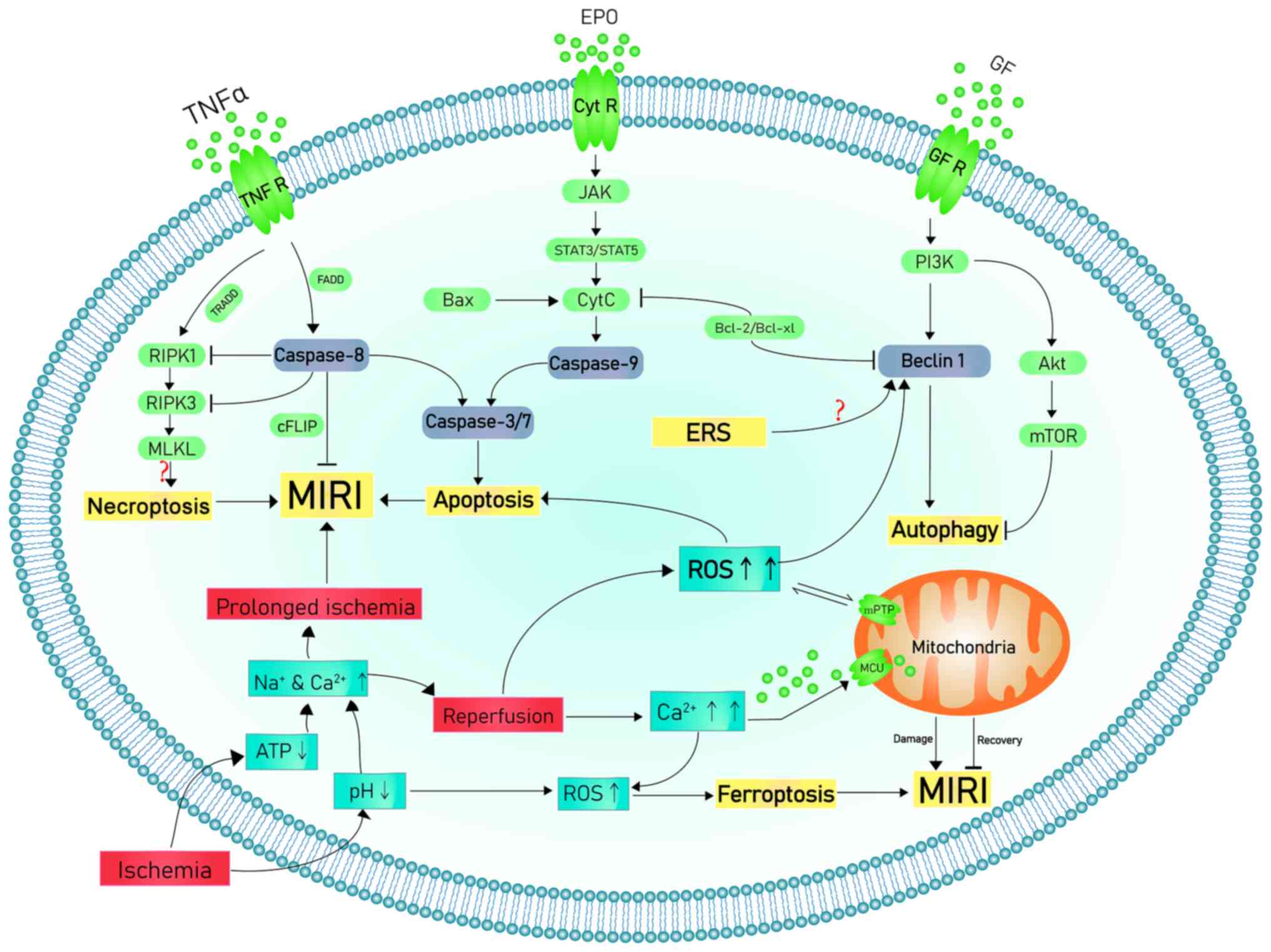
Coronary heart disease (CHD) is one of the primary
causes of mortality and disability worldwide. For instance,
according to the data obtained through the Unified Health System
database from the Ministry of Health of Brazil, the average
mortality rate for CHD increased to 78.75 during 2016-2018 in
Brazil (1). Timely restoration of
blood flow to ischemic myocardium in the early stage effectively
limits infarct size and is therefore standard treatment to prevent
death of cardiomyocytes at risk (2,3).
Evidence has indicated that during myocardial ischemia/reperfusion
(MIR), both ischemia and reperfusion cause injury to the ischemic
myocardium (4,5). Therefore, myocardial reperfusion may
further aggravate death of ischemic cardiomyocytes in patients with
myocardial infarction (MI); this is known as MIR injury (MIRI)
(3,4).
Numerous studies have researched mechanisms
underlying MIRI; the pathophysiological mechanisms of MIRI are
associated with oxidative stress, intracellular calcium overload,
energy metabolism disorder, apoptosis, endoplasmic reticulum stress
(ERS), autophagy, pyroptosis, ferroptosis and necroptosis (15,16).
Furthermore, these mechanisms are interrelated and may directly or
indirectly lead to the aggravation of cell death. The present
review summarizes research on the pathophysiological mechanisms
underlying MIRI.
During IR, especially reperfusion, ROS levels
increase due to multiple mechanisms such as increased xanthine
oxidase formation, neutrophil respiratory burst and damage of the
mitochondrial electron transport chain (24). The excessive ROS decrease membrane
fluidity, increase calcium permeability, aggravate intracellular
calcium overload and mitochondrial damage and contribute to release
of pro-apoptotic factors, such as cytochrome C (CytC) (25). In addition, oxidative reactions
between ROS and proteins cause loss of original protein structure
and function and damage of nucleic acids and chromosomes (19). In addition, ROS may trigger the
inflammatory cascade reaction and expression of adhesion molecules
that result in leukocyte aggregation, endothelial cell swelling and
the no-reflow phenomenon that refers to the incomplete and uneven
reperfusion at the microvascular level even though the proximal
artery has been re-opened after a period of ischemia (26). Oxidative stress induced by
reperfusion after ischemia is considered to be the primary
mechanism of IRI (20). Oxidative
stress is involved in ventricular remodeling by causing endothelial
dysfunction, myocardial cell injury, apoptosis and other
pathological changes that promote formation and development of
cardiomyopathy (27) and cause
cardiac dysfunction. Among potential sources of ROS, nicotinamide
adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase,
mitochondria and uncoupled nitric oxide synthase (NOS) are key
sources of reperfusion-induced oxidative stress (28). When ROS levels exceed the ability
of the cellular endogenous radical scavenging system to remove them
(29), uncontrolled ROS burst
damages the membrane and proteins and indirectly causes damage by
opening mitochondria permeability transition pore (mPTP) and
promoting activation of the apoptosis pathway. Thus, ROS
participate in myocardial injury and death through multiple
mechanisms (30).
Contraction and relaxation of myocytes are
controlled by balance of calcium. In the steady-state, calcium
influx and efflux are maintained in balance. Any increase in
calcium influx (for example, during β-adrenergic stimulation) is
met by a corresponding increase in calcium efflux (39,40).
Under normal conditions, calcium influx and efflux balance ensures
cells do not become overloaded with calcium. Sarcoplasmic reticulum
calcium content is increased when calcium influx exceeds efflux
until a new steady-state of influx and efflux is achieved (39).
After a certain period of hypoxia in cardiomyocytes,
the intracellular anaerobic metabolism may result in accumulation
of H+, decreased intracellular pH and increased levels
of intracellular Na+ via Na+/H+
exchange (NHE) (41). The
aggregation of Na+ in cardiomyocytes increases the
activity of Na+/Ca2+ exchange (NCX) proteins,
which decreases aggregation of intracellular Na+ and
transports extracellular Ca2+ into the cells, thus
increasing the concentration of Ca2+ in the cytoplasm
and causing calcium overload (42). Rapid reoxygenation increases the pH
of extracellular fluid and H+ gradient across the cell
membrane, so that the activity of NHE and NCX increases, causing
intracellular calcium overload (43). In addition to the aforementioned
NHE and NCX mechanisms, the H+/Ca2+ exchange
mechanism is also a key cause of intracellular calcium overload
(44).
