Congenital heart disease (CHD), as a collective
diagnosis for structural malformations of the heart and valves, as
well as the endothoracic great blood vessels, occurring during
embryonic development, represents the most common birth deformity
in humans, with a prevalence of ~1% among live births worldwide
(1,2). When minor cardiac structural
anomalies are included, such as aneurysm of the atrial septum and
bicuspid aortic valve (BAV), which is the most prevalent congenital
cardiovascular anomaly with an incidence of 1-2% in the population,
the overall prevalence of CHD may be as high as ~5% (2). Based on the occurrence of cardiac
lesions in certain locations, CHD is clinically classified into
>20 distinct subtypes, including ventricular septal defect
(VSD), patent ductus arteriosus (PDA), transposition of the great
arteries (TGA), double outlet of the right ventricle (DORV),
tricuspid valve atresia (TVA), atrial septal defect (ASD),
endocardial cushion defect, aortic stenosis, a right aortic arch, a
single ventricle, tetralogy of Fallot, hypoplastic left heart and
hypoplastic right heart (3-6).
Irrespective of minor CHD that may resolve spontaneously (3), major CHD may contribute to diminished
health-associated quality of life (7-9),
decreased exercise performance (10-14),
delayed neurodevelopment and brain injury (15-18),
ischemic or hemorrhagic cerebral stroke (19-21),
pulmonary arterial hypertension or Eisenmenger syndrome (22-24),
viral pneumonia (25-27),
infective endocarditis (28-30),
acute myocardial infarction (31,32),
chronic congestive heart failure (33-35),
ventricular or supraventricular arrhythmia (36-38)
and death (39-42).
Notably, CHD remains the most frequent etiology of newborn deaths
caused by birth defects, with 21.8% of the neonates who succumb to
various birth malformations having a cardiovascular abnormality
(43). Although tremendous
improvement in the outcomes of cardiac surgery and perioperative
intensive care has been achieved, which allows >90% of
CHD-infants to survive into adulthood and reach fertile age, this
results in an increasing adult population with CHD, and now the
rising number of adult patients with CHD accounts for more than
two-thirds of the overall CHD population (44). Moreover, the rates of late surgical
complication and cardiac comorbidity, and even mortality, markedly
increase in adults affected with CHD (45-47).
Despite the clinical importance, the etiologies underpinning CHD in
a considerable proportion of cases remain obscure.
In vertebrates, the heart is the first functional
organ that develops during embryonic morphogenesis, and cardiac
organogenesis undergoes an extremely complex biological process,
which is precisely mediated by a sophisticated regulatory network,
involving transcription factors, cardiac structural proteins,
signaling transducers, epigenetic modifiers and microRNAs (48). Previous studies have demonstrated
that both environmental risk factors and genetic defects may
interfere with this finely controlled developmental process, giving
rise to CHD (1,2,48-54).
The well-recognized non-inheritable pathogenic factors for CHD
include maternal viral infection, nutritional deficiency, obesity,
diabetes and decreased physical activity, as well as exposure to
toxic chemicals, therapeutic drugs and ionizing radiation during
early pregnancy (48-51).
However, accumulating evidence highlights the strong genetic basis
underpinning CHD (1,2,52-54).
Significant familial aggregation of CHD has been reported, with the
risk of CHD recurrence in the first-degree offspring of an affected
parent being between 3 and 19% depending on the distinct types of
lesion (55). In addition to
chromosomal alterations encompassing aneuploidies, microdeletions
and microduplications, pathogenetic variations in >100 genes
amply expressed in the developing heart, encompassing those
encoding sarcomeric proteins, transcription factors, chromatin
modifies and signal-transducing molecules, have been determined to
contribute to CHD (1,2,52,53,56-83).
Of these reported CHD-causative genes, the majority code for
cardiac core transcription factors, such as T-box transcription
factor (TBX)1, TBX20, TBX5, NK2 homeobox 5, GATA binding protein
(GATA)6, GATA4, GATA5, heart and neural crest derivatives expressed
(HAND)1 and HAND2(84). However,
the genetic components underpinning CHD in most cases are still
unknown.
The present study participants comprised a new
cohort of 316 index patients affected with different forms of CHD
enrolled from the Chinese Han population between March 2018 and
November 2020 at Tongji Hospital and East Hospital, Tongji
University (Shanghai, China). Clinical diagnosis and classification
of various types of CHD were made as described previously (3,67).
The relatives of the probands were also recruited when available.
Patients with known syndromic CHD or chromosomal anomaly were ruled
out from this research. Patients were diagnosed with syndromic CHD
if they manifested a distinct facial gestalt or had at least one
reported extra-cardiac malformation (85). A total of 400 unrelated volunteers
without CHD were enrolled as control individuals from the same
geographic area, who were exactly matched with the cases for sex
and ethnicity, as well as age. All research participants underwent
a comprehensive clinical assessment, as described previously
(67-69).
This research was fulfilled in compliance with the tenets of the
Declaration of Helsinki. The protocol applied to the current
investigation was approved by the Medical Ethics Committee of
Tongji Hospital, Tongji University School of Medicine [Shanghai,
China; approval no. LL(H)-09-07]. Written informed consent was
provided by the research participants or their parents prior to
commencement of sample collection.
