Introduction
Chronic kidney disease (CKD), with a global
incidence of higher than 10%, is a serious threat to human health
(1,2). Moreover, the incidence of CKD is
increasing as populations age and diseases such as diabetes,
hypertension, and obesity have become more prevalent. Apart from
health issues, CKD causes significant economic burdens to both the
family and society. CKD is characterized by renal fibrosis,
specifically, the deposition of excess extracellular matrix (ECM)
in the glomerulus and interstitial area (3). In the absence of effective treatment,
renal fibrosis eventually progresses to end-stage renal disease in
most CKD patients (4).
The endoplasmic reticulum (ER) is responsible for
protein processing and calcium storage. When overwhelmed, the ER
accumulates misfolded or unfolded proteins and the calcium balance
is disrupted, a condition known as endoplasmic reticulum stress
(ERS) (5). Various factors such as
hypoxia, drugs, oxidative stress, and abnormal protein
glycosylation can lead to the accumulation of unfolded or misfolded
proteins, ultimately triggering ERS (6). Recent studies have shown that ERS
plays an active role in the development of renal fibrosis in CKD;
however, the specific molecular mechanism is unclear (7,8).
Transforming growth factor β1 (TGF-β1), a key player in ERS, can
drive renal fibrosis (9,10). Notably, in the early stage of ERS,
an ERS-related chaperone protein, glucose-regulated protein 78
(GRP78) is upregulated and binds unfolded or misfolded proteins
inducing an unfolded protein response. This leads to the
upregulation of C/EBP homologous protein (CHOP) that induces cell
apoptosis (11). Therefore,
inhibition of TGF-β1 signaling, expression of the ERS molecular
chaperone protein, and/or directly blocking ERS may be effective
strategies for delaying renal fibrosis.
Tauroursodeoxycholic acid (TUDCA), an endogenous
bile acid derivative, is used as a liver protectant in cholestatic
liver disease (12). It is also a
potent inhibitor of the apoptosis pathway (13,14).
Research has shown that advanced glycation end products (AGEs) can
induce ERS, while TUDCA can inhibit AGE-induced ERS and, in turn,
apoptosis in a dose-dependent manner (15).
In the present study, we used small interfering RNA
(siRNA) to examine the effect of GRP78 on TGF-β1-induced renal
fibrosis. Furthermore, we explored the mechanism of TUDCA in
regulating ERS and affecting the occurrence and development of
renal fibrosis in CKD patients. Our results suggest that TUDCA can
be a potential therapy for renal fibrosis in CKD patients.
Materials and methods
Cell culture
Rat renal mesangial cells (RMCs) were routinely
cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco;
Thermo Fisher Scientific, Inc.) with or without 5% fetal bovine
serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.) at 37˚C, 95%
O2 and 5% CO2 in a humidified atmosphere.
RMCs were passaged twice a week. For the study, RMCs were treated
with recombinant human TGF-β1, and a control group was also set
up.
Cell transfection of siRNA
RMCs were seeded in 6-well plates and cultured for
24 h. Three siRNAs targeting rat GRP78 and a negative
control siRNA were purchased from Jiman Biotechnology Co. Transient
transfections were performed using Lipofectamine 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
instructions. After 48 h of transfection, GRP78 expression was
detected by western blotting and real-time fluorescent quantitative
PCR.
Real-time PCR
Cellular RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. cDNA was synthesized using the
PrimeScriptTM 1st Strand cDNA Synthesis Kit (D6110A;
Takara Bio, Inc.) following the manufacturer's instructions. The
gene fragments were then amplified and quantitated by real-time PCR
using the following conditions: 1 cycle of 95˚C for 10 min; 95˚C
for 10 sec; 60˚C for 15 sec; 72˚C for 20 sec (40 cycles), and 72˚C
for 10 min. The starting template was quantitated using the CT
value estimated by the real-time PCR recorder. GAPDH was
used as the internal control. The specific primer sequences are
listed in Table SI (also
available at URL: https://figshare.com/s/12a0cdf45471f06dc16f; DOI:
10.6084/m9.figshare.16744189).
Western blotting
Cells were harvested, lysed, and the total protein
was extracted using a protein extraction kit (P0033, Beyotime
Biotechnology), and further quantified using the Pierce™ BCA
Protein Assay Kit (Thermo Fisher Scientific, Inc.) according to the
user guide. Protein samples (15 µg) were separated on 10% SDS-PAGE
gels and transferred to PVDF membranes followed by blocking in TBST
containing 5% skimmed milk for 2 h at room temperature. Primary
antibodies were diluted as described below to working
concentrations in 1X TBST with 1% skimmed milk and incubated
overnight at 4˚C. After three washes with TBST, the membranes were
incubated with the appropriate secondary antibodies for 2 h at room
temperature with gentle rocking. Finally, the protein bands were
visualized with ECL solution for 1 min, photosensitized, and
finally analyzed using BanScan software 5.0 (ProZyme; Agilent
Technologies, Inc.). The protein levels were normalized to levels
of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The primary antibodies used were as follows: rabbit
anti-GRP78 polyclonal antibody (#3183, Cell Signaling Technology,
Inc.) diluted at 1:1,000; rabbit anti-α-SMA polyclonal antibody
(ab5694, Abcam) diluted at 1:1,000; rabbit anti-collagen type I
(COL I) monoclonal antibody (ab138492, Abcam) diluted at 1:500;
mouse anti-fibronectin monoclonal antibody (ab6328, Abcam) diluted
at 1:1,000; mouse anti-CHOP monoclonal antibody (2895, Cell
Signaling Technology, Inc.) diluted at 1:1,000; mouse anti-GAPDH
monoclonal antibody (sc-365062, Santa Cruz Biotechnology, Inc.)
