Introduction
Myhre syndrome (Online Mendelian Inheritance in Man
entry no. 139210), first observed in 1981(1), has thus far been reported in <100
patients worldwide (2). Myhre
syndrome is an autosomal dominant connective tissue disorder
affecting patients from a young age without ethnic or sex
predilection. Clinical manifestations include intrauterine growth
retardation, short stature and limited joint mobility (3). More common phenotypical traits become
recognizable in childhood, including distinct facial dysmorphic
features, such as midface hypoplasia, short palpebral fissure,
narrow mouth and prognathism, smaller dysplastic ears,
brachydactyly and clinodactyly with hyperconvex nails. Thick skin
and atypical scarring are also documented in most cases (4). These patients usually present with
hearing loss, ophthalmological manifestations, the most common
being strabismus (5). Different
cardiovascular and respiratory manifestations, including heart
failure and dyspnoea (6) due to
septal defects, aortic defects or laryngotracheal stenosis, may
also be observed, some of which may be life-threatening, while
limited intellect and autism spectrum disorder-like behaviour may
affect the capability of having normal social interactions
(7).
The diagnosis of Myhre syndrome is quite
challenging, and it is based on characteristic clinical and imaging
features and confirmed by specific molecular diagnosis.
The most common aetiology is a heterozygous mutation
in the SMAD4 gene affecting the codon for Ile500, which
encodes a tumour-suppressor protein. Thus, the TGF-β signalling
pathway is affected owing to the aforementioned manifestations
(8). In most reported cases, the
mutation appears de novo (9). A small number of Myhre syndrome
diagnoses have been associated with SMAD4 missense mutation
causing the replacement of Arg496(10).
Case report
The case reported in the present study involves an
18-year-old female patient presenting with short stature, facial
deformities, chronic muscle and joint pain, multiple allergies and
suspicion of vasculitis, without diagnosis of any autoimmune
diseases, and with a predisposition for developing recurring
choanal benign tumours causing severe nasal obstruction, dizziness
and headaches, severely affecting daily activities. Due to the
heterogeneity of symptoms, diagnosis was delayed and the patient
was finally diagnosed with Myhre syndrome following whole-exome
sequencing (WES) molecular analysis.
The patient, who is the first documented case of
Myhre syndrome in Romania, was diagnosed at the age of 17 years.
The patient was born by caesarean section after a pregnancy
associated with multiple episodes of first-trimester haemorrhage.
Both parents were aged 35 years at the time of the patient's birth.
The patient exhibited mild intrauterine growth retardation and
microcephaly. Slower weight gain and orthopnoea were observed from
the early neonatal period The patient also exhibited delayed dental
eruption, having the first teeth at 18 months of age (normal range,
6-8 months); there were no observed dental abnormalities.
Since the age of 18 months, the patient developed
several severe allergic reactions to insect bites, which required
hospitalizations and steroid treatment. By the age of 3 years, the
patient was suffering from recurrent ENT infections and vocal cord
nodules causing hoarseness, which resolved spontaneously after 1
year. At 10 years of age, the patient underwent an adenoidectomy.
The chronic muscle and joint pains were not associated with any
neurological findings.