Calcium processing and further inflow or outflow are
regulated by protein channels (45). Strict regulation of intracellular
calcium homeostasis is key for maintaining normal function and
growth of myocardial cells and disruption of intracellular calcium
homeostasis leads to ERS, aggravating ischemic injury of myocardial
cells (19). Chen et al
(46) indicated that diltiazem, a
calcium antagonist, has protective effects on ischemic myocardium,
which decreases oxidative stress, restores normal energy
metabolism, improves endothelial function and decreases apoptosis.
In addition, clinical trials have found that diltiazem exerts a
cardioprotective effect in patients who exhibit post-transplant
hypertension and vasospastic angina (47,48).
As reported in the aforementioned studies, diltiazem improves left
ventricular systolic function and coronary hemodynamics in patients
with myocardial ischemia and prevents reperfusion arrhythmia in
patients with ST segment elevation MI following treatment with PCI
(46).
Mitochondria, a key organelle responsible for
production of adenosine triphosphate (ATP) and energy metabolism in
cells, also contribute to normal physiological function of the
heart (49). Mitochondrial
integrity and loss of function in cardiomyocytes are considered to
be pathological factors underlying changes in cardiac structure and
function (49). By forming a
dynamic network, mitochondria continuously change shape by division
and fusion to meet functional needs of cardiomyocytes (50). Since cardiomyocytes consume lots of
energy, the density of mitochondria is relatively higher compared
with other types of cell (51).
The function of mitochondria is regulated by
numerous factors. Physiologically, mitochondrial Ca2+ is
an active effector, which triggers activation of mitochondrial
metabolic mechanisms that increase production of ATP. However,
during IR, cytoplasmic calcium overload leads to mitochondrial
calcium overload (52).
Ca2+ enter mitochondria primarily via the mitochondrial
calcium uniporter, a type of small conductance Ca2+
selective channel that regulates intracellular Ca2+,
production of ATP and cell death (52). The movement of Ca2+
across the mitochondrial membrane leads to amplification of
mitochondrial Ca2+ overload, causing mitochondrial
dysfunction (53). Overload of
mitochondrial Ca2+ dissipates membrane potential,
promotes opening of mPTP, a non-selective channel in the inner
mitochondrial membrane that is considered to be one of the key
participants in IRI, and leads to impaired ATP synthesis (54). The inner mitochondrial membrane is
normally impermeable to ions and proteins and is responsible for
maintaining formation of mitochondrial transmembrane potential
pores, creating non-selective channels and causing potential to
dissipate across the membrane. Our previous study found that the
degree of mPTP opening degree corresponds to change of
mitochondrial morphology, which indicates that mitochondrial
function is associated with its morphology during MIRI (50).
Disturbance of mitochondrial homeostasis contributes
to acute organ failure and cell damage following ischemia (22). The opening and closing of mPTP is
the primary mechanism for maintaining mitochondrial membrane
potential; ischemia induces mPTP closure whereas reperfusion
promotes mPTP opening (54).
During acute IR, mPTP remains closed during ischemia and opens due
to mitochondrial calcium and phosphate overload, oxidative stress
and rapid pH correction during reperfusion (55,56).
ROS levels increase in a short period of time due to IRI, resulting
in excessive cytoplasmic and mitochondrial Ca2+, which
induces opening of mPTP and depolarizes the mitochondrial membrane
potential (54). The opening of
mPTP may contribute to loss of ATP, mitochondrial swelling and CytC
release, leading to apoptosis (57). Our previous study demonstrated that
regulation of mitochondrial morphology, inhibition of mPTP opening
and maintenance of mitochondrial membrane potential protect the
myocardium against MIRI; the underlying mechanism involves
regulation of mitofusin 2 (Mfn2), optic atrophy 1 (Opa1) and
dynamin-related protein 1 (Drp1) proteins, which provides a
theoretical basis for targeted therapy (50).
Apoptosis is a form of regulated cell death that
leads to cell contraction, condensation of cytoplasm and nucleus
and formation of apoptotic bodies (58). Cytoskeletal and nuclear proteins
are degraded and cleaved by caspases during the execution phase of
apoptosis (59). Finally,
apoptotic bodies are phagocytosed by macrophages that recognize
externalized phosphatidylserine on the outer leaflet of the bilayer
(60). Because apoptotic bodies
are surrounded by cell membranes and are swallowed and digested by
phagocytes such as neutrophils, macrophages, and dendritic cells
(DCs), they eventually degrade without causing inflammation
(61). However, part of the cell
structures (e.g. cell membrane) can be preserved till the late
stage of apoptosis (62).
There are three classic apoptosis signaling
pathways: Extrinsic (death receptor), intrinsic (mitochondrial) and
ERS pathway (63).
Extrinsic pathway-induced apoptosis is triggered by
transmembrane death receptors, which are members of tumor necrosis
factor receptor (TNFR) family containing the ‘death domain’.
Through the death domain, certain ligands and associated death
receptors, including apoptosis-stimulating fragment ligand
(FasL)/FasR, TNF-α/TNFR1, TNF-related apoptosis-inducing ligand
(TRAIL)/death receptors and TRAIL/DR5, facilitate transmission of
death signals from cell surface to intracellular pathways (63). For example, binding of FasL to FasR
or TNF-α to TNFR1 causes recruitment of Fas-associated death domain
(FADD) and binding to the ligand-receptor complex (64). FADD activates pro-caspase-8
consecutively and forms a death-inducing signaling complex (DISC)
(65). DISC-activated caspase-8
leads to activation of caspase-3 and -7 to induce the apoptosis
cascade response, which results in the execution phase of apoptosis
(66,67). During of MIRI, ischemia and
reperfusion increases levels of ROS, cellular injury and
cardiovascular dysfunction, which leads to cardiomyocyte apoptosis.
Upregulation of cardiac-specific caspase-3 increases post-ischemic
infarct size and risk of mortality following MIRI (68). Furthermore, the Fas pathway serves
as a critical mediator of the cardiomyocyte apoptosis caused by
MIRI (69). In addition, TNF-α and
TRAIL levels increase during onset of reperfusion in mouse models
of MIRI (70).
The intrinsic pathway, which is activated by
hypoxia, hyperthermia and low levels of growth factors (e.g. nerve
growth factor), is triggered by mitochondria and contributes to
apoptosis. The aforementioned stimuli promote opening of mPTP and
block mitochondrial transmembrane potential, thereby increasing
release of pro-apoptotic proteins (e.g. Bax) involved in CytC and
apoptosis from the intermembrane to the cytoplasm (71). By binding and activating caspase-9,
CytC promotes formation of caspase-3 and -7. These events induced
by mitochondria are regulated by B-cell lymphoma 2 (Bcl-2) family
proteins located in the mitochondrial outer membrane, which control
the permeability of the mitochondrial membrane and regulate release
of CytC. Bcl-2 family proteins are functionally divided into pro-
and anti-apoptotic proteins (67).
Bcl-2 family proteins lead to downregulation of apoptosis mediated
by upregulation of tumor suppressor protein p53 in the nucleus or
mitochondria. When expression of p53 is inhibited, Bax and
Caspase-3 are downregulated in heart tissue in CHD (72-74).
There is also an interaction between the extrinsic and intrinsic
pathways (75). In the intrinsic
apoptosis pathway, Bax is overexpressed in ischemic myocardial
tissue and inhibition of Bax activation decreases apoptosis and
improves MIRI (76). Furthermore,
cardiac-specific overexpression of Bcl-2 markedly relieves
cardiomyocyte apoptosis and infarct size following MIRI (77).
Previous studies have indicated that apoptotic cell
death is one of the primary forms of cardiomyocyte death during
MIRI (78-84).
ER is a key site for protein synthesis, modification
and processing and its normal function serves an important role in
maintaining cellular homeostasis (85). However, adverse stimuli such as
ischemia and hypoxia lead to accumulation or misfolding of proteins
in the ER. The accumulation of unfolded protein simultaneously
activates three transmembrane stress sensors, inositol-requiring
enzyme 1 (IRE1), activating transcription factor (ATF) 6 and PERK,
known as the unfolded protein response (UPR) (86). ERS is a relatively novel pathway
regulating apoptosis that is involved in multiple physiological
functions and pathological injury, including protein folding,
intracellular Ca2+ storage, oxidative stress, hypoxia,
ischemia and lipid metabolism disorder (87). Although ERS is key for cell
survival, chronic ERS leads to apoptosis.