diluted at 1:400. The secondary antibodies used were horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG H&L (ab205718,
Abcam) and HRP-conjugated rabbit anti-mouse IgG (H+L) (ab6728;
Abcam) both diluted at 1:2,000.
Immunofluorescence
RMCs, cultured on chamber slides, were fixed with 4%
paraformaldehyde and permeabilized with PBS containing 0.1% Triton
X-100. The cells were incubated with the respective primary
antibodies described above at the following dilutions: mouse
anti-fibronectin monoclonal antibody, 1:200; rabbit anti-collagen
type I, 1:100; rabbit anti-α-SMA polyclonal antibody, 1:100.
Primary antibodies were incubated at 4˚C overnight, followed by
incubation with the corresponding secondary antibodies goat
anti-rabbit IgG-H&L conjugated to Alexa Fluor® 488
(ab150077, Abcam) diluted at a 1:200 ratio and horse anti-mouse IgG
antibody (H+L) conjugated with DyLight® 488
(DI-2488-1.5, Vector Laboratories) diluted at a 1:200 ratio for 2 h
at 37˚C. The cell nuclei were counterstained with DAPI for 1 h. The
slides were examined under a fluorescence microscope and analyzed
with Image J software v1.8.0 (National Institutes of Health).
Statistical analysis
GraphPad Prism 7.0 software (GraphPad Software,
Inc.) was used for statistical analysis. Values are expressed as
mean ± variance. The t-test was used for the statistical
calculation of real-time quantitative PCR data. Data were analyzed
using the Wilcoxon rank-sum test for paired comparisons, and the
one-way ANOVA test followed by Bonferroni or Fisher LSD post hoc
tests for multiple comparisons. P-value <0.05 denotes
statistical significance.
Results
TGF-β upregulates ERS-related
chaperone proteins and fibrotic factors
Renal fibrosis mainly involves renal mesangial
cells. Here, we examined the effect of TGF-β1 on the expression of
