Introduction
Angiogenesis is required for tumor development
(1). This process involves the
interaction between various cell types and molecules, contributing
to basement membrane degradation, endothelial cell (EC) migration
and proliferation and tube formation (2). As one of the primary malignant tumors
with high mortality rates worldwide, liver cancer is characterized
by an abundant blood supply (3).
Therefore, liver cancer angiogenesis is a therapeutic target
(4,5). New agents of anti-angiogenic therapy
are greatly required.
Sinensetin (SIN) is a polymethoxy flavone containing
five methoxy groups that is present mainly in citrus fruits
(6). Previous studies have
demonstrated that SIN can inhibit the development of several
malignant tumors, such as gallbladder adenocarcinoma, T-cell
lymphoma and gastric cancer (7-9).
A recent study revealed that SIN may be a potential anticancer drug
targeting autophagy of hepatocellular carcinoma (10). However, the effects of SIN on liver
cancer angiogenesis remain to be elucidated.
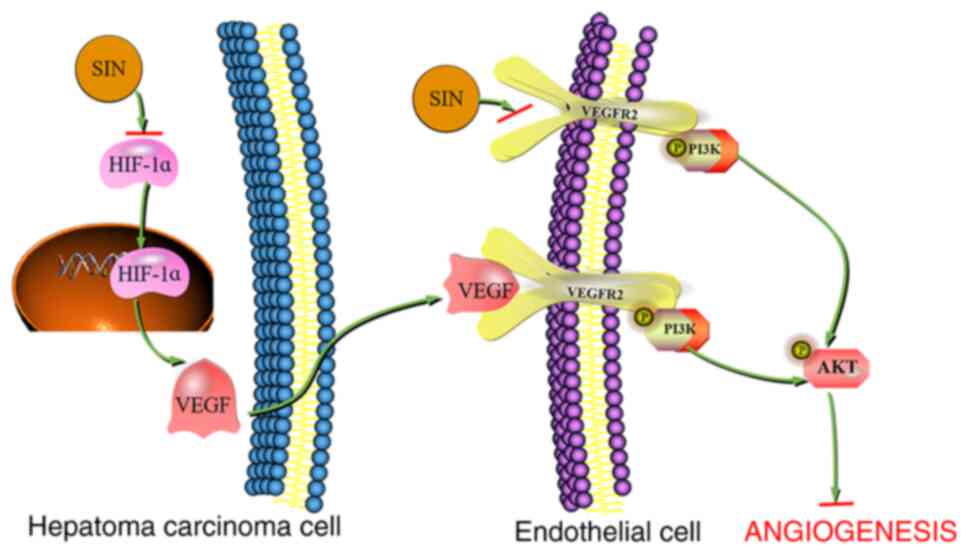
Angiogenesis is regulated by various angiogenic
factors, such as VEGF and its receptor (11), platelet-derived growth factor and
angiopoietin (12-14).
Among them, VEGFRs are the key mediators of angiogenesis (15). VEGF is mainly produced by tumor
cells, notably found in benign and malignant lesions (16). The increase in VEGF expression
induced by tumor secretion is caused by specific hypoxia-inducible
factors (HIFs) (17). As a
receptor of VEGF, VEGFR is mainly located on the EC membrane and
has the three following types: VEGFR1, VEGFR2 and VEGFR3. VEGFR2 is
mainly present in vascular ECs (18). VEGF is released by tumor cells and
can bind to VEGFR2 on ECs, causing its autophosphorylation.
Following phosphorylation at Tyr1175 of VEGFR2, the latter binds to
the p85 subunit of phosphoinositide-3-kinase (PI3K) and activates
the PI3K-AKT signaling pathway (19). However, it remains unclear whether
SIN can block the signal transduction of VEGF/VEGFR2 and eventually
lead to inhibition of angiogenesis in liver cancer.
The present study observed that SIN inhibited the
growth and angiogenesis of HepG2/C3A-derived tumors in vivo.
In vitro experiments revealed that SIN reduced VEGF through
HIF1-α in the hepatoblastoma cell line HepG2/C3A. In addition, it
was observed that SIN promoted apoptosis and inhibited
proliferation in human umbilical vein endothelial cells (HUVECs),
causing the repression of invasiveness and pro-angiogenic process
of ECs. Moreover, SIN downregulated VEGFR2 phosphorylation and the
expression levels of its downstream targets in ECs. The data
indicated that SIN may inhibit angiogenesis by regulating the
VEGF/VEGFR2/AKT signaling pathway.
Materials and methods
Chemicals and reagents
The antibody against VEGF was purchased from Novus
Biologicals. The primary antibodies including anti-VEGFR2 (cat. no.
9698), anti-phosphorylated (p)-VEGFR2 (Tyr1175) (cat. no. 3770),
anti-AKT (cat. no. 4691), anti-p-AKT (Ser473) (cat. no. 4060),
anti-platelet/endothelial cell adhesion molecule-1 (CD31) (cat. no.
77699), β-actin (cat. no. 4970), GAPDH (cat. no. 2118) and
α-tubulin (cat. no. 2125) were acquired from Cell Signaling
Technology, Inc.; anti-HIF-1α (cat. no. 20960-1-AP) was provided by
ProteinTech Group, Inc. Synthetic SIN (>98% purity; cat. no.
SS8550) was purchased from Beijing Solarbio Science &
Technology Co. Ltd. and characterized by mass spectrometry.
Recombinant human VEGF (VEGF165; cat. no. 100-20) and insulin-like
growth factor 1 (IGF-1; cat. no. 100-11) were purchased from
PeproTech, Inc. SU1498 (cat. no. HY-19326) is a selective VEGFR2
inhibitor, which was obtained from MedChemExpress.
Xenograft tumor growth assay
A total of 10 male BALB/c nude mice (age, 4 weeks;
weight, 15-20 g) purchased from Beijing Weitong Lihua Biotechnology
Co., Ltd., were used for the tumor xenograft growth assay. Mice
were housed at room temperature (22±1˚C) with 50% humidity under
special pathogen-free conditions with a 12-h light/dark schedule.
All animal studies were conducted with approval (no. 2017-106)
obtained from the Ethics Committee of Shandong Provincial
Qianfoshan Hospital. HepG2/C3A cells (ATCC) (5x107/ml)
suspended in 0.2 ml 1:1 serum-free DMEM and Matrigel (Corning,
Inc.) were implanted into the right flank of the mice (20). When the tumor size reached a volume
of 100 mm3 (approximately two weeks), tumor-bearing mice
were randomized into two groups (n=5, each group). The
experimentalgroupmice were administered with SIN (40 mg/kg)
(21) by gavage every day for 14
days and the control group received the same volume of 0.9% normal
saline for 14 days. Animal health and behavior were monitored
daily. The tumor diameter was measured by a slide caliper and the
body weight was recorded every two days. The maximum diameter of
the observed tumors was 17 mm. The tumor size was calculated with
the following formula: Length x width2 x 0.5. Tumors
exceeding 10% of the body weight of mice or became infected were
used as humane endpoints. Animals were euthanized using 30%
volume/min CO2 upon reaching experimental or humane
endpoints. None of nude mice died during the experiment. Following
14 days, the tumors were harvested and subsequently used for
western blot and immunohistochemical (IHC) analyses. The duration
of the experiment was 4 weeks from the start of the experiment.
Cell inoculation and sample collection were performed under
anesthesia using 1% (w/v) pentobarbital sodium (40 mg/kg)
injections intraperitoneally and all efforts were made to minimize
suffering, discomfort and distress.
IHC assay
The tumors were fixed overnight in 4% formaldehyde
at room temperature, and then ethanol was used for dehydration at
the conventional gradient, xylene for vitrification and paraffin
for embedding. Sections (5 µm) were created and deparaffinized with
graded xylene and rehydrated by graded ethanol. Following
heat-induced antigen retrieval in citrate buffer (pH 6.0) in a
high-pressure sterilizer at 121˚C for 10 min, the slides were
treated with 3% hydrogen peroxide to quench endogenous peroxidase
activity and subsequently incubated with 4% bovine serum albumin at
37˚C for 30 min. The tumor tissue slides were incubated with
primary anti-CD31 antibodies (1:100) at 4˚C overnight and washed
with PBS three times. HRP-labeled secondary antibody from the
MaxVision™ HRP-Polymer anti-mouse/rabbit IHC kit (ready
to use; cat. no. KIT5020; Fuzhou Maixin Biotech Co., Ltd.) was
applied and incubated for 30 min at room temperature. The slides
were then dehydrated in an ascending graded series of absolute
ethyl alcohols, cleared in xylene and cover-slipped with neutral
balsam. Following treatment with hematoxylin for 2 min at room
temperature to stain the nuclei, images were captured using a light
microscope and in ten random fields at x200 magnification.