ER and mitochondria are physically connected to form
dedicated structural domains known as mitochondria-associated ER
membranes (MAMs), which participate in key biological processes,
including lipid and Ca2+ homeostasis, mitochondrial
dynamics and associated cellular behavior, such as autophagy, ERS
and apoptosis. Gao et al (94) demonstrated the role of MAMs in
maintaining normal function of both ER and mitochondria, which are
associated with occurrence of cardiovascular disease. Another study
also suggested that GSK3β serves an important role in controlling
Ca2+ flow from the sarcoplasmic reticulum to
mitochondria via MAMs during MIRI (95).
Changes in ER oxidation lead to aberrant formation
of disulfide bonds and accumulation of peptides, thus activating
intracellular reactions called unfolded protein response (96), a form of acute response in
cardiovascular disease (97).
Stimulation of UPR leads to three primary response mechanisms,
IRE1α, PERK and ATF6, which regulate the protein folding ability of
ER (98). In the absence of PERK,
endogenous apoptosis induced by ERS is weakened due to decreased
MAM formation and inhibited ROS signal transmission to adjacent
mitochondria (99). IRE1 in MAMs
promotes effectiveness of inositol 1,4,5-trisphosphate receptor,
which is responsible for transfer of Ca2+ to
mitochondria (100). These
mechanisms connect ERS and mitochondrial function, thus affecting
the fate of cells.
Autophagy is a phenomenon in which cells digest
cytoplasm in lysosomes and is key to maintaining normal structure
and function of the heart. Autophagy, the primary function of which
is to remove and recover misfolded or damaged proteins and
organelles, is not only associated with cell survival but also with
cell death (101-103).
Apoptosis and autophagy are both adaptive responses that are key
for cell growth, survival and homeostasis (104). Specifically, apoptosis is type 1
programmed cell death, which involves early degradation of
cytoskeleton but preservation of organelles until the late phase,
whereas autophagy is type 2 cell death and involves early
degradation of organelles but preservation of cytoskeleton until
the late stage (105). However,
it has been reported that activation of autophagy does not
contribute to cell death in MIRI and may serve a protective role
(106). A study has indicated
that activation of autophagy via chloramphenicol therapy decreases
infarct size in a pig model of MIRI (106).
During myocardial ischemia, autophagy degrades
non-functional cytoplasmic proteins and provides key nutrients for
cell growth and survival, thereby inhibiting apoptosis and
necrosis. A recent study suggested that autophagy is necessary to
decrease myocardial damage following acute myocardial ischemia and
that autophagy limits activation of the NLR family pyrin
domain-containing 3 (NLRP3) inflammasome by removing damaged
mitochondria (43). However, it is
also reported that excessive autophagy during reperfusion may
aggravate the injury of the heart (107,108).
Currently, three forms of autophagy are known:
Macroautophagy, microautophagy and chaperone-mediated autophagy
(CMA) (109). In macroautophagy,
the primary sources of membranes are ER and Golgi bodies.
Autophagosomes are formed by completely including the object to be
degraded and fusing it with the lysosomal membrane before
degradation by lysosomal enzymes (110). In microautophagy, degradation
products are directly encapsulated in lysosome membranes for
degradation and digestion in lysosomes (111). In CMA, chaperone proteins are
bind to the protein to be degraded to guide transport of the
substance to lysosomes, which are digested and broken down by
enzymes. CMA pathway is a lysosomal process with obvious
selectivity, which is different from the aforementioned processes
(112).
The molecular mechanism of autophagy is primarily
composed of four pathways that involve mTOR complex 1,
AMP-activated protein kinase (AMPK), ERS and p53. mTOR and Beclin1
are autophagy-associated molecules that serve key roles in
different stages of MIRI. During the ischemic phase, mTOR acts via
AMPK/mTOR and PI3K/AKT/mTOR pathways (78), while Beclin1 is upregulated during
reperfusion (43,101,102). Beclin1 is a key autophagy protein
that regulates formation and processing of autophagosomes.
Upregulation of Beclin1 is responsible for autophagy activation
during reperfusion (113).
However, it is unclear how MIRI activates Beclin1. One potential
mechanism is its association with Bcl-2. In vitro study have
shown that Beclin1-mediated autophagy to nutritional deficiency
(e.g. a lack of amino acids and serum) of cardiomyocytes is
regulated by Bcl-2 protein (114). Moreover, ROS may also induce
Beclin1-mediated autophagy during reperfusion. During reperfusion,
high levels of ROS are not an energy crisis, but a key promoter of
autophagy. Reperfusion results in increased oxidative stress with
overexpression of Beclin1 (115,116). In addition to regulating
expression of Beclin1, ROS may also oxidize and decrease activity
of autophagy-associated proteins, leading to lipidation of light
chain 3 (LC3) as well as initiation of autophagy. As Beclin1 is
primary located in ER (117), it
remains to be determined whether reperfusion-induced ERS also
participates in upregulation of Beclin1.
Pyroptosis is a novel type of proinflammatory
programmed cell death that is caused by activation of the
NLRP3/apoptosis-associated speck-like protein (ASC)/caspase-1
pathway and high levels of interleukin-1β (IL-1β) (118). The primary biological features of
pyroptosis are dependence on caspase-1 and the accompanying
inflammatory cascade reaction. Under endogenous and exogenous
stimuli, ASC acts on pro-caspase-1 to form inflammasome and
activate pro-caspase-1(119). The
activated caspase-1 promotes activation and expression of
downstream cytokines such as IL-1β and IL-18, resulting in cell and
tissue damage (119). It is also
a key host mechanism in response to pathogens and endogenous damage
(119,120).
Compared with other modes of cell death, pyroptosis
has unique features, including activation by intracellular
inflammatory caspase, formation of membrane pores and DNA
fragmentation/destruction (67).
Pyroptosis leads to release of inflammatory cytokines and exhibits
certain characteristics of apoptosis (121) (including DNA fragmentation and
nuclear concentration) and necrosis (122) (such as loss of plasma membrane
integrity and release of intracellular content such as lactate
dehydrogenase).
Cell membrane rupture during pyroptosis is mediated
by Gasdermin D protein (GSDMD) (67). In healthy cells, GSDMD stays
functionally inactive via intramolecular interactions between its
COOH-(inhibitory) and NH2-terminal (pro-death) domains.
Hydrolysis of inflammatory caspase-1 to GSDMD produces
NH2-terminal lysate (GSDMD-NT) (123). GSDMD-NT monomers oligomerize to
form pores in the plasma membrane, leading to release of
intracellular material and contributing to cell death; however, the
underlying mechanism is unknown. GSDMD is also released in
pyroptosis without damaging adjacent cells (124). As a novel form of cell death,
pyroptosis is associated with development of the mouse model of
MIRI (125,126).
ROS produced by mitochondria are hypothesized to
serve as a signal for activation of NLRP3 inflammation downstream
of MIRI-associated mitochondrial dysfunction (127). In addition, inflammasomes are
activated by ROS production and potassium efflux with myocardial
ischemia and hypoxia, resulting in activation of caspase-1 and
cleavage of GSDMD, which aggravates MIRI (128). A study has indicated that
silencing of calpain attenuates myocardial dysfunction caused by
MIRI by suppressing activation of the NLRP3/ASC/caspase-1 axis
(125). Furthermore, inhibition
of microRNA-29a improves MIRI by targeting silent information
regulator factor 2-related enzyme 1 via suppressing oxidative
stress and NLRP3-mediated pyroptosis (129). Numerous studies have demonstrated
that pyroptosis is a key mechanism in exacerbating MIRI (130,131).
Ferroptosis, a novel and unique iron-dependent
non-apoptosis-regulated form of cell death; it is also called
iron-dependent programmed cell death due to the destruction of
glutathione-dependent antioxidant defense mechanism and
accumulation of lipid peroxides, which are caused by the increased
ROS level related to Fe2+ (132,133). Ferroptosis is characterized by
the depletion of plasma membrane polyunsaturated fatty acids,
condensed mitochondrial membrane densities, and vanishing of
mitochondria crista (126).
Studies have shown that ferroptosis is involved in regulation of
tumor (133), liver cancer
(134), Alzheimer's disease
(135), cerebral ischemic
(136) and acute kidney injury
(137) and other types of
disease.
Ferroptosis is dependent on intracellular iron and
is morphologically, biochemically and genetically distinct from
apoptosis, necrosis and autophagy (138). Iron participates in oxidative
phosphorylation of mitochondria, thereby increasing ROS levels and
ATP production (139). ROS levels
that exceed cell antioxidant capacity lead to oxidative stress,
which directly or indirectly damages large molecules such as
proteins, nucleic acids and lipids, leading to cell damage and
death (139-141).