ERS-related and fibrotic factors at the cellular level. For this,
TGF-β1 was used in a concentration- (1, 5 and 10 ng/ml) and
time-gradient (6 and 24 h) manner. We found that after 24 h of
treatment, TGF-β1 (5 and 10 ng/ml) stimulated the expression of
GRP78 and the pro-fibrotic factors COL I, FN and α-SMA were
markedly increased than in the control group (Fig. 1). Thus, we selected 5 ng/ml TGF-β1
for subsequent experiments.
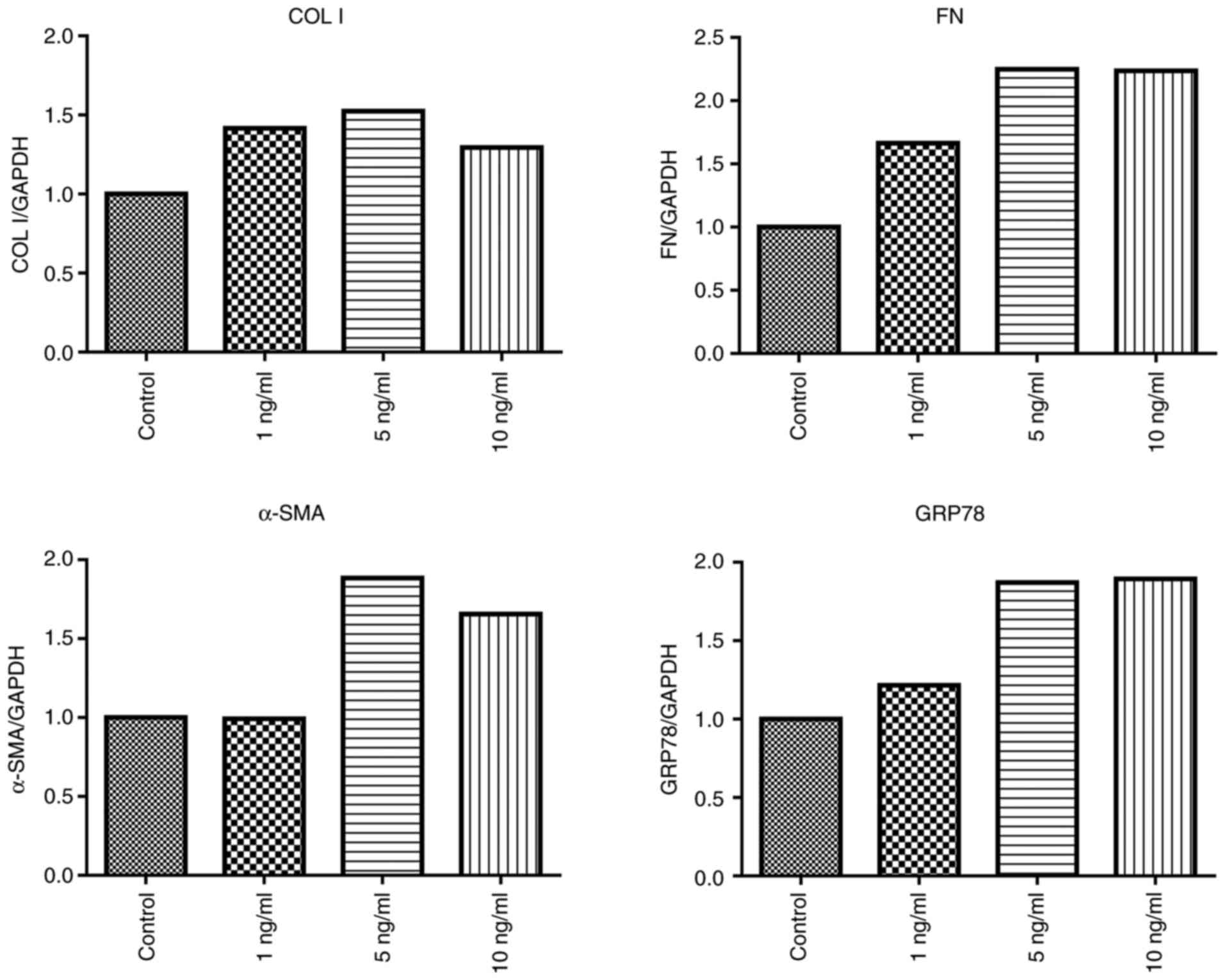 | Figure 1Effects of TGF-β1 on the expression of
COL I, FN, α-SMA and GRP78 in RMCs. RMCs were stimulated with
TGF-β1 (1, 5 and 10 ng/ml), and gene expression of COL I,
FN, α-SMA and GRP78 was assessed after 24 h by
RT-PCR. TGF-β1, transforming growth factor β1; COL I,
collagen type I; α-SMA, α-smooth muscle actin; FN,
fibronectin; GRP78, glucose-regulated protein 78; RMCs, rat
renal mesangial cells. |
Next, RMCs were stimulated with 5 ng/ml TGF-β1 for
6, 12 and 24 h. We found that the cellular expression of GRP78, the
expression of transcription factor CHOP and the expression of
fibrosis marker COL I were significantly increased after 24 h
compared to the control group (Fig.
2A). Meanwhile, TGF-β1 stimulation also observably increased
the expression of the fibrosis marker FN (Fig. 2A), although no significant
statistical significance was observed. Furthermore, the protein
levels of CHOP and GRP78 were also consistent with the PCR results,
while the changes in COL I and FN protein expression differed from
their mRNA expression (Fig. 2B).
In summary, these observations indicate that TGF-β1 treatment for
24 h significantly increased the expression of ERS-related proteins
and fibrotic factors in RMCs, corresponding to the features of
renal fibrosis.