Cell culture
HUVECs (cat. no. PCS-100-010) and HepG2/C3A (cat.
no. CRL-10741) cells were purchased from the American Type Culture
Collection. HUVECs were incubated in Endothelial Cell Medium (ECM;
ScienCell Research Laboratories, Inc.) containing 5% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% EC growth
supplement (ECGS; ScienCell Research Laboratories, Inc.) at 37˚C.
HepG2/C3A cells were incubated in Dulbecco's modified Eagle's
medium (HyClone; Cytiva) comprising 10% FBS in a hypoxic incubator
with 94% N2, 5% CO2 and 1% O2 at
37˚C.
Proliferation assay
HUVECs were harvested and seeded into 96-well plates
at a density of 5x103 cells/well and exposed to various
concentrations of SIN (3, 10, 30, 60 and 100 µM). Following culture
for 24 h, the viability of HUVECs was detected using CCK-8 (Dojindo
Molecular Technologies, Inc.). The optical density, which
represented the proliferation of HUVECs, was measured at 450 nm
using a Spectra Max 190 (Molecular Devices, LLC).
Apoptosis assay
The induction of apoptosis in HUVECs was assessed
using Annexin V-FITC and propidium iodide staining (ELabscience
Biotechnology, Inc.). Following treatment with SIN, HUVECs were
collected and resuspended in a binding buffer at a final
concentration of 1x106 cells/ml. Single cells were
incubated with 5 µl Annexin V-FITC and 5 µl PI for 15 min at room
temperature in the dark. The percentages of early and late
apoptotic cells were assessed using a FACSAria II flow cytometer
(BD Biosciences) to calculate the apoptotic rate. Data were
analyzed using FlowJo (V10; FlowJo LLC).
Wound healing assay
Migration was assessed using a scratch wound healing
assay. The cells were seeded (4x105/well) into
6-wellplates. A straight scratch was introduced in HUVEC monolayers
using a 200-µl plastic pipette tip. Following incubation for a
further 24 h in EBM containing 1% FBS, the average distance of the
cells migrating into the wound was monitored by a light microscope
(magnification, x100; Olympus Corporation). The migrated distance
was calculated using ImageJ software (version1.49p; National
Institutes of Health).
Transwell assay
Transwell inserts (Corning, Inc.) with a pore size
of 8-µm were used to assess the migratory ability of ECs. HUVECs
(1x105/well) were resuspended in 500 µl serum-free
medium with SIN (30 µM) and subsequently added to the upper plate
compartment. The medium supplemented with 10% FBS was filled to the
bottom chamber. The invaded cells were treated with 4%
paraformaldehyde for 10 min and stained with 0.1% crystal violet
(Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at room
temperature. Finally, the cells numbers in five randomly selected
fields were counted under an inverted microscope (magnification,
x100; Olympus Corporation).
Angiogenesis assay
For this assay, 96-well plates were pre-coated with
Matrigel (50 µl) for 30 min at 37˚C. SIN was dissolved in DMSO,
which was used as a control group. HUVECs were resuspended at
1-2x104 cells/ml in serum-free EBM-2 with SIN and loaded
on top of the Matrigel. Following culture for 6 h, the images were
captured using a light microscope (magnification, x40; Olympus
Corporation). Vessel morphometric parameters, including vessel
number, were quantified using ImageJ software (version1.49p;
National Institutes of Health). Tube formation was expressed as a
percentage of the control group.
Western blotting
The lysates of HUVECs were extracted using RIPA
lysis buffer. The homogenates were centrifuged at 12,000 x g for 15
min at 4˚C. The concentration levels of the protein samples were
evaluated using a bicinchoninic acid protein analysis kit (Thermo
Fisher Scientific, Inc.). Total protein (30 µg) was electrophoresed
on 7.5 and 10% SDS-PAGE gels for 1 h using an electrophoresis
apparatus (Bio-Rad Laboratories, Inc.) and transferred to PVDF
membranes (MilliporeSigma) at 280 mA for 2 h, followed by blocking
in 5% non-fat milk for 1.5 h. The membranes were incubated at 4˚C
overnight with rabbit CD31, p-VEGFR2, VEGFR2, p-AKT, AKT, β-actin
and GAPDH (1:1,000 each), HIF-1α (1:500), α-tubulin (1:3,000) and
mouse VEGF (1:1,000) antibodies. Following washing with TBS-T (0.1%
Tween-20) three times, the membranes were incubated with
HRP-conjugated secondary antibodies (ShanghaiMorui Biotechnology)
for 2 h at room temperature and developed with enhanced
chemiluminescence (ECL; MilliporeSigma) reagents. The signal
intensity was calculated using ImageJ software (version1.44p;
National Institutes of Health).
Tumor-conditioned medium (CM)
preparation
HepG2/C3A cells were plated at a density of
5x105 cells/ml in 6-well plates with DMEM containing 10%
FBS overnight. At 90% confluence, the cells were transferred from a
normoxic (21% O2) to a hypoxic (1% O2)
environment for 48 h following replacement of the medium with or
without 30 µM SIN. The collected CM was filtered through a 0.2-µm
filter (Corning, Inc.) and stored in a freezer at -80˚C.
ELISA of conditioned media
VEGF levels in HepG2/C3A CM were assessed using a
human VEGF Quantikine ELISA kit (cat. no. E-TSEL-H0026; ELabscience
Biotechnology, Inc.) following the manufacturer's protocol. The
experiment was repeated in triplicate, with five biological samples
each time.
Reverse transcription-quantitative
(RT-q) PCR
HepG2/C3A cells (2x105/ml) were treated
with or without SIN in 6-well plates under hypoxic conditions for
48 h. Total RNA in the cells was extracted using RNAiso Plus kit
(Takara Bio, Inc.). The RNA concentration levels were measured
using spectrophotometry (NanoDrop 2000; Thermo Fisher Scientific,
Inc.). Following treatment with DNAse, 1 µg RNA was
reverse-transcribed using a reverse transcriptase kit (Thermo
Fisher Scientific, Inc.). RT-qPCR was performed using SYBR-Green I
Master (Vazyme Biotech Co., Ltd.). RNA extraction, cDNA synthesis
and qPCR were performed according to the manufacturer's protocols.
The primer sequences used were obtained from a previously published
study (22) and synthesized by
Invitrogen (Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 95˚C for 30 sec, followed by 40 cycles
at 95˚C for 10 sec and 60˚C for 30 sec. The relative expression
levels of the target genes were normalized to that of GAPDH using
the 2-∆∆Cq method (23). The primer sequences used were:
Human VEGF, forward, 5'-AAAGGGAAAGGGGCAAAAACGAA-3' and reverse,
5'-AGGAACATTTACACGTCTGCGG-3'; and human GAPDH, forward,
5'-TGATGACATCAAGAAGGTGGTGAAG-3' and reverse,
5'-TCCTTGGAGGCCATGTGGGCCAT-3'. All PCR was repeated in
triplicate.
Molecular docking
The crystal structure file of VEGFR2 (PDB ID: 3VHE)
was downloaded from the Protein Data Bank (PDB) database
(http://www.rcsb.org/). Prior to docking, the
water molecules of VEGFR2 were removed and subsequently hydrogen
atoms and charges (24) were added
to the structure of this receptor. The SIN structure file was
obtained from the following website: http://zinc.docking.org/. Molecular docking was used
to predict the optimal binding site for SIN to VEGFR2 (AutoDock
4.2) (25). The docking position
of SIN on VEGFR2 was defined at the active site with a proper grid
box; the grid box size was 68x72x74; the grid center was -24.402 Å,
-0.107 Å and -4.276 Å and the grid space was 0.704 Å. The optimal
binding mode between SIN and VEGFR2 was acquired under the minimum
binding free energy conformation and the output results of AutoDock
4.2 were presented to PyMol 1.8.2.0 (Schrödinger, LLC) and
Discovery Studio 2016 Client (BIOVIA Discovery Studio Client v16;
Dassault Systèmes) software for further analysis.