A whole blood specimen was collected from each study
participant in an EDTA-coated tube and stored in a refrigerator at
-80˚C. Genomic DNA was purified from blood leucocytes by utilizing
DNA extraction reagent (Promega Corporation). The entire coding
region, as well as splicing boundaries, of the KLF13 gene
(NC_000015.10) were amplified via polymerase chain reaction (PCR)
using a DNA polymerase kit (Qiagen GmbH) and the
KLF13-specific oligonucleotide primers, as described
previously (67): Forward
5'-CCATGCGCTCACTCTTCGGT-3' and reverse, 5'-CCTTTGTCTGAGGCCGGGCT-3'
for the first party of coding exon 1 (product size, 670 bp);
forward, 5'-CGGACCTCAACCAGCAAGCG-3' and reverse,
5'-CTCCGAGAGCCAAGACCCGC-3' for the second party of coding exon 1
(product size, 569 bp); and forward, 5'-GCATGTGGGAGGGGTGTTGA-3' and
reverse, 5'-TCGTGAAACGTGTCCATCCCT-3' for coding exon 2 (product
size, 675 bp). Each mixture used for PCR was prepared in a 0.2-ml
PCR tube with a final volume of 25 µl, containing 1X Buffer (Qiagen
GmbH), 1X Q solution, a component of the HotStar Taq DNA Polymerase
kit facilitating amplification of templates with a high-degree
secondary structure or with a rich GC content by modifying the
melting behavior of DNA (Qiagen GmbH), 0.2 mM dNTPs (Qiagen GmbH),
0.5 µM forward primer, 0.5 µM reverse primer, 0.02 U/µl HotStar Taq
DNA Polymerase (Qiagen GmbH) and 0.1 µg genomic DNA. PCR was
fulfilled on a 96-well thermocycler (Bio-Rad Laboratories, Inc.).
The thermocycling conditions set for the PCR were as follows:
Initial denaturation at 95˚C for 15 min, followed by 36 cycles of
denaturation at 94˚C for 30 sec, annealing at 62˚C for 30 sec and
extension at 72˚C for 1 min, with a final extension at 72˚C for 7
min. PCR products were resolved by 1.5% agarose gel electrophoresis
and visualized after ethidium bromide staining of gels.
PCR-sequencing of extracted amplicons was conducted as described
previously (69). For a validated
KLF13 variation, the Human Gene Mutation Database (HGMD;
http://www.hgmd.cf.ac.uk/ac/index.php), Single
Nucleotide Polymorphism (SNP) datbase (https://www.ncbi.nlm.nih.gov/snp) and the Genome
Aggregation Database (gnomAD; https://gnomad.broadinstitute.org) were retrieved to
verify its novelty.
The wild-type KLF13-pcDNA3.1 plasmid (Invitrogen;
Thermo Fisher Scientific, Inc.) was constructed as described
previously (67). The mutation
discovered in the current study was introduced into wild-type
KLF13-pcDNA3.1 by site-targeted mutagenesis with a site-targeted
mutagenesis kit (Stratagene; Agilent Technologies Inc.) with the
following primers: Forward, 5'-CCCGCGGGGAGCGGCTAGCCCGGCCTCAGAC-3'
and reverse, 5'-GTCTGAGGCCGGGCTAGCCGCTCCCCGCGGG-3'. The mutant-type
KLF13-pcDNA3.1 was selected by DpnI (Takara Biotechnology Co.,
Ltd.) and was confirmed by sequencing analysis. The TBX5-pcDNA3.1
plasmid (Invitrogen; Thermo Fisher Scientific, Inc.) and the
reporter plasmid of human natriuretic peptide precursor
A-luciferase (NPPA-luc), which expresses firefly luciferase, have
been described previously (68).
The reporter plasmid of human vascular endothelial growth factor A
(VEGFA)-luc, which expresses firefly luciferase, was generated as
previously described (86).
NIH3T3 cells (Cell Bank of Type Culture Collection
of the Chinese Academy of Sciences) were seeded onto a 24-well
plate, and maintained in DMEM (Merck KGaA) containing 10% fetal
bovine serum and 1% penicillin/streptomycin (both Thermo Fisher
Scientific, Inc.), in an atmosphere of 5% CO2 at 37˚C.
NIH3T3 cells were transfected 24 h after plating with various
expression plasmids, including empty pcDNA3.1, wild-type
KLF13-pcDNA3.1 (KLF13), Glu144*-mutant KLF13-pcDNA3.1 (Glu144*),
wild-type TBX5-pcDNA3.1 (TBX5), NPPA-luc and VEGFA-luc, utilizing
the Lipofectamine® 3000 Transfection Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) as described
previously (67). The internal
control plasmid pGL4.75 (Promega Corporation), which expresses
Renilla luciferase, was co-transfected to balance
transfection efficiency. The empty pcDNA3.1 plasmid (Invitrogen;
Thermo Fisher Scientific, Inc.) was used as a negative control.
Cells were collected at 48 h post-transfection, and lysed in 0.2 ml
Reporter Lysis Buffer (Promega Corporation). The cellular lysates
were used to measure the luciferase activities of firefly and
Renilla on a luminometer (Promega Corporation), using a
dual-luciferase assay kit (Promega Corporation). The activity of
the target gene promoter was expressed as fold activation of
firefly luciferase to Renilla luciferase. For each
expression plasmid, three transfection experiments were performed
in triplicate.
Data for promoter activity are presented as the mean
± standard deviation of the original results from three
transfection experiments. Differences in promoter activities
between two groups were compared with unpaired Student's t-test.
When comparisons among multiple groups were made, one-way ANOVA
with a Tukey-Kramer HSD post-hoc test was used. A two-sided
P-value of <0.05 was used to indicate a statistically
significant difference. The statistical software used for the
analysis was SPSS version 17.0 for Windows (SPSS, Inc.).
In this investigation, a cohort of 316 unrelated
index patients suffering from various types of CHD (168 male cases
and 148 female cases, with ages ranging from 1-49 years and a mean
age of 21±9 years) was clinically investigated in contrast to a
total of 400 unrelated individuals without CHD (212 male
individuals and 188 female individuals, with ages ranging from 1-49
years and a mean age of 21±8 years). All the included patients with
CHD had echocardiographic evidence, whereas the echocardiograms of
the enrolled control subjects were normal, without evidence of
cardiovascular structural abnormalities. Among the 316 unrelated
index patients with CHD, 58 index patients reported a positive
family history of CHD, while all 400 control subjects lacked a
family history of CHD. No research participants had known
environmental risk factors predisposing them to CHD, including
maternal viral infection, nutritional deficiency, obesity, diabetes
or exposure to toxic chemicals, therapeutic drugs and ionizing
radiation during early pregnancy. Most of the patients underwent
cardiac catheterization or surgery. The clinical features of the
316 index cases with CHD are summarized in Table I.
