levels,
suggesting that GSN as a calcium-activated protein did not regulate
calcium, which is consistent with the literature (23,32).
The rearrangement of the actin network can affect
the interaction between actin and ABPs (33). The ABP GSN modulates actin filament
structure (30). The present study
revealed that the increase in GSN expression levels and the
disruption of actin dynamics were more pronounced in HTGP than in
AP, whereas this disruption was prevented by knocking down GSN,
indicating that GSN may depolymerize actin filaments in HTGP.
The role of GSN in cell-cell junctions in PDECs is
still unclear. The present data demonstrated that the protein
expression levels of GSN were increased and the cell-cell junction
protein expression was decreased in the PDEC model of HTGP, whereas
GSN silencing increased the expression levels of cell-cell junction
proteins, indicating that GSN regulated the expression of these
proteins. In addition, it has been reported that actin interacts
with multiple tight junction proteins, including ZO-1 and occluding
(34), and adherens junction
proteins, such as nectin and E-cadherin (35). Therefore, given the regulatory
effect of GSN on actin in HTGP, it was hypothesized that GSN
regulated the expression of cell-cell junction proteins via actin
depolymerization. However, additional studies are required to
elucidate the underlying mechanisms.
Endothelial barrier function depends on the
interactions of cell-cell junction proteins with the actin filament
network (36,37). However, to the best of our
knowledge, the role of GSN in epithelial barrier function has only
been studied in ischemic lung injury (38). The present results showed that the
impairment of TJs in PDECs was more severe in HTGP than in AP, and
was reduced by knocking down GSN, suggesting that GSN significantly
contributed to the maintenance of TJs in these cells. Moreover, the
increase in the permeability of PDECs was higher in HTGP than in
AP, and this effect was reduced by GSN silencing. Therefore,
considering the association between actin filaments and barrier
function (18,39), it was hypothesized that GSN may
increase cell permeability in HTGP by depolymerizing actin.
Several limitations exist in the current study and
additional work is required. Firstly, it would be beneficial to
also assess the effects of GSN overexpression to further verify the
regulatory mechanism. Secondly, a preliminary time-dependent study
of CAE was performed and it was revealed that CAE increased
apoptosis, GSN protein expression and actin filament
depolymerization in a time-dependent manner in HPDE6-C7 cells
(Fig. S3). Our future work will
assess the time-dependent effects of TG + CAE on barrier function
and actin filaments in PDECs, and the role of GSN in this
mechanism.
In conclusion, the present study indicated that GSN
may impair barrier function in PDECs in HTGP in vitro,
potentially by causing actin depolymerization. These findings
provided evidence for GSN as a novel therapeutic target for barrier
disruption in PDECs in HTGP.
Supplementary Material
Effect of TG on the viability and
apoptosis of HPDE6-C7 cells. HPDE6-C7 cells were treated with TG
(0.5, 1.0, 2.5, 5.0 and 10 mM) for 24 h. (A) Inverted biological
microscopy was used to analyze morphological changes (x100
magnification). (B) Cell viability was determined using the CCK-8
assay. **P<0.01, ***P<0.001 vs.
control. (C) Changes in cell nuclei stained with DAPI under a
fluorescence microscope (x200 magnification). The experiments were
repeated at least three times. Data are presented as the mean ±
standard deviation. TG, triglycerides.
Ultrastructure of TJs in HPDE6-C7
cells treated with CAE and TG. HPDE6-C7 cells were treated with TG
(2.5 mM), CAE (100 nM) or TG + CAE for 24 h. Ultrastructure of TJs
was analyzed by transmission electron microscopy (x80,000
magnification, red arrows indicate TJs). The experiments were
repeated at least three times. CAE, caerulein; TG, triglycerides;
TJ, tight junction.
Time-dependent assessment of the
effects of CAE on GSN expression, actin filaments and apoptosis in
HPDE6-C7 cells. HPDE6-C7 cells were treated with CAE (100 nM) for
6, 12, 24 and 48 h. (A) Changes in cell nuclei stained with DAPI
under a fluorescence microscope (x200 magnification). (B) Western
blotting and (C) semi-quantification of GSN protein expression. (D)
Changes in actin filaments, as determined by tetramethyl rhodamine
isothiocyanate-phalloidin immunofluorescence under an upright
fluorescence microscope (x1,000 magnification, white arrows
indicate actin filaments). The experiments were repeated at least
three times. Data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01. CAE, caerulein; GSN,
gelsolin.
