Introduction
It has been previously reported that ~66% of the
mortality cases associated with diabetes mellitus (DM) can be
attributed to cardiovascular diseases (1). In the absence of changes in blood
pressure and coronary artery disease, DM can alter the structure
and function of the heart by causing a condition known as ‘diabetic
cardiomyopathy’ (2). Initially,
diabetic cardiomyopathy is characterized by cardiac muscle
hypertrophy and associated diastolic dysfunction, which is
typically followed by systolic dysfunction and ultimately heart
failure (3). A maladaptive
inflammatory response has been implicated in the occurrence of
cardiac hypertrophy during diabetic cardiomyopathy (4). Previous studies have revealed that
alleviating the inflammatory response during cardiac hypertrophy is
beneficial to the survival of diabetic cardiomyopathy (5,6).
The toll-like receptor 4 (TLR4)/NF-κB signaling
pathway serves a key role in cardiac hypertrophy (7). Activity of this pathway was
previously found to be significantly promoted in the hypertrophic
myocardium (8). Gao et al
(9) previously reported that
inhibition or knockdown of TLR4/myeloid differentiation primary
response 88 (MyD88) signaling pathway attenuated inflammatory and
hypertrophic responses in transverse aortic constriction or
angiotensin-II infusion of mice and cardiomyocytes isolated from
mouse neonatal ventricles.
Resistin is a cysteine-rich polypeptide that is
mainly secreted by macrophages in humans and by adipose tissues in
rodents and humans (10). It has
been reported to be positively associated with obesity and the
development of type-2 diabetes mellitus (T2DM) (10). In addition, serum levels of
resistin were significantly higher in obese and T2DM patients
compared with those in healthy subjects (11,12).
Resistin has been previously found to be involved in mediating
inflammation, insulin resistance, cardiac hypertrophy,
hypertension, atherosclerosis, coronary artery disease and
rheumatic diseases (13-17).
Kim et al (14) previously
found that resistin overexpression could decrease myocardial
contractility, in addition to endowing primary cardiomyoblasts with
hypertrophic phenotypes to promote cardiac hypertrophy. Other
studies have also found that resistin can induce inflammation,
insulin resistance and hypertension through a TLR4-dependent
signaling pathway (13,15).
Omentin, also known as intelectin-1, is a cytokine
that is typically secreted by the adipose tissue (adipocytokine).
It is mainly expressed in omental and visceral adipose tissues in
humans (18). Physiologically,
omentin has been found to exhibit various pharmacological effects
in the cardiovascular system, with protective effects against
vascular inflammation (19),
atherogenesis (20) and myocardial
ischemia (21) among those
reported. In addition, reduced circulating levels of omentin have
been associated with increased risk of obesity-related diseases,
including metabolic syndrome and T2DM (22). Previous clinical studies have
revealed that decreased plasma concentrations of omentin are
associated with increased incidences of atherosclerosis and
ischemic heart disease (23,24).
Therefore, these previous findings of omentin aforementioned
suggest that it may serve a protective role against cardiovascular
disorders associated with metabolsim. However, the mechanistic role
of omentin in resistin-induced cardiac hypertrophy remains poorly
understood.
In the present study, the potential effects of
omentin on resistin-induced hypertrophy in H9c2 cardiomyoblasts
were investigated. The present study will investigate the
relationship of omentin, resistin and TLR4/MyD88/NF-κB pathway
through H9c2 cardiomyoblasts related experiments.
Materials and methods
Materials
Recombinant human omentin protein (cat. no.
RD172100025) was purchased from BioVendor. Recombinant human
resistin protein (450-19-25) was obtained from PeproTech, Inc.
Anti-TLR4 antibody (cat. no. ab95562) was purchased from Abcam.
Antibodies against MyD88 (cat. no. 4283), NF-κB p65 (cat. no.
8242), phosphorylated (p)-NF-κB p65 (cat. no. 3033), β-tubulin
(cat. no. 2146), p-ERK (Thr-202/Tyr-204; cat. no. 4370), and ERK
(cat. no. 4695) were obtained from Cell Signaling Technology, Inc.
Antibody against β-actin (cat. no. SAB3500350) was from
Sigma-Aldrich; Merck KGaA.
Culture of H9c2 cardiomyoblasts
H9c2 rat cardiomyoblasts were obtained from American
Type Culture Collection. H9c2 cells were cultured in DMEM (Wako
Pure Chemical Industries, Ltd.) containing 10% FBS (Zhejiang
Tianhang Biotechnology, Co., Ltd.) and 1% penicillin-streptomycin
in an atmosphere of 5% CO2 at 37˚C, consistent with
protocols described in a previous study (25). After the H9c2 cardiomyoblasts
reached 90% confluence, they were growth-arrested in FBS-free
medium for 24 h before stimulation with omentin or resistin at
37˚C.
Immunofluorescence to ascertain the
surface area of H9C2 cardiomyoblasts
After the H9c2 cardiomyoblasts were cultured in
serum-free medium for 24 h, they were treated with resistin (100
ng/ml) for 48 h with or without omentin (3, 30 and 300 ng/ml; 1 h)
pre-treatment (26-28)
at 37˚C. To determine the extent of α-actin organization within
sarcomeres, cultured H9c2 cardiomyoblasts were fixed in 100%
methanol for 10 min at 4˚C, washed with PBS three times and blocked
with 10% normal goat serum (Shanghai Yisheng Biotechnology, Co.,
Ltd.) for 30 min at room temperature. The cardiomyoblasts were then
incubated with the mouse anti-α-sarcomeric actin primary monoclonal
antibody (1:500; cat. no. 113200; Sigma-Aldrich; Merck KGaA) for 1
h at room temperature, followed by incubation with the
FITC-conjugated secondary antibody (1:1,000; cat. no. sc-2359;
Santa Cruz Biotechnology, Inc.) for 1 h at room temperature
(29). The nuclei of the H9c2
cardiomyoblasts were stained using Hoechst 33258 (1 µg/ml;
Sigma-Aldrich; Merck KGaA) for visualization for 1 h at 37˚C. A
fluorescence microscope (Carl Zeiss, AG) was used to image the
samples. The surface area of H9c2 cardiomyoblasts was measured
using ImageJ 1.49 (National Institutes of Health) from
two-dimensional images of 50 cells selected at random in 20 fields
at x400 magnification.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the H9c2
cardiomyoblasts using TRIzol® Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to manufacturer
protocols. The RNA concentration was measured using a NanoDrop™
spectrophotometer (Thermo Fisher Scientific, Inc.). Reverse
transcription was performed using a complementary DNA (cDNA)
reverse transcription kit PrimeScript RT Master Mix (cat. no.