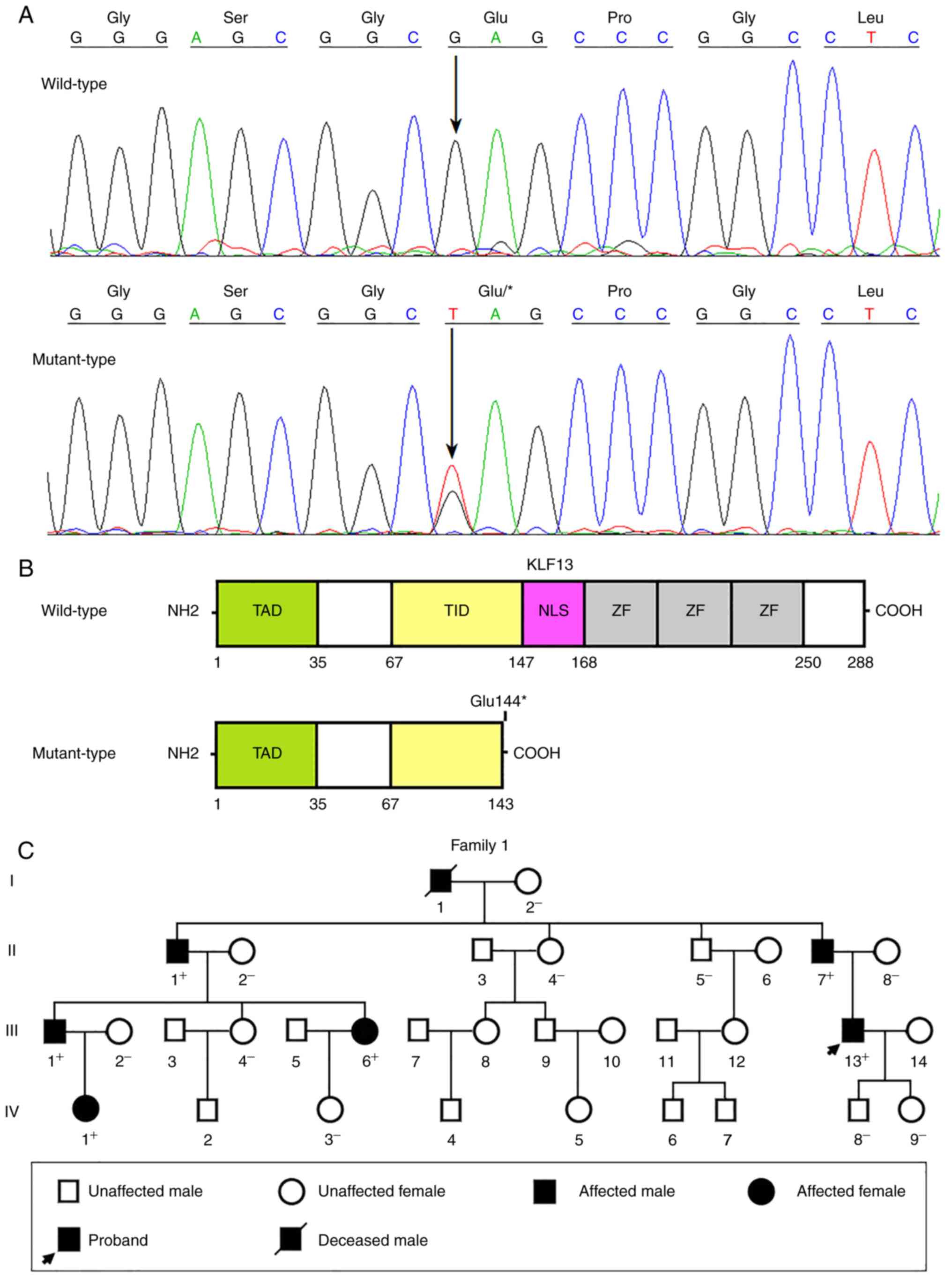
Via direct sequencing analysis of the entire coding
region and splicing donors/acceptors of KLF13 in 316 index
patients with diverse forms of CHD, a heterozygous non-synonymous
mutation, NM_015995.3: c.430G>T; p.(Glu144*), was detected in
one index patient inflicted with CHD, comprising PDA and VSD, as
well as BAV; this mutation therefore has a prevalence of ~0.32%
(1/316) in the study CHD population. The mutation carrier possessed
a CHD-positive family history, and genetic assay of the available
family members unveiled that the nonsense mutation was in
co-segregation with autosomal-dominant CHD in the whole family,
with complete penetrance for PDA, as well as BAV, and incomplete
penetrance for VSD. The nonsense mutation was neither detected in
400 control subjects nor found in the HGMD, SNP and gnomAD
databases, indicating its novelty. The sequence electropherogram
traces illustrating the KLF13 mutation in the heterozygous
status as well as its wild-type sequence in the homozygous status
are presented in Fig. 1A. The
schemas exhibiting the functional domains of both wild-type KLF13
and mutant KLF13 are provided in Fig.
1B. The pedigree structure of the studied family inflicted with
CHD is displayed in Fig. 1C. The
clinical characteristic information of the CHD-affected family
members is shown in Table II.