Statistical analysis
All results are presented as the mean ± standard
deviation (SD). Unpaired Student's t-test was applied for two-group
comparisons. Significance among multiple groups were calculated by
one-way ANOVA with Bonferroni's post-hoc test using GraphPad Prism
5 (GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
SIN inhibited the growth and
angiogenesis of HepG2/C3A-derived tumors in vivo
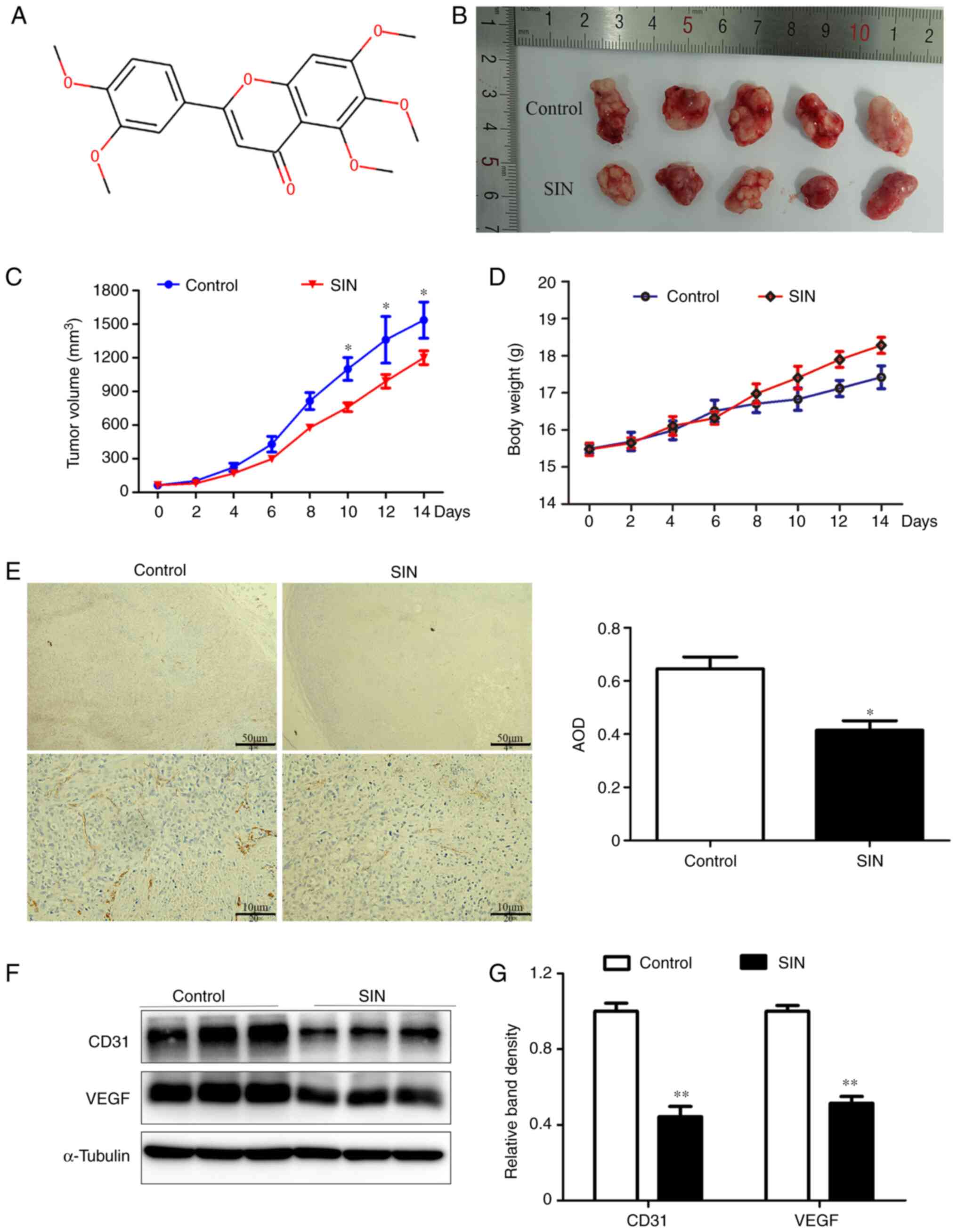
The chemical structure of SIN is displayed in
Fig. 1A. To examine whether SIN
could inhibit tumor growth in vivo, HepG2/C3A tumor models
were established in BALB/c nude mice. The analysis of the growth
curve of model mice indicated that SIN exerted an antitumor effect.
As depicted in Fig. 1B, after 14
days of treatment with SIN or saline, the tumor volume in SIN
treatment group was smaller than the control group. The difference
became more significant with increasing the drug application time
(Fig. 1C) (day 10, 12 and 14;
P<0.05). As one of cachexia-associated symptoms, the body weight
of control group increased slowly, but there were no significant
differences between the two groups (Fig. 1D). As CD31 is a marker of
neovascularization (26),
CD31expression was evaluated using IHC analysis and western
blotting. Staining for CD31 revealed that CD31 expression was
significantly lower in SIN treatment group than in the control
group (Fig. 1E; P<0.05). The
CD31 results of western blotting were also lower in SIN group,
consistent with the results of IHC staining (Fig. 1F and G; P<0.01). As an indicator of growing
tumor tissue, VEGF could promote the growth of ECs (15). Western blotting revealed that SIN
inhibited VEGF expression in HepG2/C3A-derived tumor tissues
(Fig. 1F and G).
SIN suppresses angiogenesis in vitro
by promoting apoptosis and inhibiting proliferation, migration and
tube formation of HUVECs
Under different SIN concentrations (3, 10, 30, 60
and 100 µM) for 24 h, cytotoxicity was measured using CCK8 assay.
VEGF was used as a positive control in the present study. As an
angiogenic factor secreted by tumor cells under hypoxic conditions
(15), VEGF will bind to VEGF
receptor 2 on ECs (27) and
promote proliferation, migration and survival of ECs. As shown in
Fig. 2A, SIN demonstrated a
concentration-dependent inhibitory effect on the proliferation of
HUVEC. In addition, endothelial cell proliferation induced by VEGF
can be inhibited by SIN. These results suggested that SIN inhibited
the proliferation of HUVEC. For 30, 60 and 100 µM, this effect was
significant (P<0.01). Hence, SIN at a concentration of 30 µM was
employed for subsequent experiments. To further explore the reasons
for decreased cell viability, the apoptosis levels of HUVECs were
assessed by Annexin V-FITC/PI stain. DMSO-dissolved SIN (30 µM) was
added to the experimental group. Flow cytometry results revealed
that VEGF inhibited apoptosis in HUVECs, but it was reversed by SIN
(Fig. 2B; P<0.01). As wound
healing assays and Transwell assays showed, HUVEC migration was
enhanced by VEGF compared to the control group but was
significantly attenuated in SIN-treatment group (P<0.05;
Fig. 2C; P<0.01; Fig. 2D). To further explore the potential
antiangiogenic effect of SIN, tube formation was performed in
96-well plates. The experimental results indicated that VEGF could
promote tube formation; by contrast, SIN inhibited angiogenesis
in vitro (P<0.01; Fig.
2E). The migration assays and tube formation also showed a
decreased tendency in SIN group compared to control group, but were
not statistically significant. All these data indicated that
angiogenesis was suppressed by SIN.