The ferric carrier protein transferrin and amino acid glutamine are
hypothesized to be responsible for ferroptosis (142). The cell surface transferrin
receptor and glutamine-mediated intracellular metabolic pathway
glutamine breakdown serve critical roles in cell death (143). Almost all genes involved in
ferroptosis are regulated by nuclear factor erythroid 2-related
factor 2 transcription, including genes that regulate glutathione
and NADPH regeneration, which are key for glutathione peroxidase 4
activity, lipid peroxidation and iron regulation (144).
It is hypothesized that during ischemia and early
reperfusion, cellular acidosis, internal environmental instability
and other factors promote release of ferrivalent or ferrous ions
from enzymes containing iron and sulfur clusters and activate the
iron-mediated Fenton reaction, resulting in increased generation of
ROS, which leads to oxidative stress injury and ferroptosis of
cardiomyocytes (145,146). In addition, deposition of iron in
the periinfarct and non-infarct areas has been observed in a mouse
model of MI (147). Moreover,
Baba et al (148) observed
positive iron staining in non-cardiac cells surrounding cardiac
scar.
A recent study by our group indicated that
myocardial ferroptosis (as well as apoptosis pyroptosis) is
increased during MIRI in diabetes and is attenuated by effective
treatment with antioxidant N-acetylcysteine (130). However, the interaction and
underlying mechanism of apoptosis, pyroptosis and ferroptosis in
MIRI have yet to be elucidated.
In the death receptor pathway, necrosis is induced
by activation of receptor-interacting protein kinase 3 (RIPK3), a
serine/threonine kinase. RIPK3 is typically activated by
phosphorylation of homologous RIPK1(149). RIPK3 phosphorylates and activates
pseudokinase mixed lineage kinase domain-like protein (MLKL), which
oligomyelizes and permeates the plasma membrane, leading to
necrosis (149). Necrosis is
mediated by complex IIb including RIPK1, RIPK3, and MLKL. One of
the key targets of RIPK3-induced necrosis is pseudokinase MLKL.
RIPK3-mediated phosphorylation of MLKL exposes a four-helix bundle
in MLKL that promotes cysteine-dependent tetramylation, progression
to amyloid filaments and transfer and osmosis to the plasma
membrane (150). MLKL
phosphorylation is a key for programmed necrosis. Studies have
shown that MLKL is the executor of the necrotic effect (150,151). MLKL contains an N-terminal
four-helix bundle domain connected to a C-terminal pseudokinase;
the helical bundle is the functional domain of MLKL. Under normal
conditions, RIPK3 phosphorylates MLKL, resulting in conformational
changes that counteract the autophagy effect of the kinase
homologous C-terminal region in MLKL (152). MLKL is considered to be the
primary target of necrosis but its downstream signaling pathway
requires further investigation (153).
Expression levels of necroptosis-associated
proteins, including RIPK1, RIPK3 and MLKL, have been shown to be
increased in an in vivo MIRI model (154-160).
However, reperfusion duration and species-specific susceptibility
to cell death should be considered when evaluating necroptosis
(161). A study has shown that
RIPK3 is also involved in ERS and intracellular Ca2+
overload (162), which aggravates
MIRI.
Our previous studies showed that RIP3-mediated
necroptosis is a key mechanism of enhanced inflammation and lung
tissue injury in high dose lipopolysaccharide-induced severe acute
respiratory distress syndrome in mice (153) and in cardiomyocytes with
H2O2-induced necroptotic and apoptotic cell
death (163). To the best of our
knowledge, studies regarding the role and mechanism of
RIP3-mediated necroptosis and its interaction with apoptosis and
other forms of cell death in MIRI are rare (157,164). Therefore, further studies in this
area are required.
The present review summarizes research on the
pathophysiology of MIRI, including the role of ROS in cell death
during MIRI, ischemia- and hypoxia-induced metabolic and functional
dysfunction of the electron transport chain in cardiomyocyte
mitochondria, mitochondrial disorder caused by decreased ATP
production and oxidative stress promoted by increased ROS
production caused by mitochondrial damage and electrolyte imbalance
during reperfusion. Increased ROS levels lead to cell damage and
death via autophagy, apoptosis, programmed cell death inflammation,
ferroptosis and necrosis. These interplay with one another, which
directly or indirectly leads to aggravation of the effect (Fig. 1).
To the best of our knowledge, the underlying
mechanism of MIRI has not been fully clarified. Among the known
mechanisms, explanations for Ca2+ overload, energy
metabolism disorder, oxidative stress and autophagy are relatively
complete, whereas the role of pyroptosis, necrosis and ferroptosis,
as well as downstream molecules of known signaling pathways, need
to be further investigated. Clinical trials have shown that infarct
size can be limited by non-pharmacological strategies such as
ischemic postconditioning and remote ischemic conditioning, drugs,
such as cyclosporine, insulin, glucagon-like peptide-1 agonists and
β-blockers, or stimulation of cyclic guanosine monophosphate
synthesis (165). Our clinical
studies have shown that Captopril pretreatment (166) and intraoperative use of
antioxidant therapy, such as propofol (167) and Salvia miltiorrhiza
(168) alleviate MIRI and improve
prognosis in humans. In addition, clinical treatments such as
remote ischemic preconditioning (RIPC) attenuate MIRI and improve
short-term prognosis. A key cardioprotective mechanism of RIPC is
associated with decreased opening of mPTP in the heart (36,169).
MIRI is a complex process involving multilevel and
multifactor interactions between genes, molecules, cells and
tissue. A full understanding of the pathogenesis and mechanisms
underlying development of pathophysiology may provide novel
therapeutic targets for improving the prognosis of MIRI and
decreasing mortality associated with cardiovascular disease.
Not applicable.
Funding: The present study was supported by The National Natural
Science Foundation of China (grant no. 81970247) and the Basic and
Applied Basic research of Guangdong Province (grant no.
2019A1515110732).
Not applicable.
JH and DL drafted the manuscript. LXZ, JR and DZ
collated and checked references. DL, JH and DZ prepared the figure.
LQZ and ZX edited the manuscript. Data authentication is not
applicable. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Moreira PVL, de Arruda Neta ADCP, Ferreira
SS, Ferreira FELL, de Lima RLFC, de Toledo Vianna RP, de Araújo JM,
de Alencar Rodrigues RE, da Silva Neto JM and O'Flaherty M:
Coronary heart disease and stroke mortality trends in Brazil
2000-2018. PLoS One. 16(e253639)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ,
Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, et
al: Heart disease and stroke statistics-2011 update: A report from
the American Heart Association. Circulation. 123:e18–e209.
2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ogura Y, Ouchi N, Ohashi K, Shibata R,
Kataoka Y, Kambara T, Kito T, Maruyama S, Yuasa D, Matsuo K, et al:
Therapeutic impact of follistatin-like 1 on myocardial ischemic
injury in preclinical models. Circulation. 126:1728–1738.
2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Verma S, Fedak PW, Weisel RD, Butany J,
Rao V, Maitland A, Li RK, Dhillon B and Yau TM: Fundamentals of
reperfusion injury for the clinical cardiologist. Circulation.
105:2332–2336. 2002.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
7
|
Heusch G: Myocardial stunning and
hibernation revisited. Nat Rev Cardiol. 18:522–536. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Caiazzo G, Musci RL, Frediani L, Umińska
J, Wanha W, Filipiak KJ, Kubica J and Navarese EP: State of the
art: No-Reflow phenomenon. Cardiol Clin. 38:563–573.
2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nagao K, Ooiwa K and Kanmatsuse K:
Reperfusion arrhythmia. Ryoikibetsu Shokogun Shirizu. 277–281.
1996.PubMed/NCBI(In Japanese).
|
|
10
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yang CF: Clinical manifestations and basic
mechanisms of myocardial ischemia/reperfusion injury. Ci Ji Yi Xue
Za Zhi. 30:209–215. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Javat D, Heal C, Banks J, Buchholz S and
Zhang Z: Regional to tertiary inter-hospital transfer versus
in-house percutaneous coronary intervention in acute coronary
syndrome. PLoS One. 13(e198272)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li Y, Li Y, Li B, Liu Y, Zhang J, Kuang W,
Lu J, Cao Z, Cui J, Fan Z, et al: Antiplatelet therapy with
integrated traditional Chinese and western medicine for use in
myocardial Ischemia-Reperfusion injury: A review of clinical
applications and mechanisms. Evid Based Complement Alternat Med.