In the present study, a new KLF13 mutation,
NM_015995.3: c.430G>T; p.(Glu144*), was identified in one family
suffering from PDA, BAV and VSD. The nonsense heterozygous
mutation, which co-segregated with CHD in the whole family, was
neither observed in 800 control chromosomes nor found in the
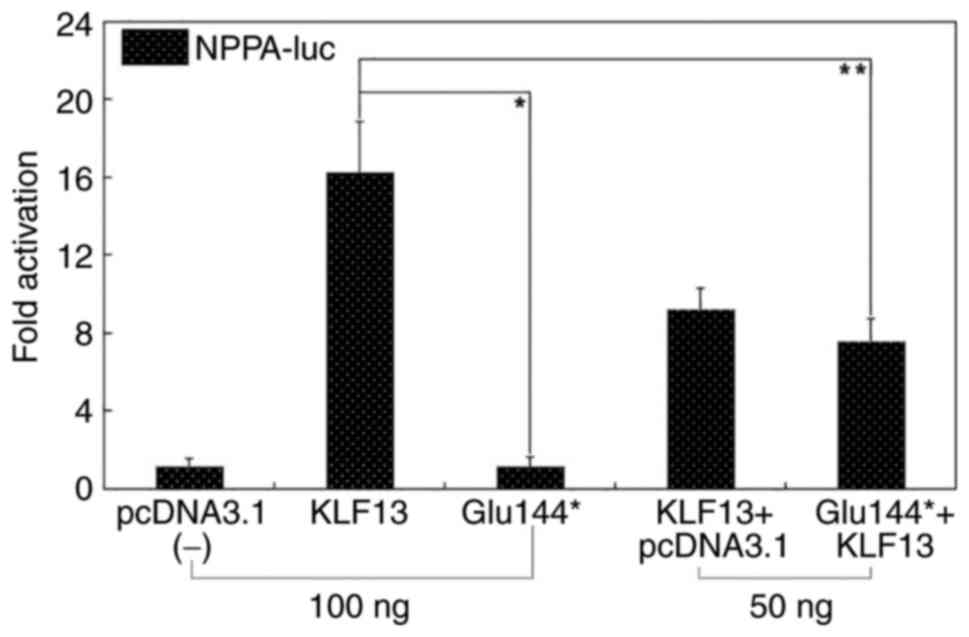
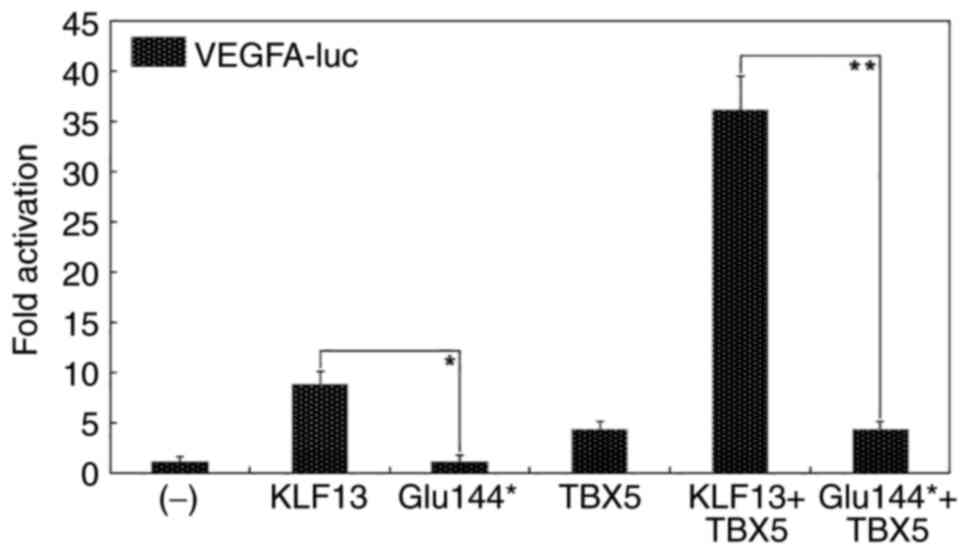
databases of HGMD, SNP and gnomAD. Functional investigations
demonstrated that the Glu144*-mutant KLF13 failed to
transcriptionally activate the promoters of NPPA and
VEGFA. Additionally, the mutation abrogated the synergistic
transcriptional activation between KLF13 and TBX5, a
well-established CHD-causative gene (68,69,84).
These findings support the fact that genetically defective
KLF13 confers an enhanced susceptibility to CHD, including
PDA, BAV and VSD. Notably, the experiments performed in one NIH3T3
cell line only failed to control for cell-dependent effects, and it
remains possible, in fact likely, that other cells may yield
different results. Hence, it is very important that additional
cells lines are used to examine the functional effect of
Glu144*-mutant KLF13 to generalize the mechanism proposed on the
basis of this work.
Not applicable.
Funding: This study received financial support from the Basic
Research Project of Shanghai, China (grant no. 20JC1418800), the
Natural Science Foundation of Shanghai, China (grant no.
18ZR1431000) and the Natural Science Foundation of Minhang
District, Shanghai, China (grant no. 2020MHZ083).
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
JW and YQY designed the investigation, and made
leading contributions to the writing of manuscript. PA, GFZ, CMZ,
YJX, JW and YQY completed the clinical investigations. PA, GFZ, JW
and YQY fulfilled the genetic and biochemical studies. JW and YQY
confirm the authenticity of all the raw data. All authors have read
and approved the final version of manuscript.
This investigation was approved by the Medical
Ethics Committee of Tongji Hospital, Tongji University School of
Medicine [Shanghai, China; approval no. LL(H)-09-07]. Written
informed consent was collected from the study individuals or their
parents prior to investigation.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Diab NS, Barish S, Dong W, Zhao S,
Allington G, Yu X, Kahle KT, Brueckner M and Jin SC: Molecular
genetics and complex inheritance of congenital heart disease. Genes
(Basel). 12(1020)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Martin LJ and Benson DW: Focused
strategies for defining the genetic architecture of congenital
heart defects. Genes (Basel). 12(827)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Benjamin EJ, Muntner P, Alonso A,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Das SR, et al: Heart disease and stroke statistics-2019
update: A report from the American Heart Association. Circulation.
139:e56–e528. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Skeffington KL, Bond AR, Bigotti MG,
AbdulGhani S, Iacobazzi D, Kang SL, Heesom KJ, Wilson MC, Stoica S,
Martin R, et al: Changes in inflammation and oxidative stress
signalling pathways in coarcted aorta triggered by bicuspid aortic
valve and growth in young children. Exp Ther Med.
20(48)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dragomir C, Manea AM, Enatescu VR,
Lacatusu AAM, Lacatusu A, Henry OI, Boia M and Ilie C: Left heart
hypoplasia operated using double pulmonary arterial banding with
double arterial duct stenting: A case report. Exp Ther Med.
20(193)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hu C, Huang S, Wu F and Ding H:
MicroRNA-219-5p participates in cyanotic congenital heart disease
progression by regulating cardiomyocyte apoptosis. Exp Ther Med.