RR036Q; Takara Bio, Inc.). The conditions of reaction were: 37˚C
for 15, 85˚C for 5 sec. The obtained cDNA was then subjected to
qPCR for measurement of mRNA expression of atrial natriuretic
peptide (ANF), B-type natriuretic peptide (BNP), β-myosin heavy
chain (β-MHC) and TLR4 using a TB Green Premix Ex Taq II (cat. no.
RR820Q; Takara Bio, Inc.). All reactions were performed using the a
Applied Biosciences 7500 system (Thermo Fisher Scientific, Inc.).
Standard procedure for two-step PCR amplification: Stage 1,
pre-degeneration, 1 cycle, 95˚C for 30 sec; stage 2, PCR reaction,
40 cycles, 95˚C for 3 sec, 60˚C for 30 sec. β-actin was used as the
internal reference. Primer sequences were procured from Sangon
Biotech Co., Ltd. The primer sequences were as follows: ANF
forward, 5'-AGGCCATATTGGAGCAAATC-3' and reverse,
5'-CATCTTCTCCTCCAGGTGGT-3'; BNP forward, 5'-GTGCTGCCCCAGATGATTCT-3'
and reverse, 5'-GCAGCTTCTGCATCGTGGAT-3'; β-MHC forward,
5'-TGCTCTACAATCTCAAGGAGAGGT-3' and reverse,
5'-TGTTGACGGTCTTACCAGCTC-3'; TLR4 forward,
5'-AAGTTATTGTGGTGGTGTCTAG-3' and reverse,
5'-GAGGTAGGTGTTTCTGCTAAG-3' and β-actin forward,
5'-GAACCCTAAGGCCAACCG-3' and reverse, 5'-TACGTACATGGCTGGGGTGT-3'.
Relative quantification of mRNA expression was analyzed using the
2-ΔΔCq method (30).
Western blotting
After the H9c2 cardiomyoblasts were treated with
resistin (100 ng/ml) for 1 or 12 h with or without pretreatment
with omentin (300 ng/ml; 1 h) at 37˚C, samples of total protein
were obtained by homogenizing H9c2 cardiomyoblasts with RIPA lysis
buffer (CoWin Biosciences). Protein concentration was determined
using the bicinchoninic acid assay before 5X Laemmli buffer was
added. An equal amount of protein (25-30 µg) was separated by 10%
SDS-PAGE, before the proteins were transferred onto PVDF membranes
(MilliporeSigma). After blockade with 0.5% non-fat milk or 5%
bovine serum albumin (Boster Biological Technology) for 2 h at room
temperature, the PVDF membranes were incubated with primary
antibodies [TLR4, MyD88, phosphorylated (p-)-NF-κB p65, NF-κB p65,
p-ERK and ERK antibodies at 1:1,000 dilution; β-tubulin and β-actin
at 1:2,000 dilution] overnight at 4˚C. After washing three times
with TBS containing 0.1% Tween 20, the PVDF membranes were
incubated with HRP-conjugated secondary antibodies (1:2,000
dilution; cat. no. 70745; Cell Signaling Technology, Inc.) for 1 h
at room temperature. Immunoreactive bands were visualized using an
enhanced chemiluminescence detection system (Bio-Techne) and the
densitometry was quantified by ImageJ 1.49 (National Institutes of
Health).
Statistical analysis
Data are presented as the mean ± SEM at least 4
independent experiments. Statistical comparisons were performed
using one-way ANOVA followed by Bonferroni's test. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analyses were conducted using Graphpad Prism 5
(GraphPad Software, Inc.).
Results
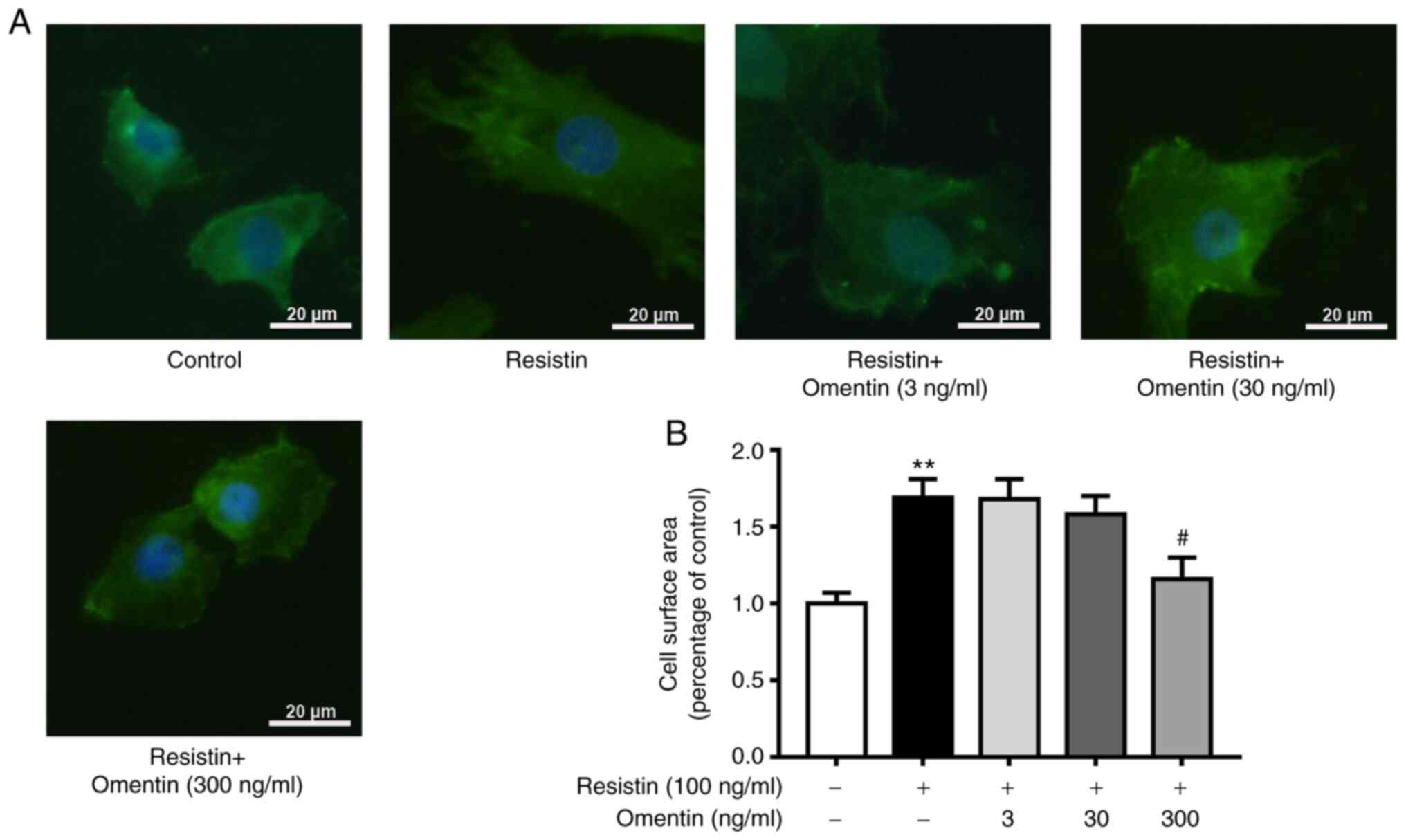
Effects of omentin on the surface area
of resistin-induced H9c2 cardiomyoblasts
Resistin has been previously shown to induce
cardiomyoblast hypertrophy (14).
