Acute lung injury (ALI), a common and devastating
respiratory disease, is characterized by trans-epithelial
neutrophil migration, an uncontrolled inflammatory response, damage
caused to lung epithelial and endothelial cells, and destruction of
the associated cell barrier (1,2).
According to the newer Berlin definition (3,4), the
concept of ‘acute lung injury’ that was used for the milder form of
acute respiratory distress syndrome (ARDS; the definition of
ARDS/ALI is provided in Table I)
(4,5) in the former definition has been
discarded, although the term ‘ALI’ is still used for the milder
form in the present review. Among the pathophysiological features
of ALI, the destruction of lung vascular integrity is one of the
most important, as this leads to the flow of protein-rich fluid
into the alveoli, the accumulation of neutrophils in the pulmonary
microvasculature, and the release of toxic mediators from activated
neutrophils (e.g., proinflammatory cytokines and proteases)
(2,6-8).
ALI may be induced by direct causes (such as inhalation injury,
serious pneumonia and drowning) or indirect causes (such as trauma,
sepsis and drug overdose) (9).
Clinically, ALI is mainly responsible for causing hypoxemia,
pulmonary edema, bilateral lung infiltration and decreased lung
compliance, which leads to the high morbidity and mortality rates
observed in intensive care units (ICU) (10,11).
Several biomarkers have been shown to be closely associated with
the high morbidity and mortality rates that are due to ALI. To
date, a number of studies have demonstrated that tumor necrosis
factor-α, interleukin (IL)-1β, IL-6, IL-8 and IL-18 are the most
closely associated with the outcome of ALI (12,13).
In addition to these cytokines, alveolar epithelial biomarkers
(including surfactant D and the receptor for advanced glycation
end-products), protein C and plasminogen activator inhibitor-1 have
been shown to be associated with the prognosis of the disease
(14-16).
Although ALI continues to garner increasing levels
of attention, few useful clinical therapeutic methods are available
for the treatment of this disease. Lung-protective mechanical
ventilation, the main clinical treatment method available, is used
to improve the breathing condition of patients, thereby increasing
their survival rate, although the mortality rate due to ALI/ARDS
remains high (17,18). In addition, neither
anti-inflammatory drugs (such as corticosteroids) nor
β-adrenoceptor agonists have been demonstrated to effectively
reduce the mortality rate of patients with ALI (19,20).
Table II (21-32)
offers a summary of the research that has been conducted on the
pharmacological treatment of ALI to date. Current research studies
have indicated that stem cell-based therapies may potentially
provide an important means of treating ALI due to their
regenerative potential, stability and safety (33). Furthermore, microRNAs (miRNAs) have
been shown to function as potential biomarkers, are a therapeutic
target in animal models of ALI, and may ultimately serve as
putative biopharmaceuticals based on studies that have been
performed from the bench to the clinic (34,35).