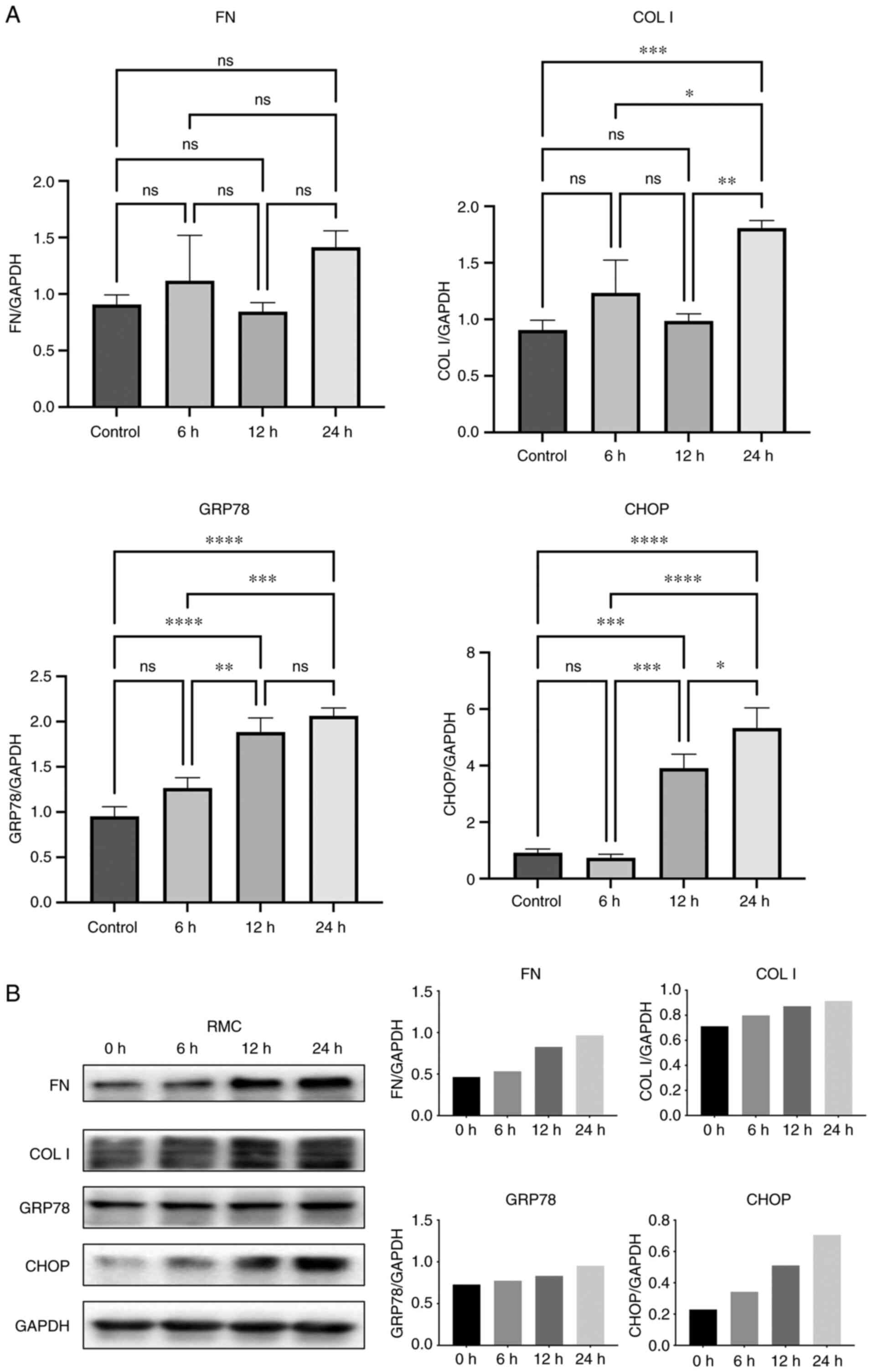 | Figure 2Effects of TGF-β 1 on the expression
of FN, COL I, GRP78s and CHOP in RMCs. (A) RMCs were stimulated
with TGF-β 1 (5 ng/ml) and gene expression of FN, COL
I, GRP78 and CHOP were assessed after 6, 12 and
24 h by RT-PCR. (B) Western blot analysis and grayscale analysis
for FN, COL I, GRP78 and CHOP. *P<0.05;
**P<0.01; ***P<0.001;
****P<0.0001; ns, P>0.05 (not significant). RMCs,
rat renal mesangial cells; CHOP; C/EBP homologous protein. FN,
fibronectin; COL I, collagen I; GRP78, glucose-regulated protein
78; CHOP, C/EBP homologous protein; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; TGF, transforming growth factor. |
Downregulation of GRP78 expression
inhibits renal fibrosis
To examine whether ERS is directly related to renal
fibrosis, we transiently transfected siRNA-GRP78 to
downregulate the GRP78 gene in RMCs. Compared with the
control group, the three siRNA sequences targeting GRP78
(siR-1, siR-2 and siR-3) markedly reduced the expression of
GRP78 (51-67%) (Fig. 3A).
Western blotting further confirmed that the protein levels of GRP78
were also reduced (Fig. 3B). For
subsequent experiments, we selected an siRNA sequence (siR-1) that
produced the maximum effect and transiently transfected RMC cells
with or without TGF-β1 stimulation. Compared with the control
group, transfection of siRNA-GRP78
(GRP78siRNA+) markedly reduced the protein levels
of GRP78 in RMCs, while also reducing the levels of the fibrosis
markers FN and α-SMA. However, compared with the TGF-β stimulation
group, transfection of siRNA-GRP78 did not significantly
downregulate the TGF-β1-induced protein levels of FN and α-SMA
(Fig. 3C). These results were
verified by immunofluorescence (Fig.
3D). This shows that the downregulation of GRP78 can inhibit
fibrosis in RMCs.