2021(7409094)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li Q, Shen L, Wang Z, Jiang HP and Liu LX:
Tanshinone IIA protects against myocardial ischemia reperfusion
injury by activating the PI3K/Akt/mTOR signaling pathway. Biomed
Pharmacother. 84:106–114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li R, Jia Z and Trush MA: Defining ROS in
biology and medicine. React Oxyg Species (Apex). 1:9–21.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
González-Montero J, Brito R, Gajardo AI
and Rodrigo R: Myocardial reperfusion injury and oxidative stress:
Therapeutic opportunities. World J Cardiol. 10:74–86.
2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kandula V, Kosuru R, Li H, Yan D, Zhu Q,
Lian Q, Ge RS, Xia Z and Irwin MG: Forkhead box transcription
factor 1: Role in the pathogenesis of diabetic cardiomyopathy.
Cardiovasc Diabetol. 15(44)2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li H, Xia Z, Chen Y, Qi D and Zheng H:
Mechanism and therapies of oxidative Stress-Mediated cell death in
ischemia reperfusion injury. Oxid Med Cell Longev.
2018(2910643)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Xia Z, Chen Y, Fan Q and Xue M: Oxidative
stress-mediated reperfusion injury: Mechanism and therapies. Oxid
Med Cell Longev. 2014(373081)2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xia Z, Chen Y, Fan Q, Xue M and Liu KX:
Oxidative stress-mediated reperfusion injury 2014. Oxid Med Cell
Longev. 2015(689416)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
García N, Zazueta C and Aguilera-Aguirre
L: Oxidative stress and inflammation in cardiovascular disease.
Oxid Med Cell Longev. 2017(5853238)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Goldhaber JI and Weiss JN: Oxygen free
radicals and cardiac reperfusion abnormalities. Hypertension.
20:118–1127. 1992.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chen Y, Liu C, Zhou P, Li J, Zhao X, Wang
Y, Chen R, Song L, Zhao H and Yan H: Coronary endothelium No-Reflow
injury is associated with ROS-Modified mitochondrial fission
through the JNK-Drp1 signaling pathway. Oxid Med Cell Longev.
2021(6699516)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pena E, Brito J, El Alam S and Siques P:
Oxidative stress, kinase activity and inflammatory implications in
right ventricular hypertrophy and heart failure under hypobaric
hypoxia. Int J Mol Sci. 21(6421)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kura B, Szeiffova BB, Kalocayova B, Sykora
M and Slezak J: Oxidative stress-responsive MicroRNAs in heart
injury. Int J Mol Sci. 21(358)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng
YL, Cheng PW, Li CY and Li CJ: Current mechanistic concepts in
ischemia and reperfusion injury. Cell Physiol Biochem.
46:1650–1667. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL,
Irwin MG and Xia Z: Hyperglycemia abrogates ischemic
postconditioning cardioprotection by impairing
AdipoR1/Caveolin-3/STAT3 signaling in diabetic rats. Diabetes.
65:942–955. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Su W, Zhang Y, Zhang Q, Xu J, Zhan L, Zhu
Q, Lian Q, Liu H, Xia ZY, Xia Z and Lei S: N-acetylcysteine
attenuates myocardial dysfunction and postischemic injury by
restoring caveolin-3/eNOS signaling in diabetic rats. Cardiovasc
Diabetol. 15(146)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jeddi S, Gheibi S, Kashfi K, Carlström M
and Ghasemi A: Protective effect of intermediate doses of hydrogen
sulfide against myocardial ischemia-reperfusion injury in obese
type 2 diabetic rats. Life Sci. 256(117855)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J,
Lei S, Liu Z, Ng KF, Wong GT, et al: N-Acetylcysteine and
allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via
adiponectin and attenuated myocardial postischemic injury in
diabetes. Free Radic Biol Med. 63:291–303. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xue R, Lei S, Xia ZY, Wu Y, Meng Q, Zhan
L, Su W, Liu H, Xu J, Liu Z, et al: Selective inhibition of PTEN
preserves ischaemic post-conditioning cardioprotection in
STZ-induced Type 1 diabetic rats: Role of the PI3K/Akt and
JAK2/STAT3 pathways. Clin Sci (Lond). 130:377–392. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu Q, Wang T, Chen S, Zhou Q, Li H, Hu N,
Feng Y, Dong N, Yao S and Xia Z: Cardiac protective effects of
remote ischaemic preconditioning in children undergoing tetralogy
of fallot repair surgery: A randomized controlled trial. Eur Heart
J. 39:1028–1037. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang TT, Shi MM, Liao XL, Li YQ, Yuan HX,
Li Y, Liu X, Ning DS, Peng YM, Yang F, et al: Overexpression of
inducible nitric oxide synthase in the diabetic heart compromises
ischemic postconditioning. J Mol Cell Cardiol. 129:144–153.
2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu Y, Paterson M, Baumgardt SL, Irwin MG,
Xia Z, Bosnjak ZJ and Ge ZD: Vascular endothelial growth factor
regulation of endothelial nitric oxide synthase phosphorylation is
involved in isoflurane cardiac preconditioning. Cardiovasc Res.
115:168–178. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Trafford AW, Díaz ME, Negretti N and
Eisner DA: Enhanced Ca2+ current and decreased Ca2+ efflux restore
sarcoplasmic reticulum Ca2+ content after depletion. Circ Res.
81:477–4784. 1997.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Eisner D, Bode E, Venetucci L and Trafford
A: Calcium flux balance in the heart. J Mol Cell Cardiol.
58:110–117. 2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chen S and Li S: The Na+/Ca²+ exchanger in
cardiac ischemia/reperfusion injury. Med Sci Monit. 18:RA161–RA165.
2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Aghaei M, Motallebnezhad M, Ghorghanlu S,
Jabbari A, Enayati A, Rajaei M, Pourabouk M, Moradi A, Alizadeh AM
and Khori V: Targeting autophagy in cardiac ischemia/reperfusion
injury: A novel therapeutic strategy. J Cell Physiol.
234:16768–16778. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Hotta Y, Ishikawa N, Ohashi N and Matsui
K: Effects of SM-20550, a selective Na+-H+ exchange inhibitor, on
the ion transport of myocardial mitochondria. Mol Cell Biochem.
219:83–90. 2001.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Talukder MA, Zweier JL and Periasamy M:
Targeting calcium transport in ischaemic heart disease. Cardiovasc
Res. 84:345–352. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chen C, Lu W, Wu G, Lv L, Chen W, Huang L,
Wu X, Xu N and Wu Y: Cardioprotective effects of combined therapy
with diltiazem and superoxide dismutase on myocardial
ischemia-reperfusion injury in rats. Life Sci. 183:50–59.
2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kook H, Hong SJ, Yang KS, Lee S, Kim JS
and Park CG: Comparison of nebivolol versus diltiazem in improving
coronary artery spasm and quality of life in patients with
hypertension and vasospastic angina: A prospective, randomized,
double-blind pilot study. PLoS One. 15(e239039)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lodha AR, Pillai A, Sheth K and Hiremath
J: A retrospective cohort study exploring diltiazem as a
pharmaco-enhancer for tacrolimus, in a post-heart transplant
setting. Clin Transplant. 34(e14100)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Bou-Teen D, Kaludercic N, Weissman D,
Turan B, Maack C, Di Lisa F and Ruiz-Meana M: Mitochondrial ROS and
mitochondria-targeted antioxidants in the aged heart. Free Radic
Biol Med. 167:109–124. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Yu J, Wu J, Xie P, Maimaitili Y, Wang J,
Xia Z, Gao F, Zhang X and Zheng H: Sevoflurane postconditioning
attenuates cardiomyocyte hypoxia/reoxygenation injury via restoring
mitochondrial morphology. Peerj. 4(e2659)2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Boengler K, Kosiol M, Mayr M, Schulz R and
Rohrbach S: Mitochondria and ageing: Role in heart, skeletal muscle
and adipose tissue. J Cachexia Sarcopenia Muscle. 8:349–369.
2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Fieni F, Johnson DE, Hudmon A and Kirichok
Y: Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature.