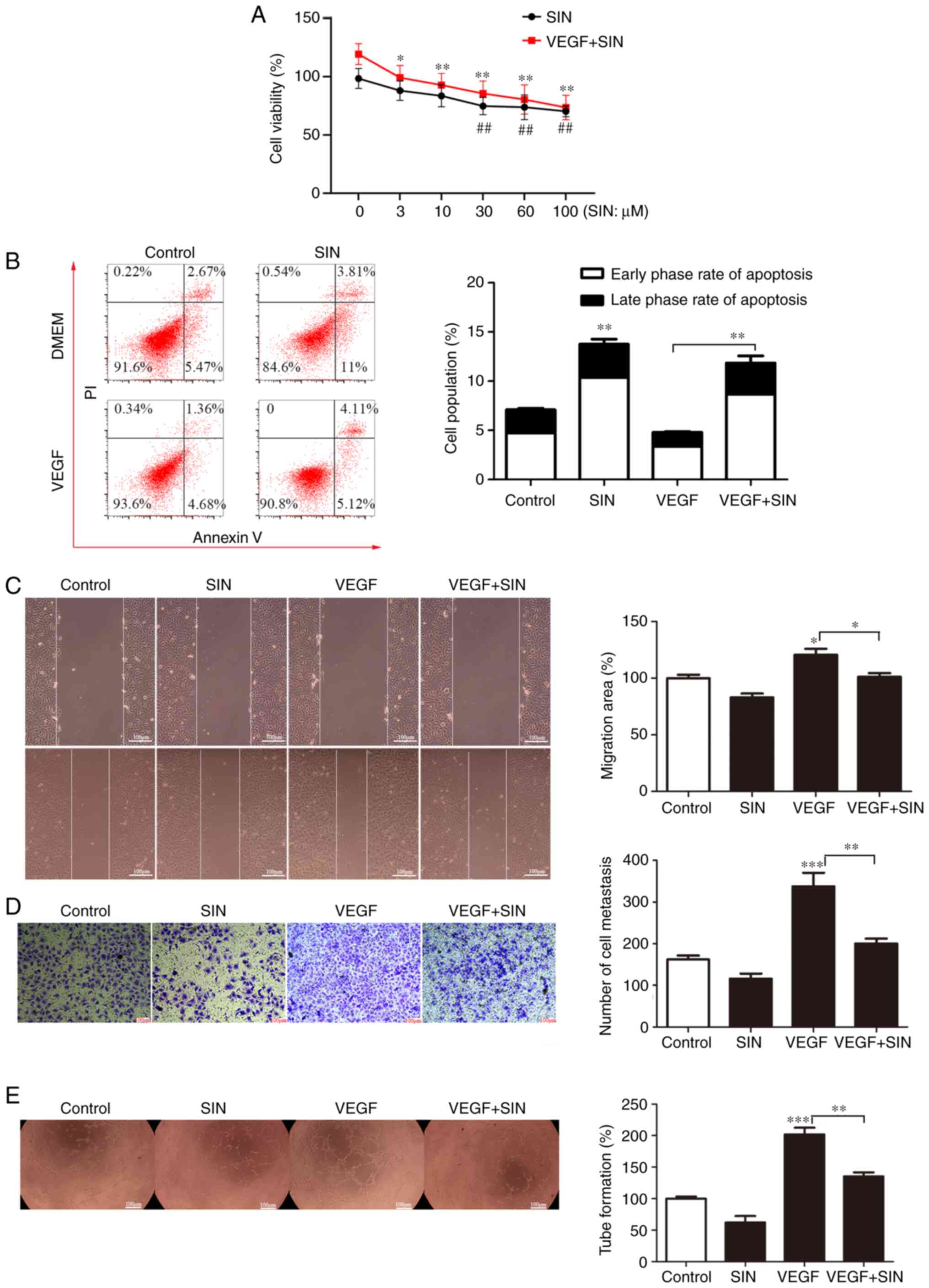 | Figure 2SIN inhibits angiogenesis in
vitro by promoting apoptosis and inhibiting HUVEC migration,
proliferation and tube formation. (A) Inhibitory effects of
different doses of SIN on the proliferation of HUVECs. The data are
presented as percentages of respective control values for cell
viability. n=8, *P<0.05, **P<0.01, VEGF
+ SIN group vs. the VEGF + vehicle group; ##P<0.01,
SIN group vs. the vehicle group. (B) Effects of SIN on induction of
HUVEC apoptosis. The cells were treated with or without VEGF and
SIN. The levels of apoptosis were assessed using flow cytometry
analysis. n=3, **P<0.01. (C) The effects of SIN on
the migration of HUVECs were assessed using the wound-healing assay
(magnification, x100). n=3, *P<0.05. (D) SIN inhibits
HUVEC migration as demonstrated by Transwell assays (magnification,
x100). n=3, **P<0.01, ***P<0.001. (E)
The angiogenic activity of HUVECs was inhibited by SIN as
determined by the tube formation assays (magnification, x40). n=8,
**P<0.01, ***P<0.001. SIN, sinensetin;
HUVEC, human umbilical vein endothelial cells; VEGF, vascular
endothelial growth factor. |
SIN suppresses angiogenesis by
inhibiting the activity of VEGF in HepG2/C3A
VEGF is a pivotal enabling factor for angiogenesis
(28), which HIF regulates
(29). HIF-1α, as a regulatory
subunit, can be increased under hypoxic conditions (30). HepG2/C3Acells were exposed to 30 µM
SIN under hypoxic conditions for 48 h and western blot analyses
revealed that HIF-1α and VEGF levels were significantly elevated
under hypoxic conditions (P<0.05) but could be reversed by SIN
(P<0.05; Fig. 3A). After
incubation with SIN (30 µM) under hypoxic conditions for 48 h, CM
of HepG2/C3A was collected to detect the secretion level of VEGF in
the supernatant. The results revealed that VEGF content in CM
increased significantly under hypoxia (P<0.001), but was
inhibited under SIN treatment (P<0.05; Fig. 3B). VEGF mRNA expression was
measured in SIN-treated HepG2/C3A cells under hypoxic conditions
and PCR results revealed that SIN demonstrated a robust VEGF
inhibitory effect under hypoxic conditions (P<0.05; Fig. 3C). As one of the tumor
cell-secreted angiogenic growth factors, VEGF is a crucial agent
for angiogenesis (31). To mimic
in vivo angiogenesis events, CM of HepG2/C3A cells were
collected to coculture with HUVECs (32) after 48 h of hypoxia exposure in a