In the present study, it was observed that resistin (100 ng/ml)
significantly increased the surface area of the H9c2
cardiomyoblasts (1.69±0.12-fold relative to control; P<0.01;
Fig. 1). Therefore, the potential
effects of omentin (3, 30 and 300 ng/ml, 1 h) pre-treatment on this
increase in the surface area of resistin-induced H9c2
cardiomyoblasts were examined. Omentin (300 ng/ml, 1 h) was found
to significantly reverse the resistin (100 ng/ml, 48 h)-induced
increases in the surface area of H9c2 cardiomyoblasts (300 ng/ml
omentin + resistin, 1.16±0.14-fold relative to control; P<0.05;
Fig. 1).
Effects of omentin on the mRNA
expression of ANF, BNP, β-MHC and TLR4 in resistin-induced H9c2
cardiomyoblasts
Re-activation of fetal genes ANF, BNP and β-MHC is a
characteristic feature of cardiac hypertrophy (31). Therefore, the present study next
assessed the potential effects of omentin (300 ng/ml, 1 h) on the
mRNA expression of ANF, BNP and β-MHC in resistin-induced H9c2
cardiomyoblasts. In addition, the effect of omentin (300 ng/ml; 1
h) on resistin-induced mRNA expression of TLR4 was also assessed in
H9c2 cardiomyoblasts. Omentin significantly reversed the
resistin-induced (100 ng/ml, 24 h) increase in expression of ANF
mRNA (resistin, 1.57±0.07-fold relative to control; omentin +
resistin, 1.21±0.06-fold relative to control; P<0.05; Fig. 2A). Omentin also significantly
reversed the resistin-induced (100 ng/ml, 24 h) increase in
expression of BNP mRNA (resistin, 1.79±0.09-fold relative to
control; omentin + resistin, 1.25±0.10-fold relative to control;
P<0.01; Fig. 2B). In addition,
omentin significantly reversed the resistin-induced (100 ng/ml, 24
h) increase in expression of β-MHC mRNA (resistin, 1.95±0.18-fold
relative to control; omentin + resistin, 1.35±0.11-fold relative to
control; P<0.05; Fig. 2C).
Omentin significantly reversed the resistin-induced (100 ng/ml, 24
h) increase in expression of TLR4 mRNA (resistin, 1.55±0.09-fold
relative to control; omentin + resistin, 1.11±0.05-fold relative to
control; P<0.05; Fig. 2D).
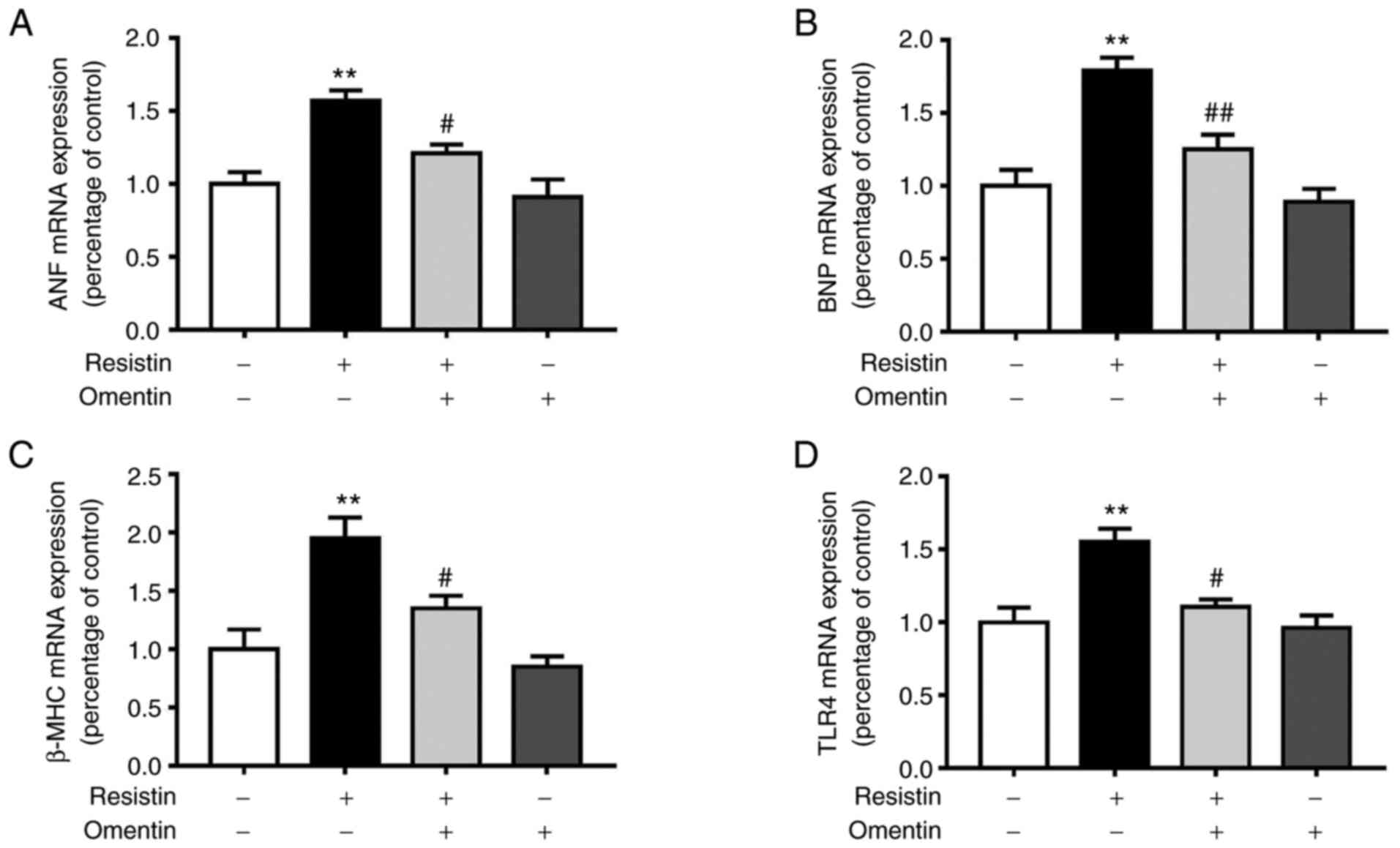 | Figure 2Effects of omentin on the mRNA
expression of ANF, BNP, β-MHC and TLR4 in resistin-induced H9c2
cardiomyoblasts. After the H9c2 cardiomyoblasts were treated with
resistin (100 ng/ml, 24 h) in the absence or presence of omentin
(300 ng/ml, 1 h), total RNA was then extracted. (A) Expression of
ANF, (B) BNP, (C) β-MHC and (D) TLR4 mRNA was measured by reverse
transcription-quantitative PCR. n=4. **P<0.01 vs.