513:E1–E2. 2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Santulli G, Xie W, Reiken SR and Marks AR:
Mitochondrial calcium overload is a key determinant in heart
failure. Proc Natl Acad Sci USA. 112:11389–11394. 2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Cheng Y, Xia Z, Han Y and Rong J: Plant
natural product formononetin protects rat cardiomyocyte h9c2 cells
against oxygen glucose deprivation and reoxygenation via inhibiting
ROS formation and promoting GSK-3β phosphorylation. Oxid Med Cell
Longev. 2016(2060874)2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Shintani-Ishida K, Inui M and Yoshida K:
Ischemia-reperfusion induces myocardial infarction through
mitochondrial Ca²+ overload. J Mol Cell Cardiol.
53:233–239. 2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Kulek AR, Anzell A, Wider JM, Sanderson TH
and Przyklenk K: Mitochondrial quality control: Role in cardiac
models of lethal ischemia-reperfusion injury. Cells.
9(214)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Chang JC, Lien CF, Lee WS, Chang HR, Hsu
YC, Luo YP, Jeng JR, Hsieh JC and Yang KT: Intermittent hypoxia
prevents myocardial mitochondrial Ca 2+ Overload and
cell death during ischemia/reperfusion: The role of reactive oxygen
species. Cells-Basel. 8(564)2019.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Obeng E: Apoptosis (programmed cell death)
and its signals-A review. Braz J Biol. 81:1133–1143.
2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Slee EA, Adrain C and Martin SJ:
Executioner caspase-3, -6, and -7 perform distinct, non-redundant
roles during the demolition phase of apoptosis. J Biol Chem.
276:7320–7326. 2001.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Schlegel RA and Williamson P:
Phosphatidylserine, a death knell. Cell Death Differ. 8:551–563.
2001.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Xu X, Lai Y and Hua ZC: Apoptosis and
apoptotic body: Disease message and therapeutic target potentials.
Biosci Rep. 39(BSR20180992)2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wajant H: The Fas signaling pathway: More
than a paradigm. Science. 296:1635–1636. 2002.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Kischkel FC, Hellbardt S, Behrmann I,
Germer M, Pawlita M, Krammer PH and Peter ME:
Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a
death-inducing signaling complex (DISC) with the receptor. EMBO J.
14:5579–5588. 1995.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Teringova E and Tousek P: Apoptosis in
ischemic heart disease. J Transl Med. 15(87)2017.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817.
2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Condorelli G, Roncarati R, Ross JJ Jr,
Pisani A, Stassi G, Todaro M, Trocha S, Drusco A, Gu Y, Russo MA,
et al: Heart-targeted overexpression of caspase3 in mice increases
infarct size and depresses cardiac function. Proc Natl Acad Sci
USA. 98:9977–9982. 2001.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Lee P, Sata M, Lefer DJ, Factor SM, Walsh
K and Kitsis RN: Fas pathway is a critical mediator of cardiac
myocyte death and MI during ischemia-reperfusion in vivo. Am J
Physiol Heart Circ Physiol. 284:H456–H463. 2003.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Jeremias I, Kupatt C, Martin-Villalba A,
Habazettl H, Schenkel J, Boekstegers P and Debatin KM: Involvement
of CD95/Apo1/Fas in cell death after myocardial ischemia.
Circulation. 102:915–920. 2000.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Kosuru R, Cai Y, Kandula V, Yan D, Wang C,
Zheng H, Li Y, Irwin MG, Singh S and Xia Z: AMPK contributes to
cardioprotective effects of pterostilbene against myocardial
ischemia-reperfusion injury in diabetic rats by suppressing cardiac
oxidative stress and apoptosis. Cell Physiol Biochem. 46:1381–1397.
2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Forini F, Kusmic C, Nicolini G, Mariani L,
Zucchi R, Matteucci M, Iervasi G and Pitto L: Triiodothyronine
prevents cardiac ischemia/reperfusion mitochondrial impairment and
cell loss by regulating miR30a/p53 axis. Endocrinology.
155:4581–4590. 2014.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Lin CL, Tseng HC, Chen RF, Chen WP, Su MJ,
Fang KM and Wu ML: Intracellular zinc release-activated
ERK-dependent GSK-3β-p53 and Noxa-Mcl-1 signaling are both involved
in cardiac ischemic-reperfusion injury. Cell Death Differ.
18:1651–1663. 2011.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhang C, Shi J, Qian L, Zhang C, Wu K,
Yang C, Yan D, Wu X and Liu X: Nucleostemin exerts anti-apoptotic
function via p53 signaling pathway in cardiomyocytes. In Vitro Cell
Dev Biol Anim. 51:1064–1071. 2015.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Li M, Wang D, He J, Chen L and Li H:
Bcl-XL: A multifunctional anti-apoptotic protein.
Pharmacol Res. 151(104547)2020.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Hochhauser E, Kivity S, Offen D, Maulik N,
Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub
V, et al: Bax ablation protects against myocardial
ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart
Circ Physiol. 284:H2351–H2359. 2003.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Chen Z, Chua CC, Ho YS, Hamdy RC and Chua
BH: Overexpression of Bcl-2 attenuates apoptosis and protects
against myocardial I/R injury in transgenic mice. Am J Physiol
Heart Circ Physiol. 280:H2313–H2320. 2001.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Zhang D, He Y, Ye X, Cai Y, Xu J, Zhang L,
Li M, Liu H, Wang S and Xia Z: Activation of autophagy inhibits
nucleotide-binding oligomerization domain-like receptor protein 3
inflammasome activation and attenuates myocardial
ischemia-reperfusion injury in diabetic rats. J Diabetes Investig.
11:1126–1136. 2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Cai Y, Ying F, Liu H, Ge L, Song E, Wang
L, Zhang D, Hoi CTE, Xia Z and Irwin MG: Deletion of Rap1 protects
against myocardial ischemia/reperfusion injury through suppressing
cell apoptosis via activation of STAT3 signaling. FASEB J.
34:4482–4496. 2020.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Peng K, Chen WR, Xia F, Liu H, Meng XW,
Zhang J, Liu HY, Xia ZY and Ji FH: Dexmedetomidine post-treatment
attenuates cardiac ischaemia/reperfusion injury by inhibiting
apoptosis through HIF-1α signalling. J Cell Mol Med. 24:850–861.
2020.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Gao S, Wang R, Dong S, Wu J, Perek B, Xia
Z, Yao S and Wang T: Inactivation of TOPK caused by hyperglycemia
blocks diabetic heart sensitivity to sevoflurane postconditioning
by impairing the PTEN/PI3K/Akt signaling. Oxid Med Cell Longev.
2021(6657529)2021.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Pang L, Cai Y, Tang EH, Yan D, Kosuru R,
Li H, Irwin MG, Ma H and Xia Z: Cox-2 inhibition protects against
hypoxia/reoxygenation-induced cardiomyocyte apoptosis via
Akt-dependent enhancement of iNOS expression. Oxid Med Cell Longev.
2016(3453059)2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Korshunova AY, Blagonravov ML, Neborak EV,
Syatkin SP, Sklifasovskaya AP, Semyatov SM and Agostinelli E:
BCL2-regulated apoptotic process in myocardial ischemia-reperfusion
injury (Review). Int J Mol Med. 47:23–36. 2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Liu J, Liu M and Chen L: Novel
pathogenesis: Regulation of apoptosis by Apelin/APJ system. Acta
Biochim Biophys Sin (Shanghai). 49:471–478. 2017.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Qi Z and Chen L: Endoplasmic reticulum
stress and autophagy. Adv Exp Med Biol. 1206:167–177.
2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Sanderson TH, Gallaway M and Kumar R:
Unfolding the unfolded protein response: Unique insights into brain
ischemia. Int J Mol Sci. 16:7133–7142. 2015.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Chen X, Wang Y, Xie X, Chen H, Zhu Q, Ge
Z, Wei H, Deng J, Xia Z and Lian Q: Heme oxygenase-1 reduces
Sepsis-Induced endoplasmic reticulum stress and acute lung injury.