serum-free medium. CM from non-SIN-treated HepG2/C3A cells was used
as the control. Tube formation was observed after 6 h CM treatment.
As shown in Fig. 3D, hypoxia
increased VEGF and promoted angiogenesis, but SIN repressed VEGF
expression and inhibited angiogenesis (P<0.001). These results
indicated that the decrease of VEGF induced by SIN in HepG2/C3A
cells contributed to angiogenesis inhibition.
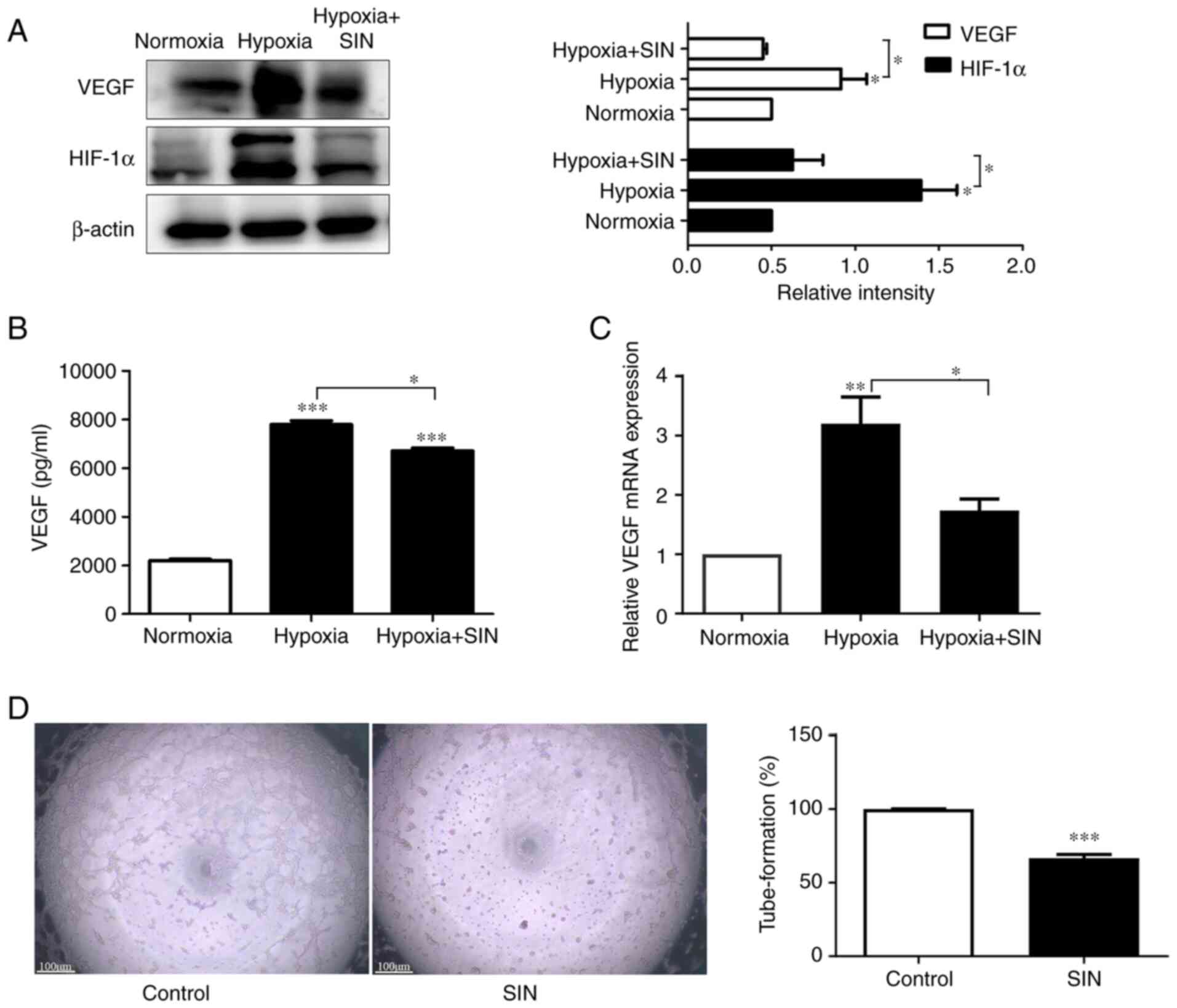 | Figure 3SIN suppresses angiogenesis by
inhibiting the activity of VEGF in HepG2/C3A cells. (A) SIN down
regulated the expression levels of VEGF and HIF-1α under hypoxic
conditions as determined by western blot analysis. n=3,
*P<0.05. (B) SIN inhibits VEGF secretion of HepG2/C3A
cells under hypoxic conditions as determined by ELISA. n=5,
*P<0.05, ***P<0.001. (C) The expression
levels of VEGF were analyzed by RT-qPCR in HepG2/C3A cells. n=3,
*P<0.05, **P<0.01. (D) HepG2/C3A cells
were pre-incubated with SIN for 48 h in a hypoxic environment and
subsequently CM was collected to assess tube formation. SIN-treated
CM inhibits the tube-formation ability of HUVEC (magnification,
x40). n=8, ***P<0.001. SIN, sinensetin; VEGF,
vascular endothelial growth factor; HIF-1α, hypoxia-inducible
factor 1α; RT-qPCR, reverse transcription-quantitative PCR; CM,
tumor-conditioned medium. |
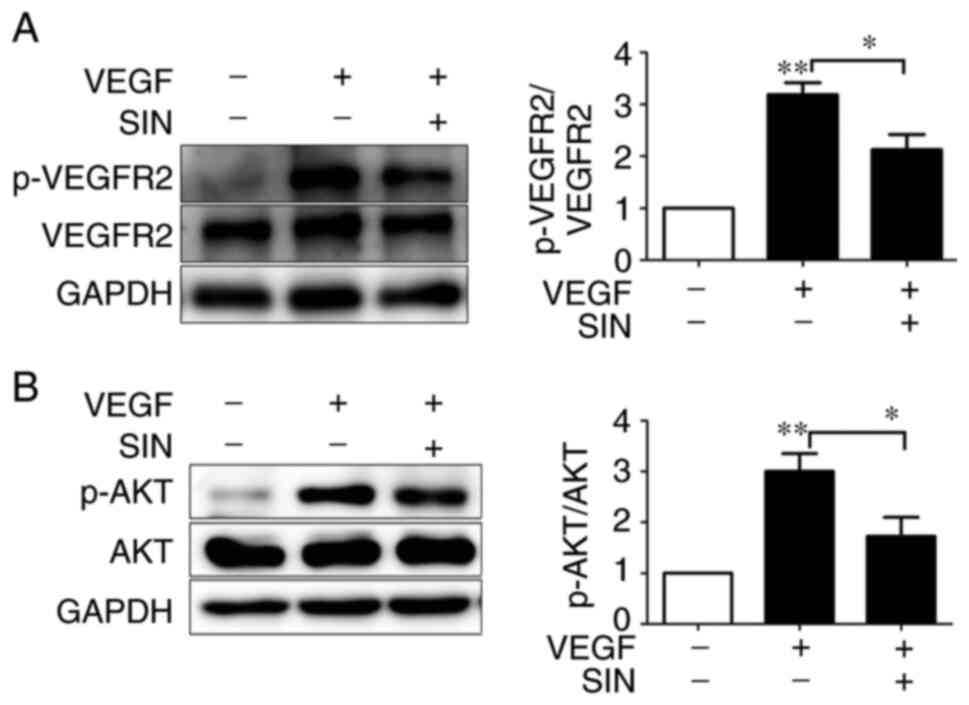
SIN inhibits phosphorylation of VEGFR2
and AKT signaling pathway
VEGF binds to VEGF receptor 2 on ECs and promotes
proliferation, migration and survival of ECs (33). To explicit the effect of SIN on
VEGFR2 protein expression, HUVECs were treated with SIN containing
exogenous VEGF or not for 30 min and then western blotting
performed. p-AKT, as a downstream signaling molecule of p-VEGFR2,
was also observed by western blotting. Phosphorylation levels of
VEGFR2 and AKT were significantly increased under VEGF treatment
(both P<0.01), while they were decreased in SIN-treated HUVEC,
even with VEGF (both P<0.05; Fig.
4A and B).
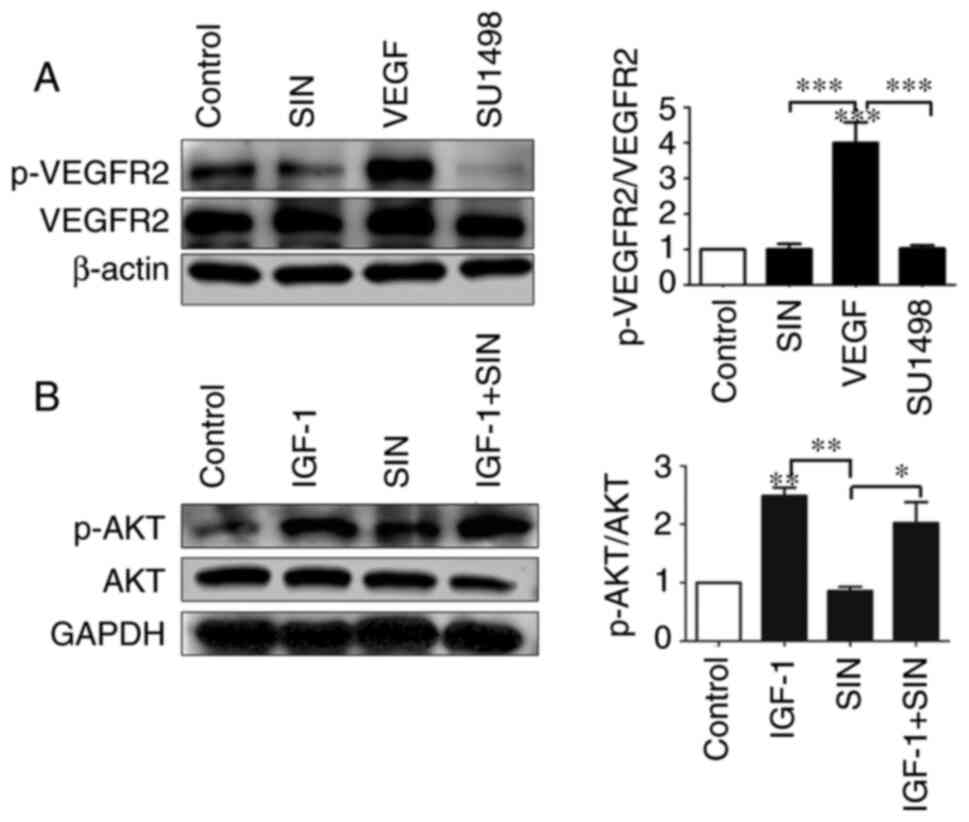
To further demonstrate the anti-angiogenesis
mechanism of SIN, whether it depended on p-VEGFR2, p-AKT, or both,
VEGF and SU1498 were used in a serum-free medium to treat HUVECs.
As a selective inhibitor of VEGFR2, SU1498 was used as a positive
control (34). Immunoblot analyses
revealed that phosphorylation of VEGFR2 was inhibited by SIN
similarly to SU1498 (Fig. 5A).