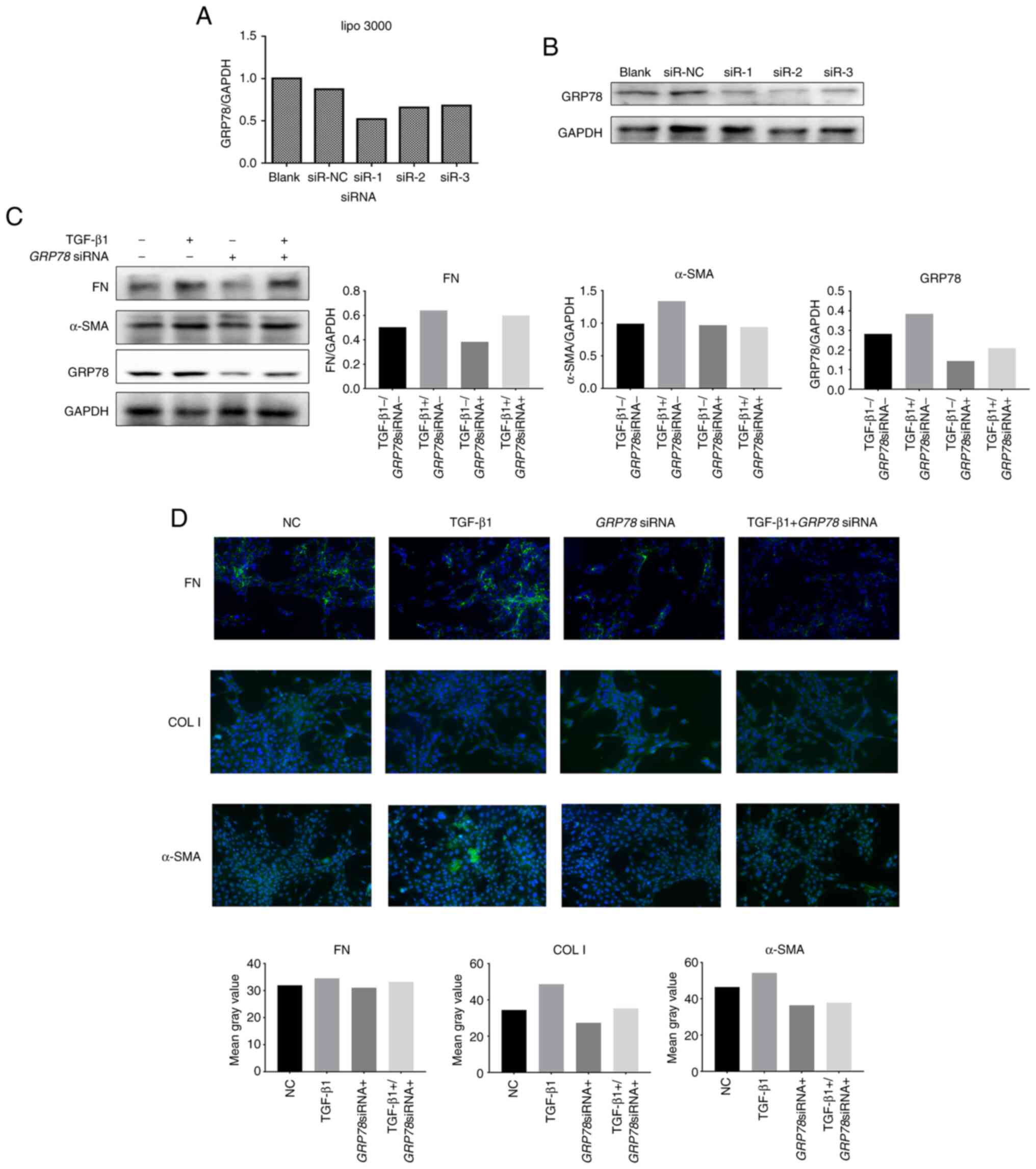 | Figure 3Silencing of GRP78 inhibits
TGF-β1-induced fibrogenesis in RMCs. (A) Three siRNAs targeting
GRP78 were individually transfected into RMCs, and the
knockdown efficiency was assessed by real-time PCR at 24 h after
transfection. (B) Western blot analysis. (C) RMCs were transiently
transfected with one siRNA (siR-1) with the highest knockdown
efficiency shown in A and then treated with (+) or without (-)
TGF-β 1 (5 ng/ml, 24 h). Gene expression was measured by real-time
PCR for FN, α-SMA and GRP78. (D)
Immunofluorescence analysis of FN, COL I and α-SMA. RMCs, rat renal
mesangial cells; FN, fibronectin; α-SMA, α-smooth muscle
actin; GRP78, glucose-regulated protein 78; COL I, collagen I;
CHOP, C/EBP homologous protein; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; TGF, transforming growth factor. |
TUDCA inhibits the pro-fibrotic effect
of TGF-β1
A serum-free medium can be used to eliminate the
influence of pro-fibrotic components in the serum. Firstly, we
examined the effect of TUDCA on ERS-related proteins in the
presence or absence of serum-containing media. The western blotting
results showed markedly differences between the two groups
(Fig. 4). In the serum-free group,
TUDCA markedly inhibited the expression of GRP78 and FN proteins in
the RMCs (Fig. 4A). The TGF-β1 (5
ng/ml)-stimulated protein levels of CHOP, GRP78 and FN were higher
than in the control group (Fig.
4B, the first column is the blank control group). However,
TUDCA intervention markedly lowered the levels of these proteins.
Furthermore, a dose-dependence experiment showed that 500 µM TUDCA
produced a stronger effect than 300 or 100 µM TUDCA (Fig. 4B).