Acknowledgements
The authors would like to thank Mrs. Xiaomin Mo
(Department of Electron Microscopy, Guangxi Medical University,
Nanning, Guangxi, China) for her assistance in transmission
electron microscopy.
Funding
Funding: The present study was supported by grants from the
Guangxi Natural Science Foundation (grant no. 2018GXNSFBA281078)
and the National Natural Science Foundation of China (grant no.
81970558).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HYY was the main contributor to the research,
performing experimental work, analyzing data and writing the
manuscript. GDT and ZHL conceived and designed the study. JLX and
QW designed experimental procedures and performed cell culture. YYQ
established an in vitro model of HTGP. SYZ performed
lentiviral-mediated RNA interference and collected experimental
data. HYY and GDT confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
de Pretis N, Amodio A and Frulloni L:
Hypertriglyceridemic pancreatitis: Epidemiology, pathophysiology
and clinical management. United European Gastroenterol J.
6:649–655. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yang AL and McNabb-Baltar J:
Hypertriglyceridemia and acute pancreatitis. Pancreatology.
20:795–800. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
De Pretis N, De Marchi G and Frulloni L:
Hypertriglyceridemic pancreatitis. Minerva Gastroenterol Dietol.
66:238–245. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Guo YY, Li HX, Zhang Y and He WH:
Hypertriglyceridemia-induced acute pancreatitis: Progress on
disease mechanisms and treatment modalities. Discov Med.
27:101–109. 2019.PubMed/NCBI
|
|
5
|
Konok GP and Thompson AG: Pancreatic
ductal mucosa as a protective barrier in the pathogenesis of
pancreatitis. Am J Surg. 117:18–23. 1969.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Quilichini E, Fabre M, Dirami T, Stedman
A, De Vas M, Ozguc O, Pasek RC, Cereghini S, Morillon L, Guerra C,
et al: Pancreatic ductal deletion of Hnf1b disrupts exocrine
homeostasis, leads to pancreatitis, and facilitates tumorigenesis.
Cell Mol Gastroenterol Hepatol. 8:487–511. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hayashi M and Novak I: Molecular basis of
potassium channels in pancreatic duct epithelial cells. Channels
(Austin). 7:432–441. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ishiguro H, Yamamoto A, Nakakuki M, Yi L,
Ishiguro M, Yamaguchi M, Kondo S and Mochimaru Y: Physiology and
pathophysiology of bicarbonate secretion by pancreatic duct
epithelium. Nagoya J Med Sci. 74:1–18. 2012.PubMed/NCBI
|
|
9
|
Maléth J and Hegyi P: Calcium signaling in
pancreatic ductal epithelial cells: An old friend and a nasty
enemy. Cell Calcium. 55:337–345. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wei B, Gong Y, Yang H, Zhou J, Su Z and
Liang Z: Role of tumor necrosis factor receptor-associated factor 6
in pyroptosis during acute pancreatitis. Mol Med Rep.
24(848)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Goldberg IJ, Cabodevilla AG, Samovski D,
Cifarelli V, Basu D and Abumrad NA: Lipolytic enzymes and free
fatty acids at the endothelial interface. Atherosclerosis. 329:1–8.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Su YR, Hong YP, Mei FC, Wang CY, Li M,
Zhou Y, Zhao KL, Yu J and Wang WX: High-Fat diet aggravates the
intestinal barrier injury via TLR4-rip3 pathway in a rat model of
severe acute pancreatitis. Mediators Inflamm.
2019(2512687)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tanaka S, Nemoto Y, Takei Y, Morikawa R,
Oshima S, Nagaishi T, Okamoto R, Tsuchiya K, Nakamura T, Stutte S
and Watanabe M: High-fat diet-derived free fatty acids impair the
intestinal immune system and increase sensitivity to intestinal
epithelial damage. Biochem Biophys Res Commun. 522:971–977.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Svitkina TM: Ultrastructure of the actin
cytoskeleton. Curr Opin Cell Biol. 54:1–8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Van Itallie CM, Tietgens AJ, Krystofiak E,
Kachar B and Anderson JM: A complex of ZO-1 and the BAR-domain
protein TOCA-1 regulates actin assembly at the tight junction. Mol
Biol Cell. 26:2769–2787. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Malinova TS and Huveneers S: Sensing of
cytoskeletal forces by asymmetric adherens junctions. Trends Cell
Biol. 28:328–341. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yonemura S: Actin filament association at
adherens junctions. J Med Invest. 64:14–19. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shakhov AS, Dugina VB and Alieva IB:
Structural features of actin cytoskeleton required for
endotheliocyte barrier function. Biochemistry (Mosc). 84:358–369.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pollard TD: Actin and actin-binding
proteins. Cold Spring Harb Perspect Biol. 8(a018226)2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Petersen OH, Tepikin AV, Gerasimenko JV,
Gerasimenko OV, Sutton R and Criddle DN: Fatty acids, alcohol and
fatty acid ethyl esters: Toxic Ca2+ signal generation
and pancreatitis. Cell Calcium. 45:634–642. 2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bürgin-Maunder CS, Brooks PR and Russell
FD: Omega-3 fatty acids modulate Weibel-Palade body degranulation
and actin cytoskeleton rearrangement in PMA-stimulated human
umbilical vein endothelial cells. Mar Drugs. 11:4435–4450.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Izadi M, Hou W, Qualmann B and Kessels MM:
Direct effects of Ca2+/calmodulin on actin filament
formation. Biochem Biophys Res Commun. 506:355–360. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Feldt J, Schicht M, Garreis F, Welss J,
Schneider UW and Paulsen F: Structure, regulation and related
diseases of the actin-binding protein gelsolin. Expert Rev Mol Med.