IGF-1, which is an activator of PI3K/AKT pathway, was used as a
negative control (35). AKT is a
downstream target protein of VEGFR2(36). After 90 min exposure to IGF (10
ng/ml), AKT phosphorylation in HUVECs significantly increased, but
it could not be reversed by SIN (Fig.
5B). The above results suggested that SIN mainly acted on
VEGFR2 to inhibit angiogenesis rather than acted directly on
AKT.
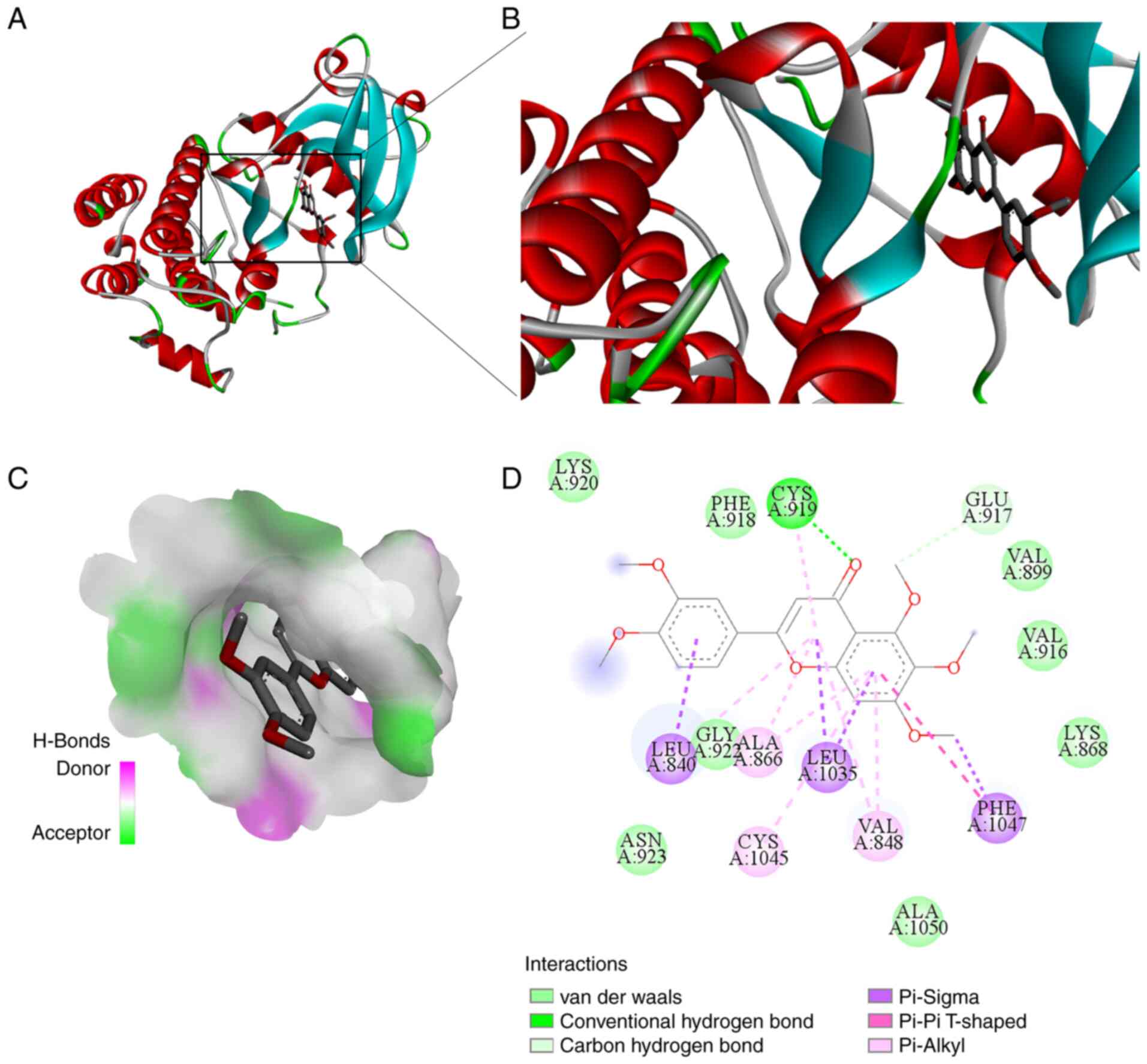
Molecule docking analysis
As one of the best theoretical methods, molecular
docking has traditionally been employed to study binding affinities
between target proteins and virtually screened ligands (37). To more effectively mimic the
internal environment, molecular docking analysis was performed
using VEGFR2 (PDB ID: 3VHE) to determine the interactions between
SIN and VEGFR2(38). Molecular
docking studies revealed that SIN occupied the active site, as
displayed in Fig. 6A-C. SIN was
surrounded by some hydrophobic residues (PHE918, VAL899, VAL916,
LEU840, GLY922, ALA866, LEU1035, PHE1047, VAL848 and ALA1050) and
some polar residues (CYS919, GLU917, LYS868, ASN923, CYS1045 and
LYS920), as depicted in Fig. 6D.
The hydrophobic force may contribute to the interactions between
SIN and VEGFR2. The hydrogen bond interactions between SIN and CYS
919 residue might decrease the hydrophilicity while increasing the
hydrophobicity of VEGFR 2. As demonstrated in Fig. 6D, the residue GLU917held SIN at the
active site through carbon-hydrogen bonds. The residue PHE1047
formed a π-π T-shaped interaction, ALA866, CYS1045 and VAL848
combined with SIN through π-alkyl interactions. Furthermore, the
residues LEU840, LEU1035 and PHE1047 held SIN at the binding pocket
via π-Sigma interactions. Additionally, the residues LYS920,
PHE918, VAL899, VAL916, LYS868, GLY922, ASN923 and ALA1050 formed
van der Waals forces between the pocket and SIN. These results
indicate that SIN inactivates the VEGF/VEGFR2 axis, resulting in
decreased AKT phosphorylation downstream and participating in its
anti-angiogenesis effect (Fig.
7).
Discussion
Chinese herbs are an essential part of Traditional
Chinese Medicine and contribute to liver cancer management in China
(39). Fructus Aurantii
Immaturus is frequently used in Traditional Chinese Medicine
and is rich in flavonoids that exhibit multiple biological effects,
including anticancer activity in human liver cancer (40). SIN is a methoxyflavone commonly
found in citrus species, exhibiting an anticancer effect on liver
cancer (10,41). Tumor growth depends on angiogenesis
to provide nutrition and oxygen to tumor cells (42). Although SIN has been shown to
promote autophagy-related hepatocellular carcinoma cell death, the
impact of this compound on angiogenesis of liver cancer has not
been confirmed.
In the present study, an in vivo animal model
study was used to indicate that SIN restrained HepG2/C3A-derived
tumor growth. A previous study (43) demonstrated that CD31 is enriched at
the EC intercellular junctions; this receptor is considered a
neoangiogenesis marker. IHC staining and western blot analysis of
CD31 revealed that SIN inhibited CD31 expression in tumor tissues,
suggesting that this compound exhibited an antiangiogenic effect in
liver cancer in vivo. Western blot analysis indicated that
SIN reduced VEGF secretion in tumor cells; this may be the
mechanism responsible for the suppression of tumor angiogenesis
caused by SIN.
Angiogenesis requires EC proliferation and migration
to appropriate positions leading to their assembly in vascular
structures (44). Therefore, the
present study investigated the antiangiogenic effect of SIN in
vitro. SIN exhibited significant cytotoxicity to HUVECs even in
the presence of VEGF, which was consistent with the study of Lam
et al (45). VEGF is a
potent regulator of angiogenesis that inhibits the induction of EC
apoptosis in new blood vessels (46). Subsequently, the induction of
apoptosis by SIN was evaluated. Flow cytometry results demonstrated
that induction of HUVEC apoptosis was maintained at a low level in
the presence of VEGF compared with that of the normal group,
whereas it was increased following treatment of the cells with SIN.
Angiogenesis relies on EC destabilization, dissociation and
migration. The present study revealed that SIN may delay HUVEC
migration, as determined by the wound healing results. The results
of the tube formation assays indicated that SIN could inhibit the
angiogenic activity of ECs induced by VEGF. Based on these results,
SIN was shown to inhibit angiogenesis, albeit with an unknown
mechanism.