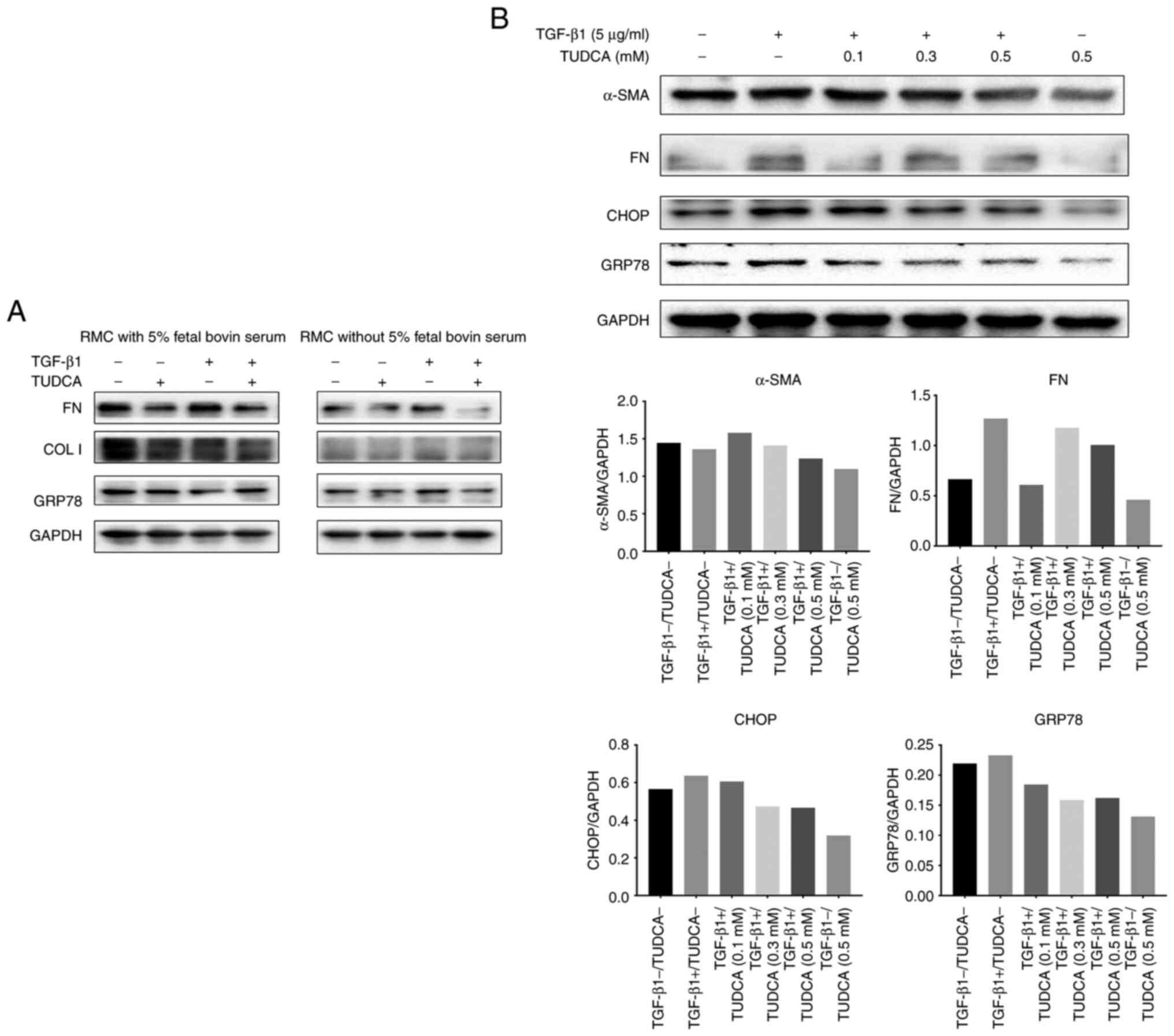 | Figure 4Effects of tauroursodeoxycholic acid
(TUDCA) on fibrogenesis in RMCs. (A) RMCs were treated with TUDCA
(0.1 mM) for 1 h, treated with (+) or without (-) TGF-β1 (5 ng/ml)
for 24 h in medium with or without 5% fetal bovine serum, and
protein expression was measured by western blot analysis for FN,
COL I and GRP78. (B) RMCs were treated with TUDCA (0.1, 0.3 and 0.5
mM) for 1 h, treated with (+) or without (-) TGF-β1 for 24 h in
medium without 5% fetal bovine serum, and protein expression was
measured by western blot analysis for FN, α-SMA, CHOP and GRP78.
FN, fibronectin; α-SMA, α-smooth muscle actin; GRP78,
glucose-regulated protein 78; COL I, collagen I; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; TGF, transforming growth
factor. |
Discussion
In recent years, the growing incidence of chronic
kidney disease (CKD) has significantly lowered the quality of life
of afflicted individuals. Since renal fibrosis cannot be reversed
and there is an absence of effective treatment, the CKD mortality
rate is also increasing. Therefore, here, we explored the key
regulatory mechanisms of renal fibrosis and used drugs to block or
delay its progress. The current mainstream view is that renal
fibrosis is caused by extracellular matrix (ECM) accumulation
involving epithelial-mesenchymal transition (EMT), transforming
growth factor (TGF)-β signal transduction, oxidative stress, and
proteinuria (16). Notably,
endoplasmic reticulum stress (ERS) is closely related to EMT, TGF-β
signal transduction, oxidative stress, and proteinuria (9). Therefore, novel drugs targeting
different intracellular ER-related pathways can essentially slow
the development of renal fibrosis.