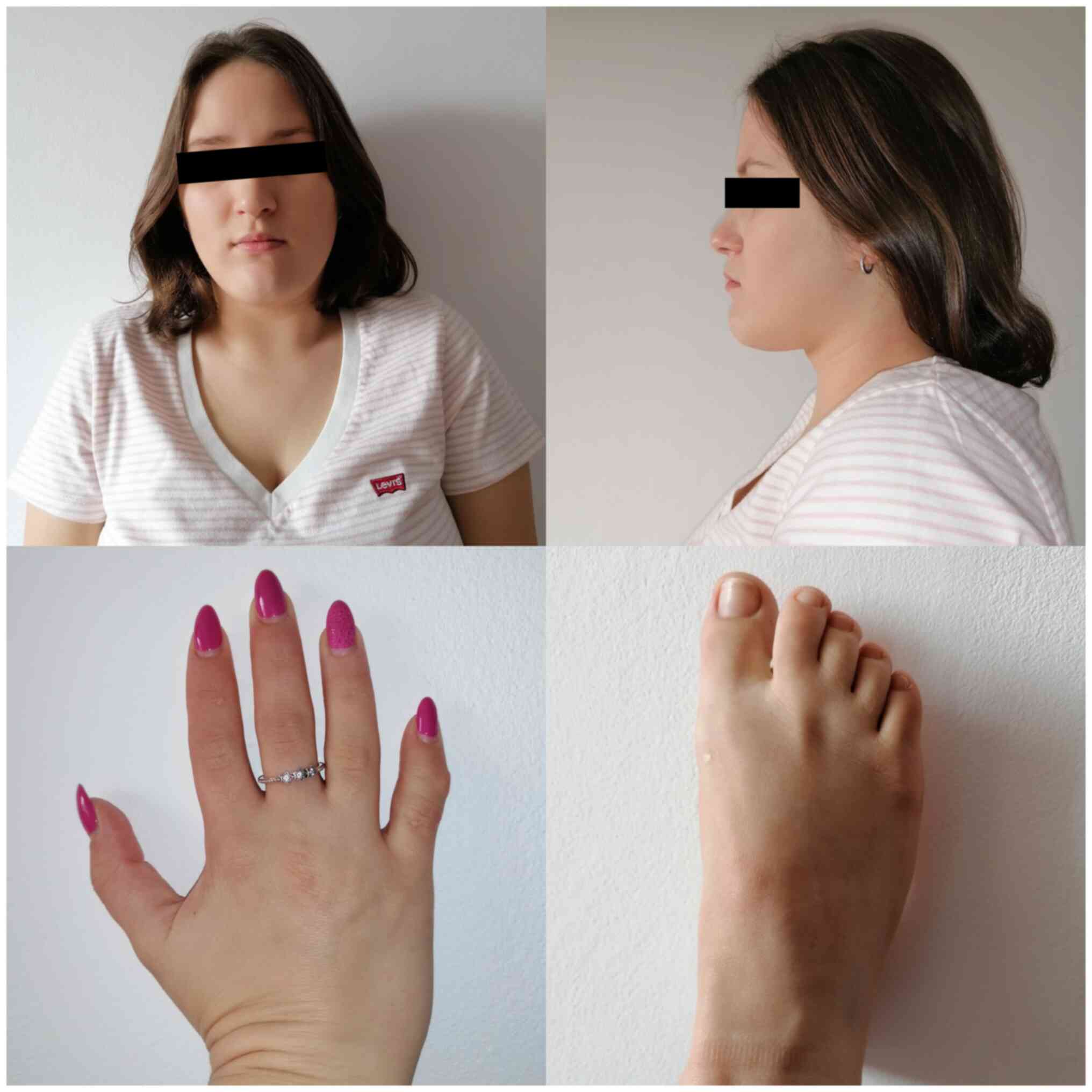
The patient exhibited a short stature at 152 cm
(mother: 178 cm, father: 182 cm), lumbar lordosis, thickened
calvarium and facial deformities, including maxillary hypoplasia,
large forehead, microstomia, thin upper lip, prominent nasal root,
narrow palpebral fissures and strabismus. The skin on the palms and
feet was thickened and stiff, and there were multiple stretch marks
and abnormal scarring on different parts of the body.
Brachydactyly, camptodactyly and clinodactyly were also be observed
in the hands. A routine clinical check-up followed by ultrasound
imaging revealed the presence of mild coarctation of the aorta, in
the absence of hemodynamically significant alterations (Fig. 1).
By the time of diagnosis, the patient had developed
facial tics, chronic constipation (despite a healthy, well-balanced
diet) and severe menstrual abnormalities (dysmenorrhea and
oligomenorrhea), which were corrected to a certain extent with
medication (oestrogen and progesterone).
Immediately prior to recommending genetic testing,
the patient presented with severe nasal obstruction, dizziness and
severe headaches. The ENT clinical and MRI examinations revealed a
bilateral circumferential choanal tumour, with a suspected
diagnosis of rhinoscleroma, or post-adenoidectomy stenosis. Surgery
was performed under general anaesthesia with orotracheal
intubation, with an apparent favourable postoperative evolution. By
the age of 12 years, the surgical follow-up showed signs of
recurrence and further investigations have been advised, but have
not been performed due to the patient's altered emotional status at
the time. Three months later, in the context of the patient's ENT
symptomatology, audiometry was also performed, revealing conductive
hearing loss.