Angiogenesis is necessary for tumor development and
provides oxygen and nutrients to tumor cells (47). Circulating VEGF levels are
increased in hepatocellular carcinoma and are linked to tumor
angiogenesis and progression (48). VEGF is a mitogen of ECs that
promotes their proliferation and migration into tumors required for
the formation of new capillaries (49). Western blot analysis revealed that
HepG2/C3A-derived VEGF was reduced by SIN. Due to the rapid growth
of tumor cells leading to hypoxia, the expression levels of HIF
increase, causing VEGF upregulation and promoting angiogenesis
(29). SIN treatment was
accompanied by reduced protein levels of HIF-1α, which may mediate
VEGF downregulation induced by SIN. To further account for these
phenomena, a tube formation experiment was used to assess whether
SIN inhibits angiogenesis via the HIF-1α-VEGF pathway.
VEGF binds to VEGFR2 on the EC membrane and
initiates angiogenesis (33).
Subsequently, VEGFR2 is phosphorylated and activates the downstream
intracellular pathway, which promotes the proliferation, migration
and survival of ECs (50). The
present study demonstrated that SIN downregulated VEGFR2
phosphorylation. The data indicated that VEGFR2 phosphorylation in
HUVECs was increased following addition of VEGF for 30 min;
however, this increase was eliminated with the addition of SIN.
When SIN was replaced by SU1498, a specific inhibitor of
VEGFR2(51), the same results were
observed. In VEGF-induced conditions, SIN specifically suppressed
the phosphorylation of VEGFR2, while this effect was not noted in
the SIN group in the absence of VEGF. This may explain why the
SIN-treated group did not differ significantly from the control
group with regard to tube formation and migratory activity.
VEGFR2 phosphorylation activates downstream
signaling pathways, such as MAPK/ERK and PI3K/AKT (36). Western blot analysis indicated that
the phosphorylation levels of ERK in HUVECs did not change
significantly following SIN treatment, suggesting that SIN-induced
inhibition of angiogenesis was independent of the MAPK/ERK pathway.
However, SIN downregulated the phosphorylation levels of AKT in
VEGF-treated cells. To further confirm that inhibition of
SIN-induced angiogenesis relies on the VEGFR2/AKT pathway, IGF-1,
which is an activator of the PI3K/AKT pathway, was added prior to
SIN treatment. Western blot analysis demonstrated that SIN did not
reverse IGF-1-induced phosphorylation of AKT. Furthermore, the
binding of SIN to VEGFR2 was simulated using molecular docking. The
molecular docking data suggested that SIN directly interacted with
VEGFR2 by various important residues, confirming that this compound
could directly bind to VEGFR2 and produce antiangiogenic effects.
Therefore, the VEGF/VEGFR2/AKT pathway may be the mechanism by
which SIN inhibits liver cancer angiogenesis.
The current study contains certain limitations.
Although it was shown that the expression levels of the HIF-1α
protein were downregulated by SIN, the detailed mechanism of this
process remains unclear. The translation of the HIF-1α protein is
considered to be an important regulatory mechanism of
HIF-1α-inhibiting compounds (52).
Therefore, further studies are required to explore the mechanisms
that are related to the protein translation of HIF-1α.
In summary, the present study demonstrated that SIN
exhibited a significant antiangiogenic effect by inhibiting the
viability of ECs and inducing apoptosis. Concomitantly, SIN
suppressed angiogenesis by inhibiting the migratory activity and
tube formation in HUVECs. SIN potently inhibited angiogenesis in
vitro by eliciting the blockade of VEGF expression in
HepG2/C3A-derived tumors and the inhibition of the VEGFR2/AKT
signaling pathway in ECs. The results of the present study may be
necessary for developing strategies that aim to prevent liver
cancer with SIN.
Acknowledgements
Not applicable.
Funding
Funding: The present study was financed in part by the
Traditional Chinese Medicine Science and Technology Development
Plan Project of Shandong Province, China (grant no. 2019-0370), the
Natural Science Foundation of Shandong Province (grant nos.
ZR2019BH007 and ZR2019MH128), Youth Science and Technology
Innovation Team Projects of Higher Learning Institutions of
Shandong Province, China (grant no. 2019KJK013) and Youth research
and innovation team of Shandong University of Traditional Chinese
Medicine, China (grant no. 17).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JLiu designed the experiments. XL, YLi and YWa
performed the experiments and prepared the article. FL, YLiu, YWu,
HR and XZ performed the animal experiments and analysis. JLia and
RZ participated in data analysis discussions. XL and JLiu confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal experiments and experimental protocols
were approved by the Ethics Committee of Qianfoshan Hospital
(Jinan, China; approval no. 2017-106; approval date: 14 December
2017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Carmeliet P and Jain RK: Principles and
mechanisms of vessel normalization for cancer and other angiogenic
diseases. Nat Rev Drug Discov. 10:417–427. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Xue C, Shao S, Yan Y, Yang S, Bai S, Wu Y,
Zhang J, Liu R, Ma H, Chai L, et al: Association between G-protein
coupled receptor 4 expression and microvessel density,
clinicopathological characteristics and survival in hepatocellular
carcinoma. Oncol Lett. 19:2609–2620. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Welker MW and Trojan J: Anti-angiogenesis
in hepatocellular carcinoma treatment: Current evidence and future
perspectives. World J Gastroenterol. 17:3075–3081. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Edeline J, Boucher E, Rolland Y, Vauléon
E, Pracht M, Perrin C, Le Roux C and Raoul JL: Comparison of tumor
response by response evaluation criteria in solid tumors (RECIST)
and modified RECIST in patients treated with sorafenib for
hepatocellular carcinoma. Cancer. 118:147–156. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhang H, Tian G, Zhao C, Han Y,
DiMarco-Crook C, Lu C, Bao Y, Li C, Xiao H and Zheng J:
Characterization of polymethoxyflavone demethylation during drying
processes of citrus peels. Food Funct. 10:5707–5717.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Huang B, Zhai M, Qin A, Wu J, Jiang X and
Qiao Z: Sinensetin flavone exhibits potent anticancer activity
against drug-resistant human gallbladder adenocarcinoma cells by
targeting PTEN/PI3K/AKT signalling pathway, induces cellular
apoptosis and inhibits cell migration and invasion. J BUON.
25:1251–1256. 2020.PubMed/NCBI
|
|
8
|
Tan KT, Lin MX, Lin SC, Tung YT, Lin SH
and Lin CC: Sinensetin induces apoptosis and autophagy in the
treatment of human T-cell lymphoma. Anticancer Drugs. 30:485–494.
2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Dong Y, Ji G, Cao A, Shi J, Shi H, Xie J
and Wu D: Effects of sinensetin on proliferation and apoptosis of
human gastric cancer AGS cells. Zhongguo Zhong Yao Za Zhi.
36:790–794. 2011.PubMed/NCBI(In Chinese).
|
|
10
|
Kim SM, Ha SE, Lee HJ, Rampogu S, Vetrivel
P, Kim HH, Venkatarame Gowda Saralamma V, Lee KW and Kim GS:
Sinensetin induces autophagic cell death through p53-related
AMPK/mTOR signaling in hepatocellular carcinoma HepG2 cells.
Nutrients. 12(2462)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Stevens M and Oltean S: Modulation of
receptor tyrosine kinase activity through alternative splicing of
ligands and receptors in the VEGF-A/VEGFR axis. Cells.
8(288)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Guo D, Wang Q, Li C, Wang Y and Chen X:
VEGF stimulated the angiogenesis by promoting the mitochondrial
functions. Oncotarget. 8:77020–77027. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yan ZX, Luo Y and Liu NF: Blockade of
angiopoietin-2/Tie2 signaling pathway specifically promotes
inflammation-induced angiogenesis in mouse cornea. Int J
Ophthalmol. 10:1187–1194. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Al-Aqtash RA, Zihlif MA, Hammad H, Nassar
ZD, Meliti JA and Taha MO: Ligand-based computational modelling of
platelet-derived growth factor beta receptor leading to new
angiogenesis inhibitory leads. Comput Biol Chem. 71:170–179.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shibuya M: Vascular endothelial growth
factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A
crucial target for anti- and pro-angiogenic therapies. Genes
Cancer. 2:1097–1105. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li S, Xu G, Gao F, Bi J and Huo R:
Expression and association of VEGF-Notch pathways in infantile
hemangiomas. Exp Ther Med. 14:3737–3743. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dulloo I, Phang BH, Othman R, Tan SY,
Vijayaraghavan A, Goh LK, Martin-Lopez M, Marques MM, Li CW, Wang
de Y, et al: Hypoxia-inducible TAp73 supports tumorigenesis by
regulating the angiogenic transcriptome. Nat Cell Biol. 17:511–523.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Schlieve CR, Mojica SG, Holoyda KA, Hou X,
Fowler KL and Grikscheit TC: Vascular endothelial growth factor
(VEGF) bioavailability regulates angiogenesis and intestinal stem
and progenitor cell proliferation during postnatal small intestinal
development. PLoS One. 11(e0151396)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nicolas S, Abdellatef S, Haddad MA,
Fakhoury I and El-Sibai M: Hypoxia and EGF stimulation regulate
VEGF expression in human glioblastoma multiforme (GBM) cells by
differential regulation of the PI3K/Rho-GTPase and MAPK pathways.