However, there remain limitations and issues that need to be
explored and resolved. Essentially, it is necessary to further
understand the mechanisms underlying the pathophysiological process
of ALI, and to identify novel therapeutic approaches to improve the
survival rate and prognosis of patients with ALI (36). As a dynamic organelle, the
mitochondrion provides an important intracellular component that
allows cells to adapt to the environment, also participating in
stress sensing. The functions of mitochondria include bioenergetic,
biosynthetic and signaling aspects (37). For example, mitochondria produce
adenosine triphosphate (ATP) via oxidative phosphorylation
(OXPHOS). They also take up intracellular Ca2+ and
relieve the effects of toxicity associated with reactive oxygen
species (ROS) (38,39). Mitochondrial dysfunction usually
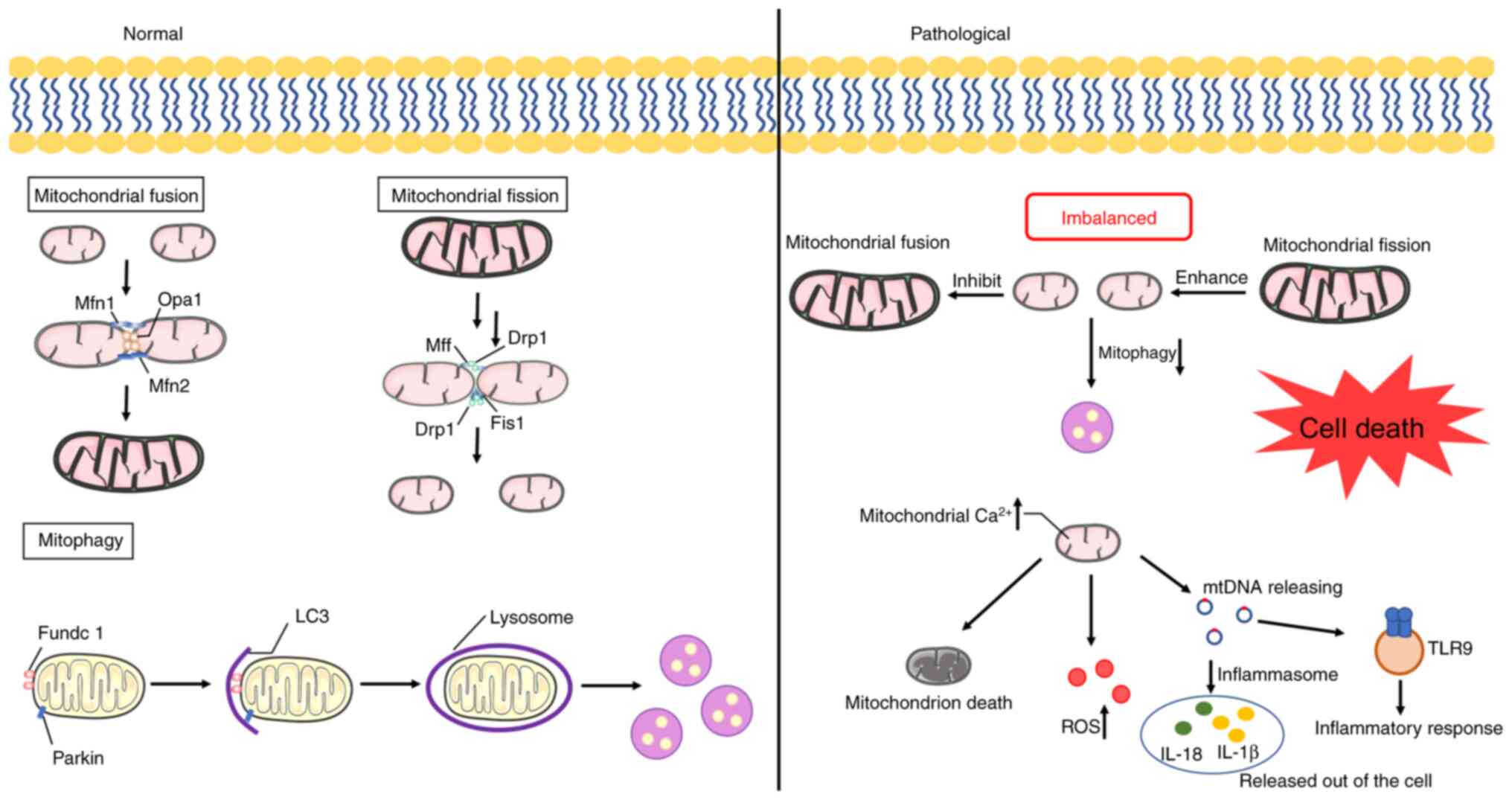
results in cell death, and even tissue damage (Fig. 1). In addition, mitochondrial
dynamics comprises one of the most critical features of
mitochondrial biology, being crucially involved in the
establishment and development of multiple types of lung disease
(40). Mitochondrial dynamics is a
quick and transient process involved in apoptosis, immunity,
cellular signaling, and the cell cycle (41). This process comprises a coordinated
cycle of fission and fusion of mitochondria that operates in order
to maintain their intracellular shape, size and distribution,
although this process differs according to the types of cells
involved, and the underlying molecular mechanism is known to be
associated with the pathogenesis of human diseases (42,43).