Routine check-up urinalysis revealed haematuria,
leukocyturia and proteinuria, leading to the diagnosis of nephrotic
syndrome. Corroborated with a lack of determined aetiology for the
ENT manifestations, vasculitis was suspected. The absence of
symptoms suggesting an autoimmune systemic inflammatory process and
the results of paraclinical investigations (negative inflammatory
markers, negative antineutrophil cytoplasmic antibodies and
anti-C1q serology) excluded the presumed diagnosis.
The ophthalmological evaluation revealed esophoria,
pseudopapillitis, myopia and astigmatism. The recommended MRI
examination for the evaluation of the optic nerve revealed no
pathological findings (Table
I).
 | Table IClinical features of the present case
in comparison with other reported cases of patients with Myhre
syndrome. |
Table I
Clinical features of the present case
in comparison with other reported cases of patients with Myhre
syndrome.
| Phenotype in Myhre
syndrome | Literature reports on
Myhre syndrome phenotype | Patient
phenotype | (Refs.) |
|---|
| Facial
deformities | Microcephaly with
thickened calvarium, narrowed palpebral fissures, midface
hypoplasia, microstomia, prognathism, hypertelorism, prominent
nasal root, cleft palate | Microcephaly,
maxillary hypoplasia, large forehead, microstomia, thin upper lip,
prominent nasal root, narrow palpebral fissures | (1,2) |
| Height, weight and
developmental abnormalities | Short stature,
obesity | Short stature (152
cm), delayed dental eruption, intrauterine growth retardation | (1-3) |
| Osteoarticular
abnormalities | Limited joint
mobility, thick calvaria, short neck, broad ribs, brachydactyly,
camptodactyly, clinodactyly | Brachydactyly,
camptodactyly, clinodactyly, chronic muscle and joint pain | (1-3) |
| Muscular
hypertrophy | + | - | |
| Neurological
involvement | Seizures, mental
retardation, cerebral ataxia | Facial tics | (1,2) |
| Cardiovascular
involvement | Septal defects,
aortic stenosis, aortic coarctation, pericardial effusion,
pericardial fibrosis | Aortic coarctation,
mitral valve regurgitation | (2) |
| Respiratory tract
involvement | Laryngotracheal
stenosis, respiratory failure, dyspnea | Orthopnea | (2) |
| Ear, nose and throat
involvement | Early-onset mixed
conductive and sensorineural deafness, voice abnormalities
(hoarseness, nasal voice) | Vocal cord nodules,
hoarseness, hearing loss, recurring bilateral choanal tumor | (1,3) |
| Genito-urinary
involvement | Cryptorchidism,
menstrual abnormalities (oligomenorrhea, metrorrhagia),
vesicoureteral reflux, primary enuresis | Menstrual
irregularities (oligomenorrhea) | |
| Gastroenterological
involvement | Severe
constipation | Chronic
constipation | (4) |
| Skin hypertrophy | Skin thickening and
stiffness | Thickened skin,
abnormal scarring, stretch marks | (2) |
| Ophthalmological
involvement | Strabismus,
astigmatism, pseudopapillitis | Esophoria,
pseudopapillitis, myopia, astigmatism | (1,2) |
| Immunological
involvement | - | Multiple
allergies | |
Given the phenotype of the patient and the lack of
similar clinical manifestations in other family members, after
informed consent was obtained from both the patient and her
parents, WES was performed. Sequence analysis of all protein-coding
genes using the Whole Exome Plus test (Blueprint Genetics)
identified a heterozygous missense variant of SMAD4
c.1498A>G, p. (Ile500Val), which is pathogenic for Myhre
syndrome. The test was associated with whole exome
deletion/duplication (copy number variation) analysis, which came
back negative. The patient was also tested for secondary findings,
whole-exome data being analysed in 59 genes following the
recommendations of the American College of Medical Genetics and
Genomics (11). No other
abnormalities were identified in the genetic analyses. The
patient's father was diagnosed with multiple rectocolic micropolyps
and was also tested for the SMAD4 mutation, as an
association was suspected, but the results of the genetic test were
negative. As the patient's mother was asymptomatic, she was not
tested for the mutation identified in the proband.