control; #P<0.05 and ##P<0.01 vs.
resistin-only. ANF, atrial natriuretic peptide; BNP, B-type
natriuretic peptide; β-MHC, β-myosin heavy chain; TLR4, toll-like
receptor 4. |
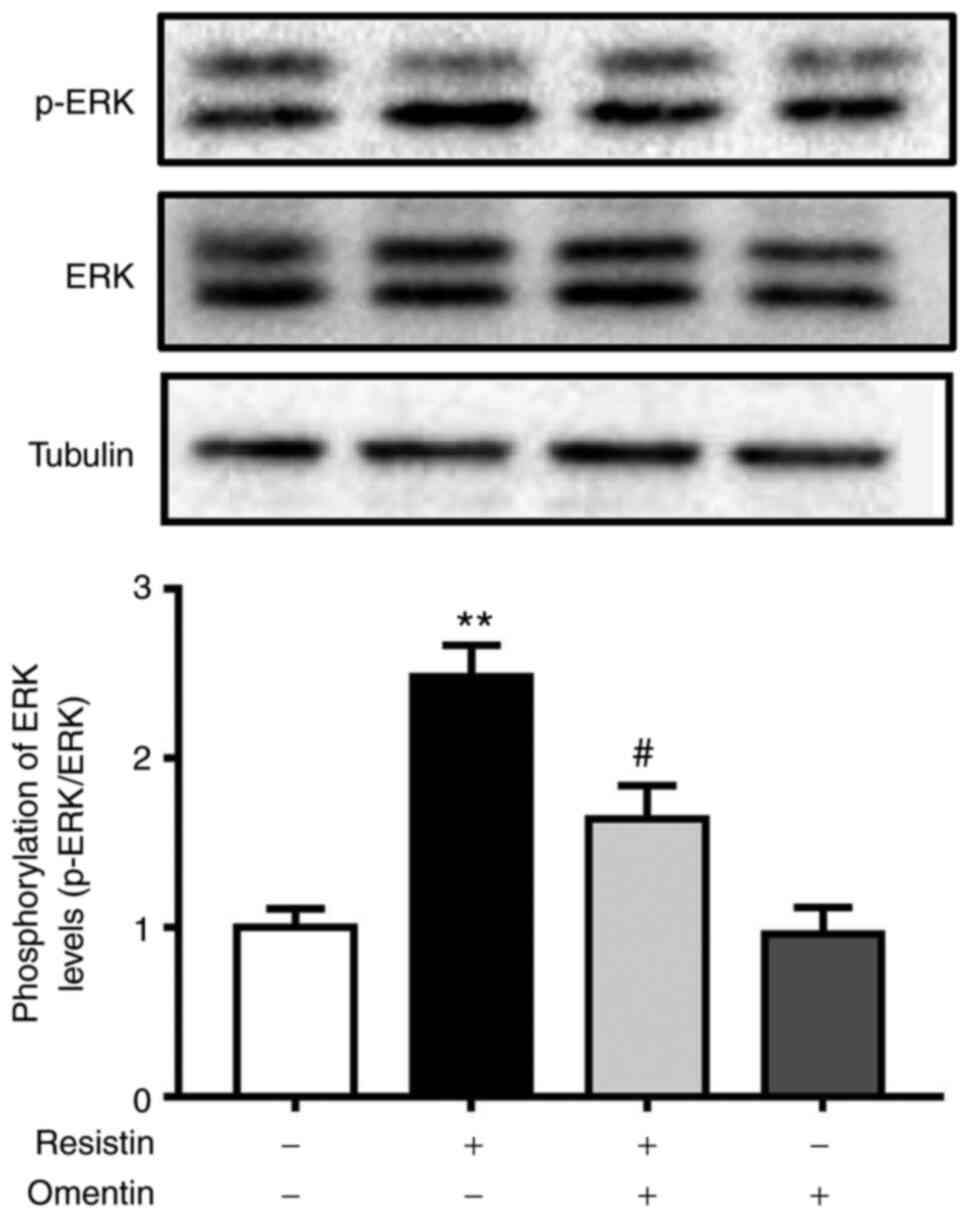
Effects of omentin on ERK
phosphorylation in resistin-induced H9c2 cardiomyoblasts
An important process in cardiomyoblast hypertrophy
is ERK activation (28,29). The present study next investigated
the effects of omentin (300 ng/ml, 1 h) on resistin-induced ERK
phosphorylation in H9c2 cardiomyoblasts by western blotting.
Stimulation of H9c2 cardiomyoblasts with resistin (100 ng/ml, 1 h)
had no effects on the protein expression of total ERK, but
significantly increased phosphorylation levels of ERK at
Thr-202/Tyr-204 and the p-ERK/ERK ratio (Fig. 3), suggesting an increase in ERK
activity due to resistin. However, omentin pre-treatment
significantly prevented the resistin-induced ERK phosphorylation
(resistin, 2.48±0.19-fold relative to control; omentin + resistin;
1.64±0.20-fold relative to control; P<0.05; Fig. 3).