21(36)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Andonian CS, Freilinger S, Achenbach S,
Ewert P, Gundlach U, Hoerer J, Kaemmerer H, Pieper L, Weyand M,
Neidenbach RC, et al: ‘Well-being paradox’ revisited: A
cross-sectional study of quality of life in over 4000 adults with
congenital heart disease. BMJ Open. 11(e049531)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Brudy L, Meyer M, Oberhoffer R, Ewert P
and Müller J: Move more-be happier? physical activity and
health-related quality of life in children with congenital heart
disease. Am Heart J. 241:68–73. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Moons P, Luyckx K, Thomet C, Budts W,
Enomoto J, Sluman MA, Lu CW, Jackson JL, Khairy P, Cook SC, et al:
Physical functioning, mental health, and quality of life in
different congenital heart defects: Comparative analysis in 3538
patients from 15 countries. Can J Cardiol. 37:215–223.
2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hayama Y, Ohuchi H, Negishi J, Iwasa T,
Sakaguchi H, Miyazaki A, Tsuda E and Kurosaki K: Effect of
stiffened and dilated ascending aorta on aerobic exercise capacity
in repaired patients with complex congenital heart disease. Am J
Cardiol. 129:87–94. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Spiesshoefer J, Orwat S, Henke C, Kabitz
HJ, Katsianos S, Borrelli C, Baumgartner H, Nofer JR, Spieker M,
Bengel P, et al: Inspiratory muscle dysfunction and restrictive
lung function impairment in congenital heart disease: Association
with immune inflammatory response and exercise intolerance. Int J
Cardiol. 318:45–51. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Meyer M, Brudy L, García-Cuenllas L, Hager
A, Ewert P, Oberhoffer R and Müller J: Current state of home-based
exercise interventions in patients with congenital heart disease: A
systematic review. Heart. 106:333–341. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xu C, Su X, Ma S, Shu Y, Zhang Y, Hu Y and
Mo X: Effects of exercise training in postoperative patients with
congenital heart disease: A systematic review and meta-analysis of
randomized controlled trials. J Am Heart Assoc.
9(e013516)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Meyer M, Brudy L, Fuertes-Moure A, Hager
A, Oberhoffer-Fritz R, Ewert P and Müller J: E-health exercise
intervention for pediatric patients with congenital heart disease:
A randomized controlled trial. J Pediatr. 233:163–168.
2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Asschenfeldt B, Evald L, Heiberg J, Salvig
C, Østergaard L, Dalby RB, Eskildsen SF and Hjortdal VE:
Neuropsychological status and structural brain imaging in adults
with simple congenital heart defects closed in childhood. J Am
Heart Assoc. 9(e015843)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kessler N, Feldmann M, Schlosser L,
Rometsch S, Brugger P, Kottke R, Knirsch W, Oxenius A, Greutmann M
and Latal B: Structural brain abnormalities in adults with
congenital heart disease: Prevalence and association with estimated
intelligence quotient. Int J Cardiol. 306:61–66. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bonthrone AF, Dimitrova R, Chew A, Kelly
CJ, Cordero-Grande L, Carney O, Egloff A, Hughes E, Vecchiato K,
Simpson J, et al: Individualized brain development and cognitive
outcome in infants with congenital heart disease. Brain Commun.
3(fcab046)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gui J, Liang S, Sun Y, Liu Y, Chen C, Wang
B, Zhong J, Yu Y and He S: Effect of perioperative
amplitude-integrated electroencephalography on neurodevelopmental
outcomes following infant heart surgery. Exp Ther Med.
20:2879–2887. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Giang KW, Mandalenakis Z, Dellborg M,
Lappas G, Eriksson P, Hansson PO and Rosengren A: Long-term risk of
hemorrhagic stroke in young patients with congenital heart disease.
Stroke. 49:1155–1162. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Giang KW, Fedchenko M, Dellborg M,
Eriksson P and Mandalenakis Z: Burden of ischemic stroke in
patients with congenital heart disease: a nationwide, case-control
study. J Am Heart Assoc. 10(e020939)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Freisinger E, Gerß J, Makowski L,
Marschall U, Reinecke H, Baumgartner H, Koeppe J and Diller GP:
Current use and safety of novel oral anticoagulants in adults with
congenital heart disease: Results of a nationwide analysis
including more than 44 000 patients. Eur Heart J. 41:4168–4177.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Diller GP, Körten MA, Bauer UM, Miera O,
Tutarel O, Kaemmerer H, Berger F and Baumgartner H: German
Competence Network for Congenital Heart Defects Investigators.
Current therapy and outcome of Eisenmenger syndrome: Data of the
German National Register for congenital heart defects. Eur Heart J.
37:1449–1455. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kaemmerer H, Gorenflo M, Huscher D,
Pittrow D, Apitz C, Baumgartner H, Berger F, Bruch L, Brunnemer E,
Budts W, et al: Pulmonary hypertension in adults with congenital
heart disease: Real-world data from the International COMPERA-CHD
Registry. J Clin Med. 9(1456)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Long L, Xiao Y, Yin X, Gao S, Zhou L and
Liu H: Expression of serum miR-27b and miR-451 in patients with
congenital heart disease associated pulmonary artery hypertension
and risk factor analysis. Exp Ther Med. 20:3196–3202.
2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Diller GP, Enders D, Lammers AE, Orwat S,
Schmidt R, Radke RM, Gerss J, De Torres Alba F, Kaleschke G, Bauer
UM, et al: Mortality and morbidity in patients with congenital
heart disease hospitalised for viral pneumonia. Heart.
107:1069–1076. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Radke RM, Frenzel T, Baumgartner H and
Diller GP: Adult congenital heart disease and the COVID-19
pandemic. Heart. 106:1302–1309. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Diller GP, Gatzoulis MA, Broberg CS,
Aboulhosn J, Brida M, Schwerzmann M, Chessa M, Kovacs AH and
Roos-Hesselink J: Coronavirus disease 2019 in adults with
congenital heart disease: A position paper from the ESC working
group of adult congenital heart disease, and the International
Society for Adult Congenital Heart Disease. Eur Heart J.