Therefore, deciphering the underlying mechanisms of mitochondrial
biology and mitophagy will help to strengthen our understanding of
these processes, leading to the development of possible new
treatments. To complete this review, the literature containing
keywords such as ‘mitochondria’ and ‘ALI’ was searched in PubMed;
most of the papers used were published in the last 5 years. The
present review will consequently first provide an outline of the
mitochondrial structure and the processes of mitochondrial biology
and mitophagy, and subsequently will summarize the current state of
play with research on the association of mitochondria with ALI,
also discussing the role of mitochondria in ALI.
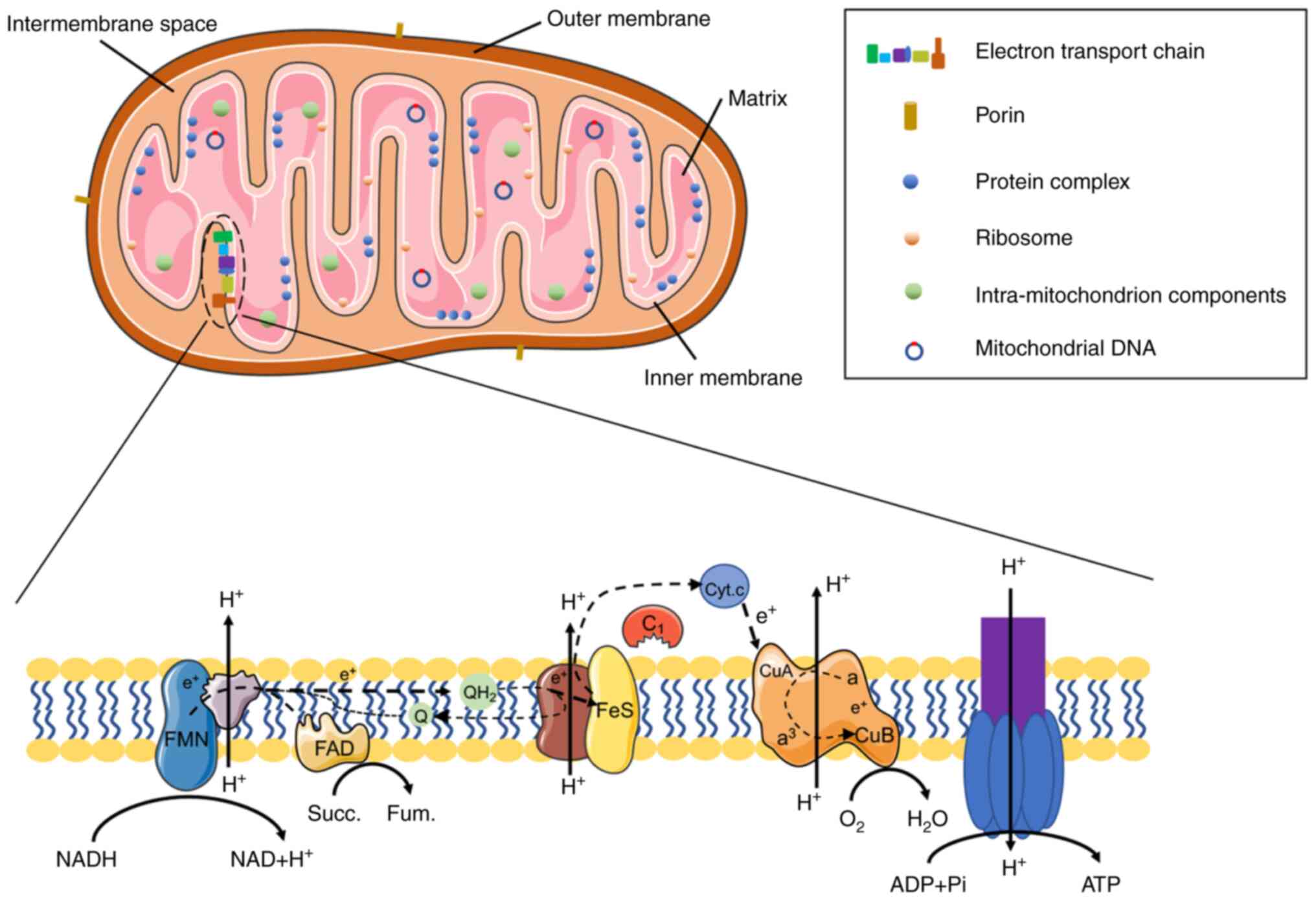
Mitochondria are double-membrane organelles that not
only have complex and special structures, but also perform numerous
functions. They exist in eukaryotic organisms and are located
around the cellular nuclei (44).