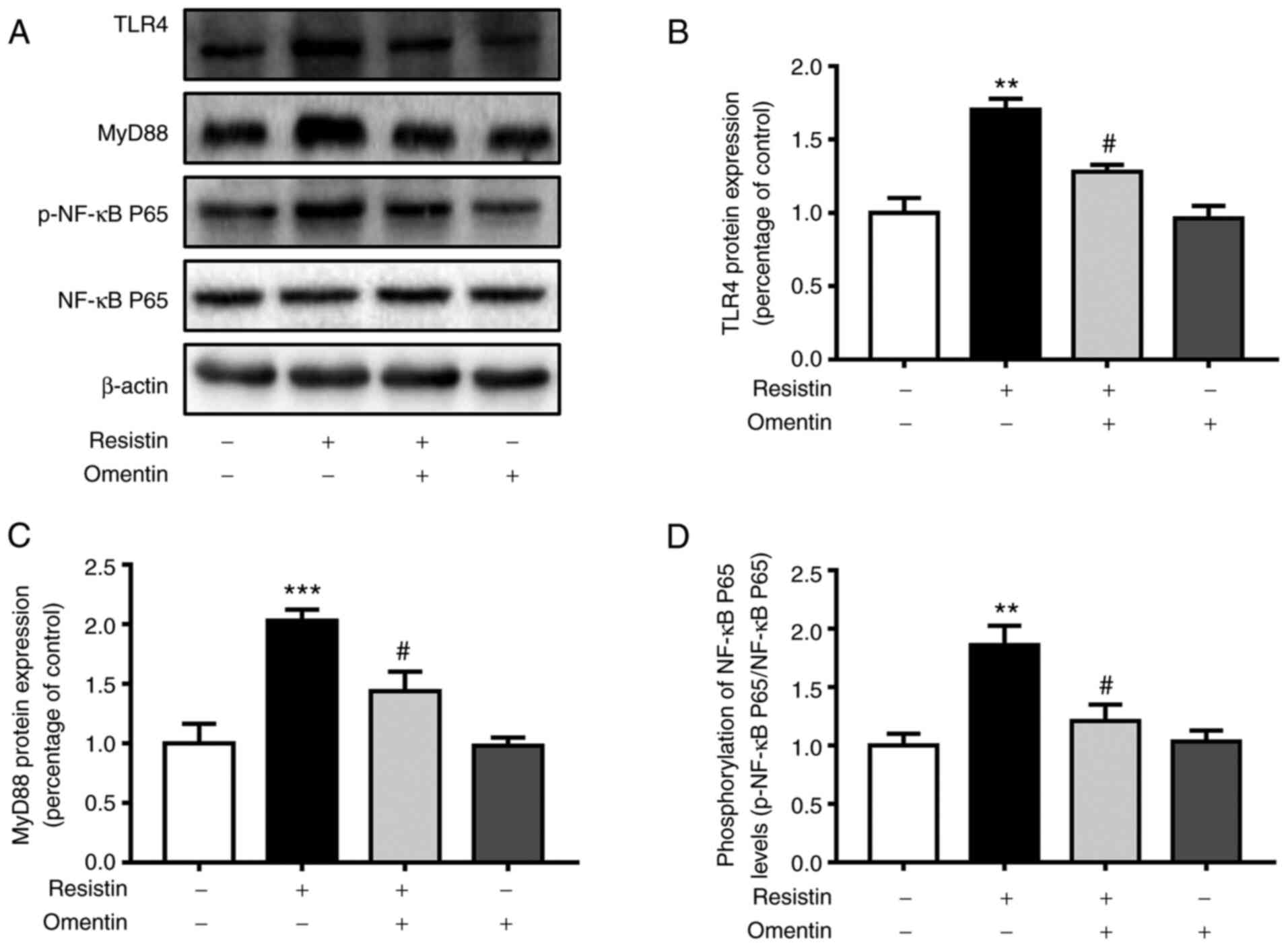
Effect of omentin on the protein
expression of TLR4, MyD88 and NF-κB p65 phosphorylation in
resistin-induced H9c2 cardiomyoblasts
The present study next assessed the effects of
omentin (300 ng/ml, 1 h) on the resistin-induced protein levels of
TLR4, MyD88, p-NF-κB p65 and NF-κB p65 in H9c2 cardiomyoblasts by
western blotting. Omentin significantly inhibited the
resistin-induced (100 ng/ml, 12 h) protein expression of TLR4
(resistin, 1.70±0.07-fold relative to control; omentin + resistin,
1.28±0.09-fold relative to control, P<0.05; Fig. 4B) in H9c2 cardiomyoblasts. Omentin
also significantly prevented the resistin-induced (100 ng/ml, 12 h)
expression of the MyD88 protein (resistin, 2.03±0.09-fold relative
to control; omentin + resistin, 1.44±0.17-fold relative to control,
P<0.05; Fig. 4C) in H9c2
cardiomyoblasts. In addition, omentin significantly reversed the
resistin-induced (100 ng/ml, 12 h) protein phosphorylation of NF-κB
p65 (resistin, 1.86±0.17-fold relative to control; omentin +
resistin, 1.21±0.14-fold relative to control, P<0.05; Fig. 4D) in H9c2 cardiomyoblasts. However,
omentin had no effects on the protein expression of total NF-κB
p65, suggesting that omentin inhibited the activity of
resistin-induced TLR4/MyD88/NF-κB signaling in H9c2
cardiomyoblasts.
Discussion
In the present study, it was demonstrated that
omentin inhibited the resistin-induced hypertrophy of H9c2
cardiomyoblasts. In addition, omentin inhibited resistin-induced
expression of TLR4, MyD88 and NF-κB p65 phosphorylation, which are
important molecular components of the TLR4/MyD88/NF-κB inflammatory
pathway (32). Omentin also
inhibited the resistin-induced re-activation of the expression of
fetal genes ANF, BNP and β-MHC, which is the characteristic feature
of cardiac hypertrophy (31).
Furthermore, it was demonstrated that omentin inhibited the
resistin-induced activation of ERK, which is an important mediator
of cardiomyoblast hypertrophy (33,34).
Taken together, these observations suggest that omentin can inhibit
the hypertrophy of resistin-induced H9c2 cardiomyoblasts by
blunting the activity of TLR4/MyD88/NF-κB signaling.
TLRs are pathogen pattern-recognition receptors and
serve as important components of the innate immune system (35). The first member of the TLR family
to be identified was TLR4(32),
which can mediate myocardial inflammation (36). In addition, the TLR4-activated
mediation of inflammatory signaling serves an important role in
myocarditis, cardiac hypertrophy, ischemia-reperfusion injury and
myocardial infarction (37-39).