42:1858–1865. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Diller GP and Baumgartner H: Endocarditis
in adults with congenital heart disease: New answers-new questions.
Eur Heart J. 38:2057–2059. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tutarel O, Alonso-Gonzalez R, Montanaro C,
Schiff R, Uribarri A, Kempny A, Grübler MR, Uebing A, Swan L,
Diller GP, et al: Infective endocarditis in adults with congenital
heart disease remains a lethal disease. Heart. 104:161–165.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cahill TJ, Jewell PD, Denne L, Franklin
RC, Frigiola A, Orchard E and Prendergast BD: Contemporary
epidemiology of infective endocarditis in patients with congenital
heart disease: A UK prospective study. Am Heart J. 215:70–77.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Fedchenko M, Mandalenakis Z, Giang KW,
Rosengren A, Eriksson P and Dellborg M: Long-term outcomes after
myocardial infarction in middle-aged and older patients with
congenital heart disease-a nationwide study. Eur Heart J.
42:2577–2586. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Orwat S and Diller GP: Congenital heart
defects as an intrinsic additional risk factor for the occurrence
and outcome of myocardial infarction. Eur Heart J. 42:2587–2589.
2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hirono K, Hata Y, Miyao N, Okabe M,
Takarada S, Nakaoka H, Ibuki K, Ozawa S, Yoshimura N, Nishida N, et
al: Left ventricular noncompaction and congenital heart disease
increases the risk of congestive heart failure. J Clin Med.
9(785)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Menachem JN, Schlendorf KH, Mazurek JA,
Bichell DP, Brinkley DM, Frischhertz BP, Mettler BA, Shah AS,
Zalawadiya S, Book W, et al: Advanced heart failure in adults with
congenital heart disease. JACC Heart Fail. 8:87–99. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Arnaert S, De Meester P, Troost E, Droogne
W, Van Aelst L, Van Cleemput J, Voros G, Gewillig M, Cools B, Moons
P, et al: Heart failure related to adult congenital heart disease:
Prevalence, outcome and risk factors. ESC Heart Fail. 8:2940–2950.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sakhi R, Kauling RM, Theuns DA,
Szili-Torok T, Bhagwandien RE, van den Bosch AE, Cuypers JAAE,
Roos-Hesselink JW and Yap SC: Early detection of ventricular
arrhythmias in adults with congenital heart disease using an
insertable cardiac monitor (EDVA-CHD study). Int J Cardiol.
305:63–69. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Casteigt B, Samuel M, Laplante L, Shohoudi
A, Apers S, Kovacs AH, Luyckx K, Thomet C, Budts W, Enomoto J, et
al: Atrial arrhythmias and patient-reported outcomes in adults with
congenital heart disease: An international study. Heart Rhythm.
18:793–800. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wasmer K, Eckardt L, Baumgartner H and
Köbe J: Therapy of supraventricular and ventricular arrhythmias in
adults with congenital heart disease-narrative review. Cardiovasc
Diagn Ther. 11:550–562. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Goldstein SA, D'Ottavio A, Spears T,
Chiswell K, Hartman RJ, Krasuski RA, Kemper AR, Meyer RE, Hoffman
TM, Walsh MJ, et al: Causes of death and cardiovascular
comorbidities in adults with congenital heart disease. J Am Heart
Assoc. 9(e016400)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Oliver JM, Gallego P, Gonzalez AE, Avila
P, Alonso A, Garcia-Hamilton D, Peinado R, Dos-Subirà L,
Pijuan-Domenech A, Rueda J, et al: Predicting sudden cardiac death
in adults with congenital heart disease. Heart. 107:67–75.
2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Vehmeijer JT, Koyak Z, Leerink JM,
Zwinderman AH, Harris L, Peinado R, Oechslin EN, Robbers-Visser D,
Groenink M, Boekholdt SM, et al: Identification of patients at risk
of sudden cardiac death in congenital heart disease: the
PRospEctiVE study on implaNTable cardIOverter defibrillator therapy
and suddeN cardiac death in Adults with Congenital Heart Disease
(PREVENTION-ACHD). Heart Rhythm. 18:785–792. 2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Williams JL, Torok RD, D'Ottavio A, Spears
T, Chiswell K, Forestieri NE, Sang CJ, Paolillo JA, Walsh MJ,
Hoffman TM, et al: Causes of death in infants and children with
congenital heart disease. Pediatr Cardiol. 42:1308–1315.
2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Virani SS, Alonso A, Aparicio HJ, Benjamin
EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng
S, Delling FN, et al: Heart disease and stroke statistics-2021
update: A report from the American Heart Association. Circulation.
143:e254–e743. 2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Diller GP, Arvanitaki A, Opotowsky AR,
Jenkins K, Moons P, Kempny A, Tandon A, Redington A, Khairy P,
Mital S, et al: Lifespan perspective on congenital heart disease
research: JACC state-of-the-art review. J Am Coll Cardiol.
77:2219–2235. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bouma BJ and Mulder BJ: Changing landscape
of congenital heart disease. Circ Res. 120:908–922. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Spector LG, Menk JS, Knight JH, McCracken
C, Thomas AS, Vinocur JM, Oster ME, St Louis JD, Moller JH and
Kochilas L: Trends in long-term mortality after congenital heart
surgery. J Am Coll Cardiol. 71:2434–2446. 2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Niwa K, Kaemmerer H and von Kodolitsch Y:
Current diagnosis and management of late complications in adult
congenital heart disease. Cardiovasc Diagn Ther. 11:478–480.
2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kalisch-Smith JI, Ved N and Sparrow DB:
Environmental risk factors for congenital heart disease. Cold
Spring Harb Perspect Biol. 12(a037234)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zhou J, Xiong Y, Dong X, Wang H, Qian Y,
Ma D and Li X: Genome-wide methylation analysis reveals
differentially methylated CpG sites and altered expression of heart
development-associated genes in fetuses with cardiac defects. Exp
Ther Med. 22(1032)2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Bigras JL: Cardiovascular risk factors in
patients with congenital heart disease. Can J Cardiol.