Cells. 8(1397)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tang Q, He X, Liao H, He L, Wang Y, Zhou
D, Ye S and Chen Q: Ultrasound microbubble contrast agent-mediated
suicide gene transfection in the treatment of hepatic cancer. Oncol
Lett. 4:970–972. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xiong YJ, Deng ZB, Liu JN, Qiu JJ, Guo L,
Feng PP, Sui JR, Chen DP and Guo HS: Enhancement of epithelial cell
autophagy induced by sinensetin alleviates epithelial barrier
dysfunction in colitis. Pharmacol Res. 148(104461)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liu F, Wang B, Li L, Dong F, Chen X, Li Y,
Dong X, Wada Y, Kapron CM and Liu J: Low-dose cadmium upregulates
VEGF expression in lung adenocarcinoma cells. Int J Environ Res
Public Health. 12:10508–10521. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang R, Liu Y, Hu X, Pan J, Gong D and
Zhang G: New insights into the binding mechanism between osthole
and β-lactoglobulin: Spectroscopic, chemometrics and docking
studies. Food Res Int. 120:226–234. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cao X, Wang S, Bi R, Tian S, Huo Y and Liu
J: Toxic effects of Cr(VI) on the bovine hemoglobin and human
vascular endothelial cells: Molecular interaction and cell damage.
Chemosphere. 222:355–363. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Okamura S, Osaki T, Nishimura K, Ohsaki H,
Shintani M, Matsuoka H, Maeda K, Shiogama K, Itoh T and Kamoshida
S: Thymidine kinase-1/CD31 double immunostaining for identifying
activated tumor vessels. Biotech Histochem. 94:60–64.
2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Holmqvist K, Cross MJ, Rolny C, Hägerkvist
R, Rahimi N, Matsumoto T, Claesson-Welsh L and Welsh M: The adaptor
protein shb binds to tyrosine 1175 in vascular endothelial growth
factor (VEGF) receptor-2 and regulates VEGF-dependent cellular
migration. J Biol Chem. 279:22267–22275. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Farhat FS, Tfayli A, Fakhruddin N, Mahfouz
R, Otrock ZK, Alameddine RS, Awada AH and Shamseddine A:
Expression, prognostic and predictive impact of VEGF and bFGF in
non-small cell lung cancer. Crit Rev Oncol Hematol. 84:149–160.
2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Semenza GL, Agani F, Booth G, Forsythe J,
Iyer N, Jiang BH, Leung S, Roe R, Wiener C and Yu A: Structural and
functional analysis of hypoxia-inducible factor 1. Kidney Int.
51:553–555. 1997.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ye J and Yuan L: Inhibition of p38 MAPK
reduces tumor conditioned medium-induced angiogenesis in
co-cultured human umbilical vein endothelial cells and fibroblasts.
Biosci Biotechno lBiochem. 71:1162–1169. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu J, Yuan L, Molema G, Regan E, Janes L,
Beeler D, Spokes KC, Okada Y, Minami T, Oettgen P and Aird WC:
Vascular bed-specific regulation of the von Willebrand factor
promoter in the heart and skeletal muscle. Blood. 117:342–351.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shibuya M: Structure and function of
VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct
Funct. 26:25–35. 2001.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Shu-Ya T, Qiu-Yang Z, Jing-Jing L, Jin Y
and Biao Y: Suppression of pathological ocular neovascularization
by a small molecule, SU1498. Biomed Pharmacother.
128(110248)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen S, Hu M, Shen M, Wang S, Wang C, Chen
F, Tang Y, Wang X, Zeng H, Chen M, et al: IGF-1 facilitates
thrombopoiesis primarily through Akt activation. Blood.
132:210–222. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huang X, Zhou G, Wu W, Ma G, D'Amore PA,
Mukai S and Lei H: Editing VEGFR2 blocks VEGF-induced activation of
Akt and tube formation. Invest Ophthalmol Vis Sci. 58:1228–1236.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hosseini-Koupaei M, Shareghi B, Saboury AA
and Davar F: Molecular investigation on the interaction of spermine
with proteinase K by multispectroscopic techniques and molecular
simulation studies. Int J Biol Macromol. 94:406–414.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu D, Cao X, Kong Y, Mu T and Liu J:
Inhibitory mechanism of sinensetin on α-glucosidase and
non-enzymatic glycation: Insights from spectroscopy and molecular
docking analyses. Int J Biol Macromol. 166:259–267. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hu B, An HM, Wang SS, Chen JJ and Xu L:
Preventive and therapeutic effects of Chinese herbal compounds
against hepatocellular carcinoma. Molecules. 21(142)2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liao CY, Lee CC, Tsai CC, Hsueh CW, Wang
CC, Chen IH, Tsai MK, Liu MY, Hsieh AT, Su KJ, et al: Novel
investigations of flavonoids as chemopreventive agents for
hepatocellular carcinoma. Biomed Res Int.
2015(840542)2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kim SM, Rampogu S, Vetrivel P, Kulkarni
AM, Ha SE, Kim HH, Lee KW and Kim GS: Transcriptome analysis of
sinensetin-treated liver cancer cells guided by biological network
analysis. Oncol Lett. 21(355)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Figueiredo CC, Pereira NB, Pereira LX,
Oliveira LAM, Campos PP, Andrade SP and Moro L: Double
immunofluorescence labeling for CD31 and CD105 as a marker for
polyether polyurethane-induced angiogenesis in mice. Histol
Histopathol. 34:257–264. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Salvucci O, Maric D, Economopoulou M,
Sakakibara S, Merlin S, Follenzi A and Tosato G: EphrinB reverse
signaling contributes to endothelial and mural cell assembly into
vascular structures. Blood. 114:1707–1716. 2009.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lam IK, Alex D, Wang YH, Liu P, Liu AL, Du
GH and Lee SM: In vitro and in vivo structure and activity
relationship analysis of polymethoxylated flavonoids: identifying
sinensetin as a novel antiangiogenesis agent. Mol Nutr Food Res.
56:945–956. 2012.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Alon T, Hemo I, Itin A, Pe'er J, Stone J
and Keshet E: Vascular endothelial growth factor acts as a survival
factor for newly formed retinal vessels and has implications for
retinopathy of prematurity. Nat Med. 1:1024–1028. 1995.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hida K, Kawamoto T, Ohga N, Akiyama K,
Hida Y and Shindoh M: Altered angiogenesis in the tumor
microenvironment. Pathol Int. 61:630–637. 2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Poon RT, Fan ST and Wong J: Clinical
implications of circulating angiogenic factors in cancer patients.
J Clin Oncol. 19:1207–1225. 2001.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Rapisarda A and Melillo G: Role of the
VEGF/VEGFR axis in cancer biology and therapy. Adv Cancer Res.
114:237–267. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jászai J and Schmidt MHH: Trends and
challenges in tumor anti-angiogenic therapies. Cells.
8(1102)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Boguslawski G, McGlynn PW, Harvey KA and
Kovala AT: SU1498, an inhibitor of vascular endothelial growth
factor receptor 2, causes accumulation of phosphorylated ERK
kinases and inhibits their activity in vivo and in vitro. J Biol
Chem. 279:5716–5724. 2004.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang J, Cao J, Weng Q, Wu R, Yan Y, Jing
H, Zhu H, He Q and Yang B: Suppression of hypoxia-inducible factor
1α (HIF-1α) by tirapazamine is dependent on eIF2α phosphorylation
rather than the mTORC1/4E-BP1 pathway. PLoS One.
5(e13910)2010.PubMed/NCBI View Article : Google Scholar
|