Discussion
Diagnosis of rare diseases is often challenging,
particularly when there is no family history, and the symptoms are
heterogeneous and do not fall within a characteristic phenotypic
spectrum.
In Myhre syndrome, symptoms tend to go unnoticed in
early childhood and become noticeable in adolescence, as in the
present case. Diagnosing the condition was even more challenging,
as there is no other case of Myhre syndrome reported in Romania.
After several seemingly unrelated symptoms and several medical
consults, molecular diagnosis was performed using WES, identifying
the mutation in the SMAD4 gene.
The key element in Myhre syndrome is the TGF-β
signalling pathway that controls important biological cell
processes, including cell proliferation, apoptosis and cell lineage
coordination. As SMAD4 represents a coactivator of TGF-β,
dysregulation of SMAD4 function has pleiotropic effects,
which are in line with the major features of Myhre syndrome
resulting from the perturbation of the developmental pathways
controlled by TGF-β/bone morphogenetic protein signalling during
embryonic development, including skeletal axis patterning, tissue
specification and organogenesis. The most common molecular defect
in Myhre syndrome is represented by the restricted 3 amino acid
spectrum of Ile500 mutations (threonine, valine and methionine)
found in almost all patients, albeit with no clear
genotype-phenotype correlations (9).
Recurrent ear infections possibly leading to hearing
loss were previously reported in several patients with Myhre
syndrome (11). SMAD4 gene
mutations are known to be associated with tumorigenesis, as the
mutated variant was identified in both malignant and benign lesions
(12). However, the predisposition
to developing ENT tumours has not yet been reported in Myhre
syndrome, to the best of our knowledge. The patient reported in the
present case exhibited severe nasal obstruction due to the
development of bilateral circumferential choanal tumours,
associated with dizziness and intense headaches, requiring surgical
intervention. After the tumour was removed, the patient's condition
improved significantly, but further investigations showed early
signs of recurrence.
Different skeletal abnormalities and facial
deformities are often observed, and the developmental delay occurs
in all Myhre syndrome patients (13). The particularity of our case is the
delayed dental eruption: The first teeth appeared in our patient at
18 months of age, but no dental abnormalities were found
otherwise.
The clinical picture of our patient is predominated
by ENT-related pathology and severe allergic manifestations. The
SMAD4 protein is an important mediator of TGF-β signalling, which
is known to serve an important role in inducing and maintaining the
inflammatory response, and its correlation with allergic asthma and
immunomodulation has already been documented (12,13).
Although allergic reactions have not been reported in Myhre
syndrome patients to date (14),
our patient experienced severe reactions to insect bites from a
very young age and has a documented allergy to vitamin K, pollen,
mould and other particles. The patient's symptoms have become
milder over time, but she required several hospitalizations and
corticosteroid treatment due to severe allergic reactions during
childhood.
As in the present case, patients with Myhre syndrome
develop this disorder due to a de novo SMAD4 pathogenic
variant. According to SMAD4 expression in the phenotype,
Myhre syndrome has a 50% recurrence risk, although fertility and
reproduction have not been assessed. If the SMAD4 pathogenic
variant is absent in the parents, the risk to sibs is presumed to
be slightly higher compared with that of the general population,
but <1% as a possibility of parental germline mosaicism;
therefore, if a SMAD4 pathogenic variant has been identified
in an affected family member, prenatal testing or preimplantation
genetic testing can be recommended.
In conclusion, Myhre syndrome is a rare autosomal
dominant connective tissue disorder caused by a germline mutation
in the SMAD4 gene. Common signs include short stature,
skeletal abnormalities, limited joint mobility, characteristic
facial features, intellectual and behavioural abnormalities. We
herein report the case of an 18-year-old female patient with short
stature, brachydactyly/clinodactyly, facial dysmorphia, thickened
skin, ENT complications, coarctation of the aorta and SMAD4
c.1498A>G, p. (Ile500Val) mutation, with a late diagnosis. An
atypical finding in the present case was severe allergies. Although
the patient's clinical symptoms are chronic and persistent, the
major discomfort is mental. The late diagnosis, lack of medical
information, as well as the lack of adequate medical knowledge have
led to generalized anxiety that ultimately required psychological
intervention.