In the present study, we first determined the
optimal stimulation concentration and time of TGF-β1 required for
fibrosis characteristics in renal mesangial cells (RMCs). Then,
siRNAs were employed to examine the effect of glucose-regulated
protein 78 (GRP78) on TGF-β1-mediated fibrosis in RMCs.
Furthermore, we found that tauroursodeoxycholic acid (TUDCA)
downregulated GRP78 and CHOP to regulate ERS which, in turn,
delayed TGF-β1-mediated renal fibrosis. We showed that siRNA
targeting GRP78 inhibited TGF-β1-induced fibrosis in RMCs
suggesting a key role for ERS in fibrosis development.
Interestingly, TUDCA could inhibit the expression of the
ERS-related proteins GRP78 and C/EBP homologous protein (CHOP),
thereby inhibiting TGF-β1-induced fibrosis in RMCs. This suggests
that TUDCA could be a potential therapeutic drug for CKD renal
fibrosis. In the present study, western blot analysis was used as
the main analysis method. However, western blot analysis can only
be used for identifying proteins qualitatively or semi-quantifying
protein amounts roughly. For future research, more repetitive
experiments and animal models are needed to confirm the conclusions
of the present study.
Renal fibrosis is a complex process characterized by
fibroblast proliferation and ECM accumulation (17,18).
Mesangial cells are one of the main cell types that produce
mesangial matrix (MM) components. Under pathological conditions,
RMCs are activated leading to excessive proliferation and ECM
secretion. This eventually causes the fibrosis of glomeruli. In
addition, mesangial cells also secrete various inflammatory
factors, adhesion molecules, chemokines, and enzymes, all of which
facilitate glomerular fibrosis (19,20).
Several studies have shown that RMCs play an important role in the
pathogenesis of glomerular fibrosis, which ultimately causes
glomerular sclerosis.
TGF-β1 promotes the proliferation of mesangial cells
and also significantly downregulates the ECM degradation via MMP
antagonists (tissue inhibitors of metalloproteinases, TIMPs). This
aggravates the accumulation of glomerular ECM, causing severe renal
fibrosis, further leading to glomerular sclerosis, and ultimately
worsening renal function (21,22).
We showed that silencing of GRP78 can inhibit fibrosis in
RMCs. A previous study using the angiotensin II reperfusion model
reported that inhibition of ERS reduced the activity of TGF-β1
which, in turn, reduced myocardial hypertrophy and fibrosis
(23). This is consistent with our
findings. In addition, inhibition of GRP78 in human and mouse lung
fibroblasts was shown to downregulate fibrosis markers such as
collagen and α-SMA (24).
Notably, in this study, inhibiting the expression of
GRP78 alone could not significantly downregulate the TGF-β1-induced
levels of FN and α-SMA. However, TUDCA could downregulate both
GRP78 and CHOP in ERS through signaling pathways such as AGEs,
which are the end-products of glycosylation and significantly
inhibit the TGF-β1-induced expression of RMC fibrosis markers (FN,
α-SMA). A previous study showed that downregulation of CHOP reduced
unilateral ureteral ligation-induced mouse renal tubular cell
apoptosis and renal fibrosis via inhibition of the
HMGB1/TLR4/NFκB/IL-1β signaling pathway and downstream
TGF-β1/Smad2/3 signaling (25). In
addition, the absence of CHOP was found to reduce the recruitment
of pro-fibrotic factors, oxidative stress, and inflammatory cells
including macrophages (26).
Therefore, we speculate that CHOP could be an important player in
renal fibrosis involving ERS-related pathways. However, there could
be other related factors/mechanisms promoting renal fibrosis that
need to be further investigated to provide a theoretical basis for
the development of new drugs.
In summary, we confirmed that the ERS-related
transcription factor CHOP may be involved in TGF-β1-induced renal
fibrosis, supporting the findings of previous studies. In addition,
TUDCA, a potential drug candidate for CKD, can delay the process of
renal mesangial cell fibrosis by inhibiting TGF-β1-induced
accumulation of ECM.
Supplementary Material
Primer sequences of GAPDH, collagen I,
α-SMA, FN, GRP78 and CHOP genes.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Science and
Technology Commission of Shanghai Municipality (grant no.
19ZR1400100).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
LL was responsible for the conceptualization of the
research design, performing experiments and formal analysis and
wrote the original draft. ZYG was responsible for the
conceptualization of the research design, performing experiments
and formal analysis. JW was responsible for the conceptualization
of the research design, performing experiments, formal analysis,
and writing, reviewing and editing the manuscript. PPF conducted
part of the experiments and the formal analysis. YFJ was
responsible for the conceptualization of the research design, and
writing, reviewing and editing the manuscript. CGX was responsible
for funding acquisition, conceptualization of the research design,
methodology, performing experiments, and writing, reviewing and
editing the manuscript. All authors have read and approved the
final manuscript, and agree to be accountable for all aspects of
the research in ensuring that the accuracy or integrity (in
particulary the data provided) of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Clinical Research
Ethics Committee of Eastern Hepatobiliary Surgery Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Webster AC, Nagler EV, Morton RL and
Masson P: Chronic kidney disease. Lancet. 389:1238–1252.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Djudjaj S and Boor P: Cellular and
molecular mechanisms of kidney fibrosis. Mol Aspects Med. 65:16–36.