A number of studies have previously shown that production of
proinflammatory factors is regulated by the TLR4/MyD88/NF-κB
signaling pathway, which in turn induces inflammation in the
myocardial tissues and causing injury (40,41).
In addition, it has been reported that resistin is involved in
myocardial inflammation through a TLR4-associated pathway to cause
myocardial tissue injury (13-15,42,43).
Cardiac hypertrophy is an adaptive change of the
myocardium to increase the volume or pressure loads (44). However, this physiological
adaptation is frequently accompanied with pathological changes
(31). Activation of several
intracellular signaling pathways is closely associated with the
occurrence and development of cardiac hypertrophy. Han et al
(45) found TLR4 activation could
initiate myocardial remodeling. In another study, Ehrentraut et
al (46) previously found that
TLR4 antagonists could reduce cardiac hypertrophy in mice.
Activation of the TLR4/MyD88/NF-κB signaling pathway has been
demonstrated to increase the expression of a number of the
proinflammatory cytokines, such as TNF-α and IL-6, which involved
in the inflammatory response to cause myocardial injury (40,41).
In addition, cardiac hypertrophy and myocardial inflammation were
stifled effectively by inhibiting the activity of the TLR4
signaling pathway (46,47). In particular, inhibiting the
expression of NF-κB p65 which is one of components in the NF-κB
signaling pathway was able to reduce the myocardial inflammatory
response, inhibit the development of cardiac hypertrophy and reduce
the risk of heart failure in the transgenic mouse (48). The present study demonstrated that
resistin could significantly activate the TLR4/MyD88/NF-κB
signaling pathway in H9c2 cardiomyoblasts, to induce an increase in
the surface area of H9c2 cardiomyoblasts.
Omentin is a newly identified adipocytokine that was
shown to exert an anti-inflammatory effect (49,50).
Previous studies have demonstrated that various cardiovascular
diseases, including carotid atherosclerosis and coronary artery
disease, manifest with reduced plasma concentrations of omentin
(23,24,51).
Genre et al (52) found
that low serum omentin levels were associated with cardiovascular
risk factors, including obesity and high atherosclerosis indices,
in patients with axial spondyloarthritis. In addition, a number of
previous studies have demonstrated high plasma concentrations of
omentin to be associated with superior outcomes in patients with
acute heart failure or coronary heart disease (49,53).
Ma et al (8) found that
inhibition of TLR4 pathway can inhibit myocardial hypertrophy in
mice. In the present study, H9c2 cardiomyoblasts were first
pre-treated with omentin (3, 30 and 300 ng/ml) and then found that
300 ng/ml omentin prevented the resistin-induced hypertrophy of
H9c2 cardiomyoblasts. However, omentin at 3 and 30 ng/ml could not,
which are consistent with the cardiovascular benefits of increasing
the human serum omentin concentration (53). In addition, it was found that
omentin inhibited the mRNA and protein expression of TLR4 induced
by resistin in H9c2 cardiomyoblasts. Omentin also inhibited the
protein expression of MyD88 and phosphorlylation of NF-κB p65 after
resistin stimulation in H9c2 cardiomyoblasts. Therefore, the
ability of omentin to attenuate cardiomyoblast hypertrophy is
likely due to its ability to inhibit the expression of components
in the TLR4/MyD88/NF-κB signaling pathway in H9c2 cardiomyoblasts.
Collectively, these data suggest that omentin-mediated inhibition
of the TLR4/MyD88/NF-κB signaling pathway may represent a common
pathway that leads to the beneficial actions of omentin in the
cardiovascular system.
ERK activation is an important process in
cardiomyoblast hypertrophy (33,34).
Inhibition of TLR4 was shown to reduce ERK activity in various cell
types, including cardiomyoblasts (44,45).
This suggests that inhibition of TLR4 may reduce cardiomyoblasts
hypertrophy. The present study demonstrated that stimulation of
H9c2 cardiomyoblasts with resistin led to increased ERK activity.
However, pre-treatment of the H9c2 cardiomyoblasts with omentin
inhibited ERK activation in response to resistin treatment.
Reactivation of fetal myocardial genes, including
ANF, BNP and β-MHC, is a characteristic feature of cardiac
hypertrophy (31). The present
study found that resistin could enhance the expression of these
fetal myocardial genes in H9c2 cardiomyoblasts, whilst omentin
could inhibit this resistin-induced expression of myocardial fetal
genes in H9c2 cardiomyoblasts. Therefore, inhibition of
resistin-induced cardiomyoblast hypertrophy by omentin may be
associated with the inhibition of the expression of myocardial
fetal genes.
A number of limitations are associated with the
present study. The findings from the present study would require
verification in vivo. In addition, agonists of the
TLR4/MyD88/NF-κB signaling pathway or TLR4 knockdown would need be
used to clarify if the inhibition of omentin on resistin-induced
hypertrophy of cardiomyoblasts can be reversed. The extent of p65
activation in the nucleus would also require further confirmation
in subsequent studies. Furthermore, measurements of the expression
of inflammatory factors following omentin treatment will need to be
performed. It would also be of interest to investigate the
potential effects of omentin on resistin-induced oxidative stress
(27).
To conclude, the present study showed that omentin
can inhibit resistin-induced hypertrophy of H9c2 cardiomyoblasts
through inhibition of the TLR4/MyD88/NF-κB signaling pathway. These
results suggest that omentin may be an attractive therapeutic
target against resistin-induced cardiac hypertrophy.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from Shanxi
Cardiovascular Hospital Incentive Plan Fund (grant no.
XYS20170101), Natural Science Foundation of Shanxi Province, China
(grant no. 201901D111362), Natural Science Foundation of Shanxi
Province, China (grant no. 201701D121162), Scientific Research
Foundation of Health Commission of Shanxi Province, China (grant
no. 201601093) and Zhejiang Medical and Health Research Projects,
China (grant nos. 2018KY915 and 2019KY793).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YD designed the current study. YD and XY wrote the
manuscript. XY established the hypertrophic model of H9c2
cardiomyoblasts. XY and MG performed H9c2 cardiomyoblasts
immunofluorescence staining. XY and JY performed RT-qPCR. LW and PY
performed western blotting. XY performed statistical analysis. YD
and XY confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Low Wang CC, Hess CN, Hiatt WR and
Goldfine AB: Clinical update: Cardiovascular disease in diabetes
mellitus: Atherosclerotic cardiovascular disease and heart failure
in type 2 diabetes mellitus-mechanisms, management, and clinical
considerations. Circulation. 133:2459–2502. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Boudina S and Abel ED: Diabetic
cardiomyopathy revisited. Circulation. 115:3213–3223.