Mediators Inflamm. 2018(9413876)2018.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhang YM, Wang CY, Zheng FC, Gao FF, Chen
YC, Huang ZQ, Xia ZY, Irwin MG, Li WQ, Liu XP, et al: Effects of
N-n-butyl haloperidol iodide on the rat myocardial sarcoplasmic
reticulum Ca(2+)-ATPase during ischemia/reperfusion. Biochem
Biophys Res Commun. 425:426–430. 2012.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Liu Y, Baumgardt SL, Fang J, Shi Y, Qiao
S, Bosnjak ZJ, Vásquez-Vivar J, Xia Z, Warltier DC, Kersten JR and
Ge ZD: Transgenic overexpression of GTP cyclohydrolase 1 in
cardiomyocytes ameliorates post-infarction cardiac remodeling. Sci
Rep. 7(3093)2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Su RY, Geng XY, Yang Y and Yin HS:
Nesfatin-1 inhibits myocardial ischaemia/reperfusion injury through
activating Akt/ERK pathway-dependent attenuation of endoplasmic
reticulum stress. J Cell Mol Med. 25:5050–5059. 2021.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Vekich JA, Belmont PJ, Thuerauf DJ and
Glembotski CC: Protein disulfide isomerase-associated 6 is an
ATF6-inducible ER stress response protein that protects cardiac
myocytes from ischemia/reperfusion-mediated cell death. J Mol Cell
Cardiol. 53:259–267. 2012.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Glembotski CC: Roles for ATF6 and the
sarco/endoplasmic reticulum protein quality control system in the
heart. J Mol Cell Cardiol. 71:11–15. 2014.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Dai B, Qiao L, Zhang M, Cheng L, Zhang L,
Geng L, Shi C, Zhang M, Sui C, Shen W, et al: LncRNA AK054386
functions as a ceRNA to sequester miR-199 and induce sustained
endoplasmic reticulum stress in hepatic reperfusion injury. Oxid
Med Cell Longev. 2019(8189079)2019.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Gao P, Yan Z and Zhu Z:
Mitochondria-Associated endoplasmic reticulum membranes in
cardiovascular diseases. Front Cell Dev Biol.
8(604240)2020.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Zhou J, Ahmad F, Parikh S, Hoffman NE,
Rajan S, Verma VK, Song J, Yuan A, Shanmughapriya S, Guo Y, et al:
Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe
resulting in fatal dilated cardiomyopathy. Circ Res. 118:1208–1222.
2016.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17(327)2016.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Zhang G, Wang X, Gillette TG, Deng Y and
Wang ZV: Unfolded protein response as a therapeutic target in
cardiovascular disease. Curr Top Med Chem. 19:1902–1917.
2019.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Song S, Tan J, Miao Y and Zhang Q:
Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement
of UPR and the core autophagy machinery. J Cell Physiol.
233:3867–3874. 2018.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A
and Agostinis P: PERK is required at the ER-mitochondrial contact
sites to convey apoptosis after ROS-based ER stress. Cell Death
Differ. 19:1880–1891. 2012.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Carreras-Sureda A, Jaña F, Urra H, Durand
S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos-Fernández E,
Sassano ML, et al: Non-canonical function of IRE1α determines
mitochondria-associated endoplasmic reticulum composition to
control calcium transfer and bioenergetics. Nat Cell Biol.
21:755–767. 2019.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Shi B, Ma M, Zheng Y, Pan Y and Lin X:
MTOR and Beclin1: Two key autophagy-related molecules and their
roles in myocardial ischemia/reperfusion injury. J Cell Physiol.
234:12562–12568. 2019.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Wang S, Wang C, Yan F, Wang T, He Y, Li H,
Xia Z and Zhang Z: N-Acetylcysteine attenuates diabetic myocardial
ischemia reperfusion injury through inhibiting excessive autophagy.
Mediators Inflamm. 2017(9257291)2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S,
Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu
YP, Acevedo-Arozena A, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy (4th
edition)1. Autophagy. 17:1–382. 2021.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Ouyang C, You J and Xie Z: The interplay
between autophagy and apoptosis in the diabetic heart. J Mol Cell
Cardiol. 71:71–80. 2014.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688.
2005.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Sala-Mercado JA, Wider J, Undyala VV,
Jahania S, Yoo W, Mentzer RJ, Gottlieb RA and Przyklenk K: Profound
cardioprotection with chloramphenicol succinate in the swine model
of myocardial ischemia-reperfusion injury. Circulation. 122 (11
Suppl):S179–S184. 2010.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Wang Y, Zhou L, Su W, Huang F, Zhang Y,
Xia ZY, Xia Z and Lei S: Selective inhibition of PKCβ2 restores
ischemic Postconditioning-Mediated cardioprotection by modulating
autophagy in diabetic rats. J Diabetes Res.
2020(2408240)2020.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Chen Z, Hu Z, Lu Z, Cai S, Gu X, Zhuang H,
Ruan Z, Xia Z, Irwin MG, Feng D and Zhang L: Differential microRNA
profiling in a cellular hypoxia reoxygenation model upon
posthypoxic propofol treatment reveals alterations in autophagy
signaling network. Oxid Med Cell Longev.
2013(378484)2013.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Abdrakhmanov A, Gogvadze V and Zhivotovsky
B: To Eat or to Die: Deciphering selective forms of autophagy.
Trends Biochem Sci. 45:347–364. 2020.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Cong Y, Dinesh KN, Mauthe M, Verlhac P and
Reggiori F: Manipulation of selective macroautophagy by pathogens
at a glance. J Cell Sci. 133(jcs240440)2020.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Schuck S: Microautophagy-distinct
molecular mechanisms handle cargoes of many sizes. J Cell Sci.
133(jcs246322)2020.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Yang Q, Wang R and Zhu L:
Chaperone-Mediated autophagy. Adv Exp Med Biol. 1206:435–452.
2019.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Zhu H and He L: Beclin 1 biology and its
role in heart disease. Curr Cardiol Rev. 11:229–237.
2015.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Brady NR, Hamacher-Brady A, Yuan H and
Gottlieb RA: The autophagic response to nutrient deprivation in the
hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic
reticulum calcium stores. FEBS J. 274:3184–3197. 2007.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181.
2012.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ and Diwan A: Autophagy is impaired in cardiac
ischemia-reperfusion injury. Autophagy. 8:1394–1396.
2012.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Manganelli V, Matarrese P, Antonioli M,
Gambardella L, Vescovo T, Gretzmeier C, Longo A, Capozzi A,
Recalchi S, Riitano G, et al: Raft-like lipid microdomains drive
autophagy initiation via AMBRA1-ERLIN1 molecular association within
MAMs. Autophagy. 17:2528–2548. 2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z,
Zhang Z and Dai Z: Propofol directly induces caspase-1-dependent
macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell
Death Dis. 10(542)2019.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ball DP, Taabazuing CY, Griswold AR, Orth
EL, Rao SD, Kotliar IB, Vostal LE, Johnson DC and Bachovchin DA:
Caspase-1 interdomain linker cleavage is required for pyroptosis.
Life Sci Alliance. 3(e202000664)2020.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Toldo S, Mauro AG, Cutter Z and Abbate A:
Inflammasome, pyroptosis, and cytokines in myocardial
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
315:H1553–H1568. 2018.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Tsuchiya K: Inflammasome-associated cell
death: Pyroptosis, apoptosis, and physiological implications.
Microbiol Immunol. 64:252–269. 2020.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Frank D and Vince JE: Pyroptosis versus
necroptosis: Similarities, differences, and crosstalk. Cell Death
Differ. 26:99–114. 2019.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158.
2016.PubMed/NCBI View Article : Google Scholar
|
|
124
|
He WT, Wan H, Hu L, Chen P, Wang X, Huang
Z, Yang ZH, Zhong CQ and Han J: Gasdermin D is an executor of
pyroptosis and required for interleukin-1β secretion. Cell Res.
25:1285–1298. 2015.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Yue RC, Lu SZ, Luo Y, Wang T, Liang H,
Zeng J, Liu J and Hu HX: Calpain silencing alleviates myocardial
ischemia-reperfusion injury through the NLRP3/ASC/Caspase-1 axis in
mice. Life Sci. 233(116631)2019.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Djulbegovic MB and Uversky VN:
Ferroptosis-an iron- and disorder-dependent programmed cell death.
Int J Biol Macromol. 135:1052–1069. 2019.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Liu Q, Zhang D, Hu D, Zhou X and Zhou Y:
The role of mitochondria in NLRP3 inflammasome activation. Mol
Immunol. 103:115–124. 2018.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Ding S, Liu D, Wang L, Wang G and Zhu Y:
Inhibiting MicroRNA-29a protects myocardial Ischemia-Reperfusion
injury by targeting SIRT1 and suppressing oxidative stress and
NLRP3-Mediated pyroptosis pathway. J Pharmacol Exp Ther.