2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sun YB, Qu X, Caruana G and Li J: The
origin of renal fibroblasts/myofibroblasts and the signals that
trigger fibrosis. Differentiation. 92:102–107. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hazzan AD, Halinski C, Agoritsas S,
Fishbane S and DeVita MV: Epidemiology and challenges to the
management of advanced CKD. Adv Chronic Kidney Dis. 23:217–221.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fernández A, Ordóñez R, Reiter RJ,
González-Gallego J and Mauriz JL: Melatonin and endoplasmic
reticulum stress: Relation to autophagy and apoptosis. J Pineal
Res. 59:292–307. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yoshida H: ER stress and diseases. FEBS J.
274:630–658. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Taniguchi M and Yoshida H: Endoplasmic
reticulum stress in kidney function and disease. Curr Opin Nephrol
Hypertens. 24:345–350. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cybulsky AV: Endoplasmic reticulum stress,
the unfolded protein response and autophagy in kidney diseases. Nat
Rev Nephrol. 13:681–696. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ke B, Zhu N, Luo F, Xu Y and Fang X:
Targeted inhibition of endoplasmic reticulum stress: New hope for
renal fibrosis (review). Mol Med Rep. 16:1014–1020. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bernales S, Papa FR and Walter P:
Intracellular signaling by the unfolded protein response. Annu Rev
Cell Dev Biol. 22:487–508. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kaplan MM and Gershwin ME: Primary biliary
cirrhosis. N Engl J Med. 353:1261–1273. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Amaral JD, Viana RJ, Ramalho RM, Steer CJ
and Rodrigues CM: Bile acids: Regulation of apoptosis by
ursodeoxycholic acid. J Lipid Res. 50:1721–1734. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Malo A, Krüger B, Seyhun E, Schäfer C,
Hoffmann RT, Göke B and Kubisch CH: Tauroursodeoxycholic acid
reduces endoplasmic reticulum stress, trypsin activation, and
acinar cell apoptosis while increasing secretion in rat pancreatic
acini. Am J Physiol Gastrointest Liver Physiol. 299:G877–G886.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen Y, Liu CP, Xu KF, Mao XD, Lu YB, Fang
L, Yang JW and Liu C: Effect of taurine-conjugated ursodeoxycholic
acid on endoplasmic reticulum stress and apoptosis induced by
advanced glycation end products in cultured mouse podocytes. Am J
Nephrol. 28:1014–1022. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xiao W, Fan Y, Wang N, Chuang PY, Lee K
and He JC: Knockdown of RTN1A attenuates ER stress and kidney
injury in albumin overload-induced nephropathy. Am J Physiol Renal
Physiol. 310:F409–F415. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ma TT and Meng XM: TGF-β/Smad and renal
fibrosis. Adv Exp Med Biol. 1165:347–364. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ke B, Fan C, Yang L and Fang X: Matrix
metalloproteinases-7 and kidney fibrosis. Front Physiol.
8(21)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhao JH: Mesangial cells and renal
fibrosis. Adv Exp Med Biol. 1165:165–194. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu Y: Renal fibrosis: New insights into
the pathogenesis and therapeutics. Kidney Int. 69:213–217.
2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Baricos WH, Cortez SL, Deboisblanc M and
Xin S: Transforming growth factor-beta is a potent inhibitor of
extracellular matrix degradation by cultured human mesangial cells.
J Am Soc Nephrol. 10:790–795. 1999.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hubchak SC, Sparks EE, Hayashida T and
Schnaper HW: Rac1 promotes TGF-beta-stimulated mesangial cell type
I collagen expression through a PI3K/Akt-dependent mechanism. Am J
Physiol Renal Physiol. 297:F1316–F1323. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kassan M, Galán M, Partyka M, Saifudeen Z,
Henrion D, Trebak M and Matrougui K: Endoplasmic reticulum stress
is involved in cardiac damage and vascular endothelial dysfunction
in hypertensive mice. Arterioscler Thromb Vasc Biol. 32:1652–1661.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Roberson EC, Tully JE, Guala AS, Reiss JN,
Godburn KE, Pociask DA, Alcorn JF, Riches DW, Dienz O,
Janssen-Heininger YM and Anathy V: Influenza induces endoplasmic
reticulum stress, caspase-12-dependent apoptosis, and c-Jun
N-terminal kinase-mediated transforming growth factor-β release in
lung epithelial cells. Am J Respir Cell Mol Biol. 46:573–581.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y,
Cheng J, Song J, Yu Q, Zhang S, et al: Chop deficiency prevents
UUO-induced renal fibrosis by attenuating fibrotic signals
originated from Hmgb1/TLR4/NFκB/IL-1β signaling. Cell Death Dis.
6(e1847)2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang Y, Liu L, Naik I, Braunstein Z, Zhong
J and Ren B: Transcription factor C/EBP homologous protein in
health and diseases. Front Immunol. 8(1612)2017.PubMed/NCBI View Article : Google Scholar
|