2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jia G, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jia G, DeMarco VG and Sowers JR: Insulin
resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat
Rev Endocrinol. 12:144–153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Di Luigi L, Corinaldesi C, Colletti M,
Scolletta S, Antinozzi C, Vannelli GB, Giannetta E, Gianfrilli D,
Isidori AM, Migliaccio S, et al: Phosphodiesterase type 5 inhibitor
sildenafil decreases the proinflammatory chemokine CXCL10 in human
cardiomyocytes and in subjects with diabetic cardiomyopathy.
Inflammation. 39:1238–1252. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tan Y, Zhang Z, Zheng C, Wintergerst KA,
Keller BB and Cai L: Mechanisms of diabetic cardiomyopathy and
potential therapeutic strategies: Preclinical and clinical
evidence. Nat Rev Cardiol. 17:585–607. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xiao Z, Kong B, Yang H, Dai C, Fang J, Qin
T and Huang H: Key player in cardiac hypertrophy, emphasizing the
role of toll-like receptor 4. Front Cardiovasc Med.
7(579036)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ma D, Zhang J, Zhang Y, Zhang X, Han X,
Song T, Zhang Y and Chu L: Inhibition of myocardial hypertrophy by
magnesium isoglycyrrhizinate through the TLR4/NF-κB signaling
pathway in mice. Int Immunopharmacol. 55:237–244. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gao W, Wang H, Zhang L, Cao Y, Bao JZ, Liu
ZX, Wang LS, Yang Q and Lu X: Retinol-binding protein 4 induces
cardiomyocyte hypertrophy by activating TLR4/MyD88 pathway.
Endocrinology. 157:2282–2293. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jamaluddin MS, Weakley SM, Yao Q and Chen
C: Resistin: Functional roles and therapeutic considerations for
cardiovascular disease. Br J Pharmacol. 165:622–632.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Steppan CM, Bailey ST, Bhat S, Brown EJ,
Banerjee RR, Wright CM, Patel HR, Ahima RS and Lazar MA: The
hormone resistin links obesity to diabetes. Nature. 409:307–312.
2001.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Gerber M, Boettner A, Seidel B, Lammert A,
Bär J, Schuster E, Thiery J, Kiess W and Kratzsch J: Serum resistin
levels of obese and lean children and adolescents: Biochemical
analysis and clinical relevance. J Clin Endocrinol Metab.
90:4503–4509. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pine GM, Batugedara HM and Nair MG: Here,
there and everywhere: Resistin-like molecules in infection,
inflammation, and metabolic disorders. Cytokine. 110:442–451.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kim M, Oh JK, Sakata S, Liang I, Park W,
Hajjar RJ and Lebeche D: Role of resistin in cardiac contractility
and hypertrophy. J Mol Cell Cardiol. 45:270–280. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jiang Y, Lu L, Hu Y, Li Q, An C, Yu X, Shu
L, Chen A, Niu C, Zhou L and Yang Z: Resistin induces hypertension
and insulin resistance in mice via a TLR4-dependent pathway. Sci
Rep. 6(22193)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ohmori R, Momiyama Y, Kato R, Taniguchi H,
Ogura M, Ayaori M, Nakamura H and Ohsuzu F: Associations between
serum resistin levels and insulin resistance, inflammation, and
coronary artery disease. J Am Coll Cardiol. 46:379–380.
2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Filková M, Haluzík M, Gay S and Senolt L:
The role of resistin as a regulator of inflammation: Implications
for various human pathologies. Clin Immunol. 133:157–170.
2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang RZ, Lee MJ, Hu H, Pray J, Wu HB,
Hansen BC, Shuldiner AR, Fried SK, McLenithan JC and Gong DW:
Identification of omentin as a novel depot-specific adipokine in
human adipose tissue: Possible role in modulating insulin action.
Am J Physiol Endocrinol Metab. 290:E1253–E1261. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yamawaki H, Kuramoto J, Kameshima S, Usui
T, Okada M and Hara Y: Omentin, a novel adipocytokine inhibits
TNF-induced vascular inflammation in human endothelial cells.
Biochem Biophys Res Commun. 408:339–343. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Watanabe K, Watanabe R, Konii H, Shirai R,
Sato K, Matsuyama TA, Ishibashi-Ueda H, Koba S, Kobayashi Y, Hirano
T and Watanabe T: Counteractive effects of omentin-1 against
atherogenesis†. Cardiovasc Res. 110:118–128. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kataoka Y, Shibata R, Ohashi K, Kambara T,
Enomoto T, Uemura Y, Ogura Y, Yuasa D, Matsuo K, Nagata T, et al:
Omentin prevents myocardial ischemic injury through AMP-activated
protein kinase- and Akt-dependent mechanisms. J Am Coll Cardiol.
63:2722–2733. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pan HY, Guo L and Li Q: Changes of serum
omentin-1 levels in normal subjects and in patients with impaired
glucose regulation and with newly diagnosed and untreated type 2
diabetes. Diabetes Res Clin Pract. 88:29–33. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Shibata R, Takahashi R, Kataoka Y, Ohashi
K, Ikeda N, Kihara S, Murohara T and Ouchi N: Association of a
fat-derived plasma protein omentin with carotid artery intima-media
thickness in apparently healthy men. Hypertens Res. 34:1309–1312.
2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhong X, Zhang HY, Tan H, Zhou Y, Liu FL,
Chen FQ and Shang DY: Association of serum omentin-1 levels with
coronary artery disease. Acta Pharmacol Sin. 32:873–878.
2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Luo JW, Zheng X, Cheng GC, Ye QH, Deng YZ
and Wu L: Resistin-induced cardiomyocyte hypertrophy is inhibited
by apelin through the inactivation of extracellular
signal-regulated kinase signaling pathway in H9c2 embryonic rat
cardiomyocytes. Biomed Rep. 5:473–478. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ou HC, Lee WJ, Wu CM, Chen JF and Sheu WH:
Aspirin prevents resistin-induced endothelial dysfunction by
modulating AMPK, ROS, and Akt/eNOS signaling. J Vasc Surg.
55:1104–1115. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cheleschi S, Gallo I, Barbarino M,
Giannotti S, Mondanelli N, Giordano A, Tenti S and Fioravanti1 A:
MicroRNA mediate visfatin and resistin induction of oxidative
stress in human osteoarthritic synovial fibroblasts Via NF-κB
pathway. Int J Mol Sci. 20(5200)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Matsuo K, Shibata R, Ohashi K, Kambara T,
Uemura Y, Hiramatsu-Ito M, Enomoto T, Yuasa D, Joki Y, Ito M, et
al: Omentin functions to attenuate cardiac hypertrophic response. J
Mol Cell Cardiol. 79:195–202. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hardt SE, Tomita H, Katus HA and Sadoshima
J: Phosphorylation of eukaryotic translation initiation factor
2Bepsilon by glycogen synthase kinase-3beta regulates
beta-adrenergic cardiac myocyte hypertrophy. Circ Res. 94:926–935.