The current case report is proof that a
multidisciplinary approach is essential for the diagnosis of rare
genetic diseases. Phenotype heterogeneity requires, in most cases,
extensive molecular testing in order to reach a definitive
diagnosis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
AC and MSM performed genetic consult and
counselling; II, AC and ZCB have seen and confirm the authenticity
of the raw data; DM and RSC performed the interpretation of genetic
test results. All the authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written consent for the
publication of the case details and any associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Myhre SA, Ruvalcaba RH and Graham CB: A
new growth deficiency syndrome. Clin Genet. 20:1–5. 1981.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Caputo V, Bocchinfuso G, Castori M,
Traversa A, Pizzuti A, Stella L, Grammatico P and Tartaglia M:
Novel SMAD4 mutation causing Myhre syndrome. Am J Med Genet A.
164A:1835–1840. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Caputo V, Cianetti L, Niceta M, Carta C,
Ciolfi A, Bocchinfuso G, Carrani E, Dentici ML, Biamino E, Belligni
E, et al: A restricted spectrum of mutations in the SMAD4
tumor-suppressor gene underlies Myhre syndrome. Am J Hum Genet.
90:161–169. 2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Asakura Y, Muroya K, Sato T, Kurosawa K,
Nishimura G and Adachi M: First case of a Japanese girl with Myhre
syndrome due to a heterozygous SMAD4 mutation. Am J Med Genet A.
158A:1982–1986. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Starr LJ, Lindor NM and Lin AE: Myhre
Syndrome. In: GeneReviews®. University of Washington,
Seattle, Seattle WA, 2017. https://www.ncbi.nlm.nih.gov/books/NBK425723/.
|
|
6
|
Titomanlio L, Marzano MG, Rossi E,
D'Armiento M, De Brasi D, Vega GR, Andreucci MV, Orsini AV, Santoro
L and Sebastio G: Case of Myhre syndrome with autism and peculiar
skin histological findings. Am J Med Genet. 103:163–165.
2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Michot C, Le Goff C, Mahaut C, Afenjar A,
Brooks AS, Campeau PM, Destree A, Di Rocco M, Donnai D, Hennekam R,
et al: Myhre and LAPS syndromes: Clinical and molecular review of
32 patients. Eur J Hum Genet. 2:1272–1277. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Whiteford ML: Myhre Syndrome. In: NORD
Guide to Rare Disorders. Lippincott Williams & Wilkins.
Philadelphia, PA, p225, 2003.
|
|
9
|
Lazzereschi D, Nardi F, Turco A, Ottini L,
D'Amico C, Mariani-Costantini R, Gulino A and Coppa A: A complex
pattern of mutations and abnormal splicing of Smad4 is present in
thyroid tumours. Oncogene. 24:5344–5354. 2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Le Goff C, Michot C and Cormier-Daire V:
Myhre syndrome. Clin Genet. 85:503–513. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Miller DT, Lee K, Chung WK, Gordon AS,
Herman GE, Klein TE, Stewart DR, Amendola LM, Adelman K, Bale SJ,
et al: ACMG SF v3.0 list for reporting of secondary findings in
clinical exome and genome sequencing: A policy statement of the
American College of Medical Genetics and Genomics (ACMG). Genet
Med. 23:1381–1390. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tirado-Rodriguez B, Ortega E,
Segura-Medina P and Huerta-Yepez S: TGF-β: An important mediator of
allergic disease and a molecule with dual activity in cancer
development. J Immunol Res. 2014(318481)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Garavelli L, Maini I, Baccilieri F,
Ivanovski I, Pollazzon M, Rosato S, Iughetti L, Unger S,
Superti-Furga A and Tartaglia M: Natural history and
life-threatening complications in Myhre syndrome and review of the
literature. Eur J Pediatr. 175:1307–1315. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
McGowan R, Gulati R, McHenry P, Cooke A,
Butler S, Keng WT, Murday V, Whiteford M, Dikkers FG,
Sikkema-Raddatz B, et al: Clinical features and respiratory
complications in Myhre syndrome. Eur J Med Genet. 54:e553–e559.
2011.PubMed/NCBI View Article : Google Scholar
|