36:1458–1466. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Helle E and Priest JR: Maternal obesity
and diabetes mellitus as risk factors for congenital heart disease
in the offspring. J Am Heart Assoc. 9(e011541)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Saliba A, Figueiredo AC, Baroneza JE,
Afiune JY, Pic-Taylor A, Oliveira SF and Mazzeu JF: Genetic and
genomics in congenital heart disease: A clinical review. J Pediatr
(Rio J). 96:279–288. 2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Shabana NA, Shahid SU and Irfan U: Genetic
contribution to congenital heart disease (CHD). Pediatr Cardiol.
41:12–23. 2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Majumdar U, Yasuhara J and Garg V: In vivo
and in vitro genetic models of congenital heart disease. Cold
Spring Harb Perspect Biol. 13(a036764)2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Loffredo CA, Chokkalingam A, Sill AM,
Boughman JA, Clark EB, Scheel J and Brenner JI: Prevalence of
congenital cardiovascular malformations among relatives of infants
with hypoplastic left heart, coarctation of the aorta, and
d-transposition of the great arteries. Am J Med Genet A.
124A:225–230. 2004.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Arya P, Wilson TE, Parent JJ, Ware SM,
Breman AM and Helm BM: An adult female with 5q34-q35.2 deletion: A
rare syndromic presentation of left ventricular non-compaction and
congenital heart disease. Eur J Med Genet.
63(103797)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Evangelidou P, Kousoulidou L, Salameh N,
Alexandrou A, Papaevripidou I, Alexandrou IM, Ketoni A, Ioannidou
C, Christophidou-Anastasiadou V, Tanteles GA, et al: An unusual
combination of an atypical maternally inherited novel 0.3 Mb
deletion in Williams-Beuren region and a de novo 22q11.21
microduplication in an infant with supravalvular aortic stenosis.
Eur J Med Genet. 63(104084)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Szot JO, Campagnolo C, Cao Y, Iyer KR,
Cuny H, Drysdale T, Flores-Daboub JA, Bi W, Westerfield L, Liu P,
et al: Bi-allelic mutations in NADSYN1 cause multiple organ defects
and expand the genotypic spectrum of congenital NAD deficiency
disorders. Am J Hum Genet. 106:129–136. 2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chen CA, Crutcher E, Gill H, Nelson TN,
Robak LA, Jongmans MC, Pfundt R, Prasad C, Berard RA, Fannemel M,
et al: The expanding clinical phenotype of germline ABL1-associated
congenital heart defects and skeletal malformations syndrome. Hum
Mutat. 41:1738–1744. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hsieh A, Morton SU, Willcox JAL, Gorham
JM, Tai AC, Qi H, DePalma S, McKean D, Griffin E, Manheimer KB, et
al: EM-mosaic detects mosaic point mutations that contribute to
congenital heart disease. Genome Med. 12(42)2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kolomenski JE, Delea M, Simonetti L,
Fabbro MC, Espeche LD, Taboas M, Nadra AD, Bruque CD and Dain L: An
update on genetic variants of the NKX2-5. Hum Mutat. 41:1187–1208.
2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Liu H, Giguet-Valard AG, Simonet T,
Szenker-Ravi E, Lambert L, Vincent-Delorme C, Scheidecker S, Fradin
M, Morice-Picard F, Naudion S, et al: Next-generation sequencing in
a series of 80 fetuses with complex cardiac malformations and/or
heterotaxy. Hum Mutat. 41:2167–2178. 2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lin JI, Feinstein TN, Jha A, McCleary JT,
Xu J, Arrigo AB, Rong G, Maclay LM, Ridge T, Xu X, et al: Mutation
of LRP1 in cardiac neural crest cells causes congenital heart
defects by perturbing outflow lengthening. Commun Biol.
3(312)2020.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Sutani A, Shima H, Hijikata A, Hosokawa S,
Katoh-Fukui Y, Takasawa K, Suzuki E, Doi S, Shirai T, Morio T, et
al: WDR11 is another causative gene for coloboma, cardiac anomaly
and growth retardation in 10q26 deletion syndrome. Eur J Med Genet.
63(103626)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Le Fevre A, Baptista J, Ellard S, Overton
T, Oliver A, Gradhand E and Scurr I: Compound heterozygous Pkd1l1
variants in a family with two fetuses affected by heterotaxy and
complex Chd. Eur J Med Genet. 63(103657)2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Li W, Li B, Li T, Zhang E, Wang Q, Chen S
and Sun K: Identification and analysis of KLF13 variants in
patients with congenital heart disease. BMC Med Genet.
21(78)2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wang SS, Wang TM, Qiao XH, Huang RT, Xue
S, Dong BB, Xu YJ, Liu XY and Yang YQ: KLF13 loss-of-function
variation contributes to familial congenital heart defects. Eur Rev
Med Pharmacol Sci. 24:11273–11285. 2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Zhang Y, Sun YM, Xu YJ, Zhao CM, Yuan F,
Guo XJ, Guo YH, Yang CX, Gu JN, Qiao Q, et al: A new TBX5
loss-of-function mutation contributes to congenital heart defect
and atrioventricular block. Int Heart J. 61:761–768.
2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Jiang WF, Xu YJ, Zhao CM, Wang XH, Qiu XB,
Liu X, Wu SH and Yang YQ: A novel TBX5 mutation predisposes to
familial cardiac septal defects and atrial fibrillation as well as
bicuspid aortic valve. Genet Mol Biol. 43(e20200142)2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Wang C, Lv H, Ling X, Li H, Diao F, Dai J,
Du J, Chen T, Xi Q, Zhao Y, et al: Association of assisted
reproductive technology, germline de novo mutations and congenital
heart defects in a prospective birth cohort study. Cell Res.
31:919–928. 2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lahrouchi N, Postma AV, Salazar CM, De
Laughter DM, Tjong F, Piherová L, Bowling FZ, Zimmerman D, Lodder
EM, Ta-Shma A, et al: Biallelic loss-of-function variants in PLD1
cause congenital right-sided cardiac valve defects and neonatal
cardiomyopathy. J Clin Invest. 131(e142148)2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Audain E, Wilsdon A, Breckpot J,
Izarzugaza JM, Fitzgerald TW, Kahlert AK, Sifrim A, Wünnemann F,
Perez-Riverol Y, Abdul-Khaliq H, et al: Integrative analysis of
genomic variants reveals new associations of candidate
haploinsufficient genes with congenital heart disease. PLoS Genet.