2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sugden PH and Clerk A: Cellular mechanisms
of cardiac hypertrophy. J Mol Med (Berl). 76:725–746.
1998.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Akira S and Takeda K: Toll-like receptor
signaling. Nat Rev Immunol. 4:499–511. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Yue TL, Gu JL, Wang C, Reith AD, Lee JC,
Mirabile RC, Kreutz R, Wang Y, Maleeff B, Parsons AA and Ohlstein
EH: Extracellular signal-regulated kinase plays an essential role
in hypertrophic agonists, endothelin-1 and phenylephrine-induced
cardiomyocyte hypertrophy. J Biol Chem. 275:37895–37901.
2000.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tanaka K, Honda M and Takabatake T: Redox
regulation of MAPK pathways and cardiac hypertrophy in adult rat
cardiac myocyte. J Am Coll Cardiol. 37:676–685. 2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lundberg AM, Ketelhuth DF, Johansson ME,
Gerdes N, Liu S, Yamamoto M, Akira S and Hansson GK: Toll-like
receptor 3 and 4 signalling through the TRIF and TRAM adaptors in
haematopoietic cells promotes atherosclerosis. Cardiovasc Res.
99:364–373. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Molteni M, Gemma S and Rossetti C: The
role of toll-like receptor 4 in infectious and noninfectious
inflammation. Mediators Inflamm. 2016(6978936)2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chimenti C, Verardo R, Scopelliti F,
Grande C, Petrosillo N, Piselli P, De Paulis R and Frustaci A:
Myocardial expression of Toll-like receptor 4 predicts the response
to immunosuppressive therapy in patients with virus-negative
chronic inflammatory cardiomyopathy. Eur J Heart Fail. 19:915–925.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Soraya H, Clanachan AS, Rameshrad M,
Maleki-Dizaji N, Ghazi-Khansari M and Garjani A: Chronic treatment
with metformin suppresses toll-like receptor 4 signaling and
attenuates left ventricular dysfunction following myocardial
infarction. Eur J Pharmacol. 737:77–84. 2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lu M, Tang F, Zhang J, Luan A, Mei M, Xu
C, Zhang S, Wang H and Maslov LN: Astragaloside IV attenuates
injury caused by myocardial ischemia/reperfusion in rats via
regulation of toll-like receptor 4/nuclear factor-κB signaling
pathway. Phytother Res. 29:599–606. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang J, Zhang J, Yu P, Chen M, Peng Q,
Wang Z and Dong N: Remote ischaemic preconditioning and sevoflurane
postconditioning synergistically protect rats from myocardial
injury induced by ischemia and reperfusion partly via inhibition
TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 41:22–32.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ma SR and Xie XW: NLRC5 deficiency
promotes myocardial damage induced by high fat diet in mice through
activating TLR4/NF-κB. Biomed Pharmacother. 91:755–766.
2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Silswal N, Singh AK, Aruna B, Mukhopadhyay
S, Ghosh S and Ehtesham NZ: Human resistin stimulates the
pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by
NF-kappaB-dependent pathway. Biochem Biophys Res Commun.
334:1092–1101. 2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tarkowski A, Bjersing J, Shestakov A and
Bokarewa MI: Resistin competes with lipopolysaccharide for binding
to toll-like receptor 4. J Cell Mol Med. 14:1419–1431.
2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kang YJ: Cardiac hypertrophy: A risk
factor for QT-prolongation and cardiac sudden death. Toxicol
Pathol. 34:58–66. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Han J, Ye S, Zou C, Chen T, Wang J, Li J,
Jiang L, Xu J, Huang W, Wang Y and Liang G: Angiotensin II causes
biphasic STAT3 activation through TLR4 to initiate cardiac
remodeling. Hypertension. 72:1301–1311. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ehrentraut H, Weber C, Ehrentraut S,
Schwederski M, Boehm O, Knuefermann P, Meyer R and Baumgarten G:
The toll-like receptor 4-antagonist eritoran reduces murine cardiac
hypertrophy. Eur J Heart Fail. 13:602–610. 2011.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Smeets PJ, Teunissen BE, Planavila A, de
Vogel-van den Bosch H, Willemsen PH, van der Vusse GJ and van
Bilsen M: Inflammatory pathways are activated during cardiomyocyte
hypertrophy and attenuated by peroxisome proliferator-activated
receptors PPARalpha and PPARdelta. J Biol Chem. 283:29109–29118.
2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gupta S, Young D, Maitra RK, Gupta A,
Popovic ZB, Yong SL, Mahajan A, Wang Q and Sen S: Prevention of
cardiac hypertrophy and heart failure by silencing of NF-kappaB. J
Mol Biol. 375:637–649. 2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tan BK, Adya R and Randeva HS: Omentin: A
novel link between inflammation, diabesity, and cardiovascular
disease. Trends Cardiovasc Med. 20:143–148. 2010.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Rao SS, Hu Y, Xie PL, Cao J, Wang ZX, Liu
JH, Yin H, Huang J, Tan YJ, Luo J, et al: Omentin-1 prevents
inflammation-induced osteoporosis by downregulating the
pro-inflammatory cytokines. Bone Res. 6(9)2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yoo HJ, Hwang SY, Hong HC, Choi HY, Yang
SJ, Seo JA, Kim SG, Kim NH, Choi KM, Choi DS and Baik SH:
Association of circulating omentin-1 level with arterial stiffness
and carotid plaque in type 2 diabetes. Cardiovasc Diabetol.
10(103)2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Genre F, Rueda-Gotor J, Remuzgo-Martínez
S, Pulito-Cueto V, Corrales A, Mijares V, Lera-Gómez L, Portilla V,
Expósito R, Mata C, et al: Omentin: A biomarker of cardiovascular
risk in individuals with axial spondyloarthritis. Sci Rep.
10(9636)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhou JP, Tong XY, Zhu LP, Luo JM, Luo Y,
Bai YP, Li CC and Zhang GG: Plasma omentin-1 level as a predictor
of good coronary collateral circulation. J Atheroscler Thromb.
24:940–948. 2017.PubMed/NCBI View Article : Google Scholar
|