372:128–135. 2020.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang
Z and Yao W: Diabetes aggravates myocardial ischaemia reperfusion
injury via activating Nox2-related programmed cell death in an
AMPK-dependent manner. J Cell Mol Med. 24:6670–6679.
2020.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Popov SV, Maslov LN, Naryzhnaya NV,
Mukhomezyanov AV, Krylatov AV, Tsibulnikov SY, Ryabov VV, Cohen MV
and Downey JM: The role of pyroptosis in ischemic and reperfusion
injury of the heart. J Cardiovasc Pharmacol Ther. 26:562–574.
2021.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176.
2016.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Yan N and Zhang J: Iron metabolism,
ferroptosis, and the links with alzheimer's disease. Front
Neurosci. 13(1443)2019.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Weiland A, Wang Y, Wu W, Lan X, Han X, Li
Q and Wang J: Ferroptosis and its role in diverse brain diseases.
Mol Neurobiol. 56:4880–4893. 2019.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Hu Z, Zhang H, Yang SK, Wu X, He D, Cao K
and Zhang W: Emerging role of ferroptosis in acute kidney injury.
Oxid Med Cell Longev. 2019(8010614)2019.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Yu H, Guo P, Xie X, Wang Y and Chen G:
Ferroptosis, a new form of cell death, and its relationships with
tumourous diseases. J Cell Mol Med. 21:648–657. 2017.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen Species-Induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019(5080843)2019.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Stoyanovsky DA, Tyurina YY, Shrivastava I,
Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV,
Vladimirov YA, et al: Iron catalysis of lipid peroxidation in
ferroptosis: Regulated enzymatic or random free radical reaction?
Free Radic Biol Med. 133:153–1561. 2019.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Zhao WK, Zhou Y, Xu TT and Wu Q:
Ferroptosis: Opportunities and challenges in myocardial
Ischemia-Reperfusion injury. Oxid Med Cell Longev.
2021(9929687)2021.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Ge ZD, Lian Q, Mao X and Xia Z: Current
status and challenges of NRF2 as a potential therapeutic target for
diabetic cardiomyopathy. Int Heart J. 60:512–520. 2019.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Williams RE, Zweier JL and Flaherty JT:
Treatment with deferoxamine during ischemia improves functional and
metabolic recovery and reduces reperfusion-induced oxygen radical
generation in rabbit hearts. Circulation. 83:1006–1014.
1991.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Drossos G, Lazou A, Panagopoulos P and
Westaby S: Deferoxamine cardioplegia reduces superoxide radical
production in human myocardium. Ann Thorac Surg. 59:169–172.
1995.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Omiya S, Hikoso S, Imanishi Y, Saito A,
Yamaguchi O, Takeda T, Mizote I, Oka T, Taneike M, Nakano Y, et al:
Downregulation of ferritin heavy chain increases labile iron pool,
oxidative stress and cell death in cardiomyocytes. J Mol Cell
Cardiol. 46:59–66. 2009.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Baba Y, Higa JK, Shimada BK, Horiuchi KM,
Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H and
Matsui T: Protective effects of the mechanistic target of rapamycin
against excess iron and ferroptosis in cardiomyocytes. Am J Physiol
Heart Circ Physiol. 314:H659–H668. 2018.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Murphy JM, Czabotar PE, Hildebrand JM,
Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D,
Webb AI, et al: The pseudokinase MLKL mediates necroptosis via a
molecular switch mechanism. Immunity. 39:443–453. 2013.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y,
Ma J, Chen W, Zhang Y, Zhou X, et al: Mlkl knockout mice
demonstrate the indispensable role of Mlkl in necroptosis. Cell
Res. 23:994–1006. 2013.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Li L, Tong A, Zhang Q, Wei Y and Wei X:
The molecular mechanisms of MLKL-dependent and MLKL-independent
necrosis. J Mol Cell Biol. 13:3–14. 2021.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie
W, Feng Y, Socorburam T, Wu G, Xia Z and Wu Q: Receptor interacting
protein 3-Mediated necroptosis promotes Lipopolysaccharide-Induced
inflammation and acute respiratory distress syndrome in mice. PLoS
One. 11(e155723)2016.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Oberst A, Dillon CP, Weinlich R, McCormick
LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS and Green DR:
Catalytic activity of the caspase-8-FLIP(L) complex inhibits
RIPK3-dependent necrosis. Nature. 471:363–367. 2011.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Dillon CP, Weinlich R, Rodriguez DA,
Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F,
Gong YN, et al: RIPK1 blocks early postnatal lethality mediated by
caspase-8 and RIPK3. Cell. 157:1189–1202. 2014.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Dannappel M, Vlantis K, Kumari S,
Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T,
Hermance N, et al: RIPK1 maintains epithelial homeostasis by
inhibiting apoptosis and necroptosis. Nature. 513:90–94.
2014.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv
F, Liu Y, Zheng W, Shang H, Zhang J, et al: CaMKII is a RIP3
substrate mediating ischemia- and oxidative stress-induced
myocardial necroptosis. Nat Med. 22:175–182. 2016.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Bai J, Wang Q, Qi J, Yu H, Wang C, Wang X,
Ren Y and Yang F: Promoting effect of baicalin on nitric oxide
production in CMECs via activating the PI3K-AKT-eNOS pathway
attenuates myocardial ischemia-reperfusion injury. Phytomedicine.
63(153035)2019.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Zhou H, Li D, Zhu P, Ma Q, Toan S, Wang J,
Hu S, Chen Y and Zhang Y: Inhibitory effect of melatonin on
necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway
attenuates cardiac microvascular ischemia-reperfusion injury. J
Pineal Res. 65(e12503)2018.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Yang J, Zhang F, Shi H, Gao Y, Dong Z, Ma
L, Sun X, Li X, Chang S, Wang Z, et al: Neutrophil-derived advanced
glycation end products-Nε-(carboxymethyl) lysine promotes
RIP3-mediated myocardial necroptosis via RAGE and exacerbates
myocardial ischemia/reperfusion injury. FASEB J. 33:14410–14422.
2019.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Ying L, Benjanuwattra J, Chattipakorn SC
and Chattipakorn N: The role of RIPK3-regulated cell death pathways
and necroptosis in the pathogenesis of cardiac
ischaemia-reperfusion injury. Acta Physiol (Oxf).
231(e13541)2021.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S,
Li Y, Zhou H and Chen Y: Ripk3 promotes ER stress-induced
necroptosis in cardiac IR injury: A mechanism involving calcium
overload/XO/ROS/mPTP pathway. Redox Biol. 16:157–168.
2018.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Yin W, Wang C, Peng Y, Yuan W, Zhang Z,
Liu H, Xia Z, Ren C and Qian J: Dexmedetomidine alleviates
H2O2-induced oxidative stress and cell
necroptosis through activating of α2-adrenoceptor in H9C2 cells.
Mol Biol Rep. 47:3629–3639. 2020.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Liu X, Zhang D, Dong X, Zhu R, Ye Y, Li L
and Jiang Y: Pharmacological activation of CB2 receptor protects
against ethanol-induced myocardial injury related to
RIP1/RIP3/MLKL-mediated necroptosis. Mol Cell Biochem. 474:1–14.
2020.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Garcia-Dorado D, Rodríguez-Sinovas A,
Ruiz-Meana M and Inserte J: Protection against myocardial
ischemia-reperfusion injury in clinical practice. Rev Esp Cardiol
(Engl Ed). 67:394–404. 2014.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Tian Y, Li H, Liu P, Xu JM, Irwin MG, Xia
Z and Tian G: Captopril pretreatment produces an additive
cardioprotection to isoflurane preconditioning in attenuating
myocardial ischemia reperfusion injury in rabbits and in humans.
Mediators Inflamm. 2015(819232)2015.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Huang Z, Zhong X, Irwin MG, Ji S, Wong GT,
Liu Y, Xia ZY, Finegan BA and Xia Z: Synergy of isoflurane
preconditioning and propofol postconditioning reduces myocardial
reperfusion injury in patients. Clin Sci (Lond). 121:57–69.
2011.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Xia Z, Gu J, Ansley DM, Xia F and Yu J:
Antioxidant therapy with Salvia miltiorrhiza decreases plasma
endothelin-1 and thromboxane B2 after cardiopulmonary bypass in
patients with congenital heart disease. J Thorac Cardiovasc Surg.
126:1404–1410. 2003.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Heusch G: Molecular basis of
cardioprotection: Signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015.PubMed/NCBI View Article : Google Scholar
|