17(e1009679)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Zheng SQ, Chen HX, Liu XC, Yang Q and He
GW: Genetic analysis of the CITED2 gene promoter in isolated and
sporadic congenital ventricular septal defects. J Cell Mol Med.
25:2254–2261. 2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Fu F, Li R, Lei TY, Wang D, Yang X, Han J,
Pan M, Zhen L, Li J, Li FT, et al: Compound heterozygous mutation
of the ASXL3 gene causes autosomal recessive congenital heart
disease. Hum Genet. 140:333–348. 2021.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Hou C, Zheng J, Liu W, Xie L, Sun X, Zhang
Y, Xu M, Li Y and Xiao T: Identification and characterization of a
novel ELN mutation in congenital heart disease with pulmonary
artery stenosis. Sci Rep. 11(14154)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Helm BM, Landis BJ and Ware SM: Genetic
evaluation of inpatient neonatal and infantile congenital heart
defects: New findings and review of the literature. Genes (Basel).
12(1244)2021.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Massadeh S, Albeladi M, Albesher N,
Alhabshan F, Kampe KD, Chaikhouni F, Kabbani MS, Beetz C and
Alaamery M: Novel autosomal recessive splice-altering variant in
PRKD1 is associated with congenital heart disease. Genes (Basel).
12(612)2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Musfee FI, Agopian AJ, Goldmuntz E,
Hakonarson H, Morrow BE, Taylor DM, Tristani-Firouzi M, Watkins WS,
Yandell M and Mitchell LE: Common variation in cytoskeletal genes
is associated with conotruncal heart defects. Genes (Basel).
12(655)2021.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Meerschaut I, Vergult S, Dheedene A,
Menten B, De Groote K, De Wilde H, Muiño Mosquera L, Panzer J,
Vandekerckhove K, Coucke PJ, et al: A reassessment of copy number
variations in congenital heart defects: Picturing the whole genome.
Genes (Basel). 12(1048)2021.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Yadav ML, Jain D, Neelabh Agrawal D, Kumar
A and Mohapatra B: A gain-of-function mutation in CITED2 is
associated with congenital heart disease. Mutat Res.
822(111741)2021.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Basel-Salmon L, Ruhrman-Shahar N, Barel O,
Hagari O, Marek-Yagel D, Azulai N, Bazak L, Svirsky R, Reznik-Wolf
H, Lidzbarsky GA, et al: Biallelic variants in ETV2 in a family
with congenital heart defects, vertebral abnormalities and preaxial
polydactyly. Eur J Med Genet. 64(104124)2021.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Huang S, Wu Y, Chen S, Yang Y, Wang Y,
Wang H, Li P, Zhuang J and Xia Y: Novel insertion mutation
(Arg1822_Glu1823dup) in MYH6 coiled-coil domain causing familial
atrial septal defect. Eur J Med Genet. 64(104314)2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Zhao L, Jiang WF, Yang CX, Qiao Q, Xu YJ,
Shi HY, Qiu XB, Wu SH and Yang YQ: SOX17 loss-of-function variation
underlying familial congenital heart disease. Eur J Med Genet.
64(104211)2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Li YJ and Yang YQ: An update on the
molecular diagnosis of congenital heart disease: Focus on
loss-of-function mutations. Expert Rev Mol Diagn. 17:393–401.
2017.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Sifrim A, Hitz MP, Wilsdon A, Breckpot J,
Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan
GJ, et al: Distinct genetic architectures for syndromic and
nonsyndromic congenital heart defects identified by exome
sequencing. Nat Genet. 48:1060–1065. 2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Darwich R, Li W, Yamak A, Komati H,
Andelfinger G, Sun K and Nemer M: KLF13 is a genetic modifier of
the Holt-Oram syndrome gene TBX5. Hum Mol Genet. 26:942–954.
2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Song A, Patel A, Thamatrakoln K, Liu C,
Feng D, Clayberger C and Krensky AM: Functional domains and
DNA-binding sequences of RFLAT-1/KLF13, a Krüppel-like
transcription factor of activated T lymphocytes. J Biol Chem.
277:30055–30065. 2002.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Lavallée G, Andelfinger G, Nadeau M,
Lefebvre C, Nemer G, Horb ME and Nemer M: The Kruppel-like
transcription factor KLF13 is a novel regulator of heart
development. EMBO J. 25:5201–5213. 2006.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Zhao W, Wang J, Shen J, Sun K, Zhu J, Yu
T, Ji W, Chen Y, Fu Q and Li F: Mutations in VEGFA are associated
with congenital left ventricular outflow tract obstruction. Biochem
Biophys Res Commun. 396:483–488. 2010.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Greenway SC, McLeod R, Hume S, Roslin NM,
Alvarez N, Giuffre M, Zhan SH, Shen Y, Preuss C, Andelfinger G, et
al: Exome sequencing identifies a novel variant in ACTC1 associated
with familial atrial septal defect. Can J Cardiol. 30:181–187.
2014.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Matsson H, Eason J, Bookwalter CS, Klar J,
Gustavsson P, Sunnegårdh J, Enell H, Jonzon A, Vikkula M, Gutierrez
I, et al: Alpha-cardiac actin mutations produce atrial septal
defects. Hum Mol Genet. 17:256–265. 2008.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Martin KM, Metcalfe JC and Kemp PR:
Expression of Klf9 and Klf13 in mouse development. Mech Dev.
103:149–151. 2001.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Gordon AR, Outram SV, Keramatipour M,
Goddard CA, Colledge WH, Metcalfe JC, Hager-Theodorides AL,
Crompton T and Kemp PR: Splenomegaly and modified erythropoiesis in
KLF13-/- mice. J Biol Chem. 283:11897–11904.
2008.PubMed/NCBI View Article : Google Scholar
|