20(e7)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chan MW, El Sayegh TY, Arora PD,
Laschinger CA, Overall CM, Morrison C and McCulloch CA: Regulation
of intercellular adhesion strength in fibroblasts. J Biol Chem.
279:41047–41057. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kim KM, Adyshev DM, Kása A, Zemskov EA,
Kolosova IA, Csortos C and Verin AD: Putative protein partners for
the human CPI-17 protein revealed by bacterial two-hybrid
screening. Microvasc Res. 88:19–24. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kojima T, Yamaguchi H, Ito T, Kyuno D,
Kono T, Konno T and Sawada N: Tight junctions in human pancreatic
duct epithelial cells. Tissue Barriers. 1(e24894)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Naydenov NG, Baranwal S, Khan S, Feygin A,
Gupta P and Ivanov AI: Novel mechanism of cytokine-induced
disruption of epithelial barriers: Janus kinase and protein kinase
D-dependent downregulation of junction protein expression. Tissue
Barriers. 1(e25231)2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sakakibara S, Maruo T, Miyata M, Mizutani
K and Takai Y: Requirement of the F-actin-binding activity of
l-afadin for enhancing the formation of adherens and tight
junctions. Genes Cells. 23:185–199. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lee M and Kang EH: Molecular dynamics
study of interactions between polymorphic actin filaments and
gelsolin segment-1. Proteins. 88:385–392. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Feng S, Wei Q, Hu Q, Huang X, Zhou X, Luo
G, Deng M and Lü M: Research progress on the relationship between
acute pancreatitis and calcium overload in acinar cells. Dig Dis
Sci. 64:25–38. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Nag S, Larsson M, Robinson RC and Burtnick
LD: Gelsolin: The tail of a molecular gymnast. Cytoskeleton
(Hoboken). 70:360–384. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Kang B, Jo S, Baek J, Nakamura F, Hwang W
and Lee H: Role of mechanical flow for actin network organization.
Acta Biomater. 90:217–224. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Odenwald MA, Choi W, Buckley A,
Shashikanth N, Joseph NE, Wang Y, Warren MH, Buschmann MM, Pavlyuk
R, Hildebrand J, et al: ZO-1 interactions with F-actin and occludin
direct epithelial polarization and single lumen specification in 3D
culture. J Cell Sci. 130:243–259. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Troyanovsky RB, Indra I, Chen CS, Hong S
and Troyanovsky SM: Cadherin controls nectin recruitment into
adherens junctions by remodeling the actin cytoskeleton. J Cell
Sci. 128:140–149. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
McRae M, LaFratta LM, Nguyen BM, Paris JJ,
Hauser KF and Conway DE: Characterization of cell-cell junction
changes associated with the formation of a strong endothelial
barrier. Tissue Barriers. 6(e1405774)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sluysmans S, Vasileva E, Spadaro D, Shah
J, Rouaud F and Citi S: The role of apical cell-cell junctions and
associated cytoskeleton in mechanotransduction. Biol Cell.
109:139–161. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Becker PM, Kazi AA, Wadgaonkar R, Pearse
DB, Kwiatkowski D and Garcia JG: Pulmonary vascular permeability
and ischemic injury in gelsolin-deficient mice. Am J Respir Cell
Mol Biol. 28:478–484. 2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bogatcheva NV and Verin AD: The role of
cytoskeleton in the regulation of vascular endothelial barrier
function. Microvasc Res. 76:202–207. 2008.PubMed/NCBI View Article : Google Scholar
|