According to the current prevailing theory, it is considered that
mitochondria were derived from bacteria that formed new symbiotic
cells in combination with proto-eukaryotic cells, a fact that would
explain how the structure of a mitochondrion is similar to that of
a bacterium (45). A mitochondrion
is composed of an inner membrane, an outer membrane, the
intermembranous space, the aqueous spaces and the mitochondrial
matrix (46,47). The mitochondrial outer membrane is
permeable to molecules <5,000 Da in size that are able to enter
the mitochondrion through the channel proteins (48), whereas the mitochondrial inner
membrane is only minimally permeable to molecules and ions, and
OXPHOS is localized to the inner membrane (49,50).
Over 1,000 different types of protein reside in the spaces of the
mitochondrion, including the protein complexes from eukaryotic
organisms or bacteria, and approximately 500 of them are localized
in the human mitochondrial matrix (51-53).
In cells, most proteins are translated in the cytoplasm, and are
subsequently transported into the mitochondria via the translocase
of the outer membrane and the inner membrane (54,55).
In addition to proteins, the mitochondrion contains its own genome
in the mitochondrial DNA (mtDNA), which is a 16-kb circular
molecule that encodes associated electron transport chain (ETC)
proteins, rRNAs and tRNAs (56).
The ETC comprises the enzyme complexes I-IV, cytochrome c
and coenzyme Q (57) (Fig. 2). The status of mtDNA and the
associated nuclear-encoded proteins exert an influence on the
health, fertility and lifetime of organisms (58-61).
Furthermore, the level of compatibility between mtDNA and nuclear
genes has been shown to influence the genetic divergence (62,63).
The main function of the mitochondrion is to produce ATP via
OXPHOS, and to provide energy for the cells (64). Mitochondria can also function as a
protein-protein signaling platform that helps to maintain the
balance among several metabolic pathways, including the
tricarboxylic acid cycle (58,65,66).
Metabolites (ROS, cytochrome c and succinate) produced by
mitochondria are essential for cellular signal transduction, and
mitochondrion-associated signaling significantly contributes
towards the maintenance of cellular and body health (67,68).
Mitochondrial dynamics refers to the reshaping,
rebuilding and recycling process of mitochondria, which is mainly
divided into mitochondrial fusion and mitochondrial fission
(69). The mitochondrial fusion
process mainly occurs in the early S and G1 phases of the cell
cycle, in order to ensure the normal functioning of cellular
respiration and ATP production for the synthesis of proteins
(70). By contrast, the process of
mitochondrial fusion ensures that material exchange in the
mitochondrion and the removal of damaged intra-mitochondrial
molecules can occur, which helps to repair defective mitochondria
and further protect the mitochondrion from the process of
engulfment during mitophagy in the cell (71,72).
This fusion process includes the respective fusion events of both
the outer mitochondrial membrane (OMM) and the inner mitochondrial
membrane (IMM) (73), and the
sequence of events in the predominant molecular mechanism are as
follows. First, trans-complexes of mitofusins are formed through
dimerization of the transmembrane dynamin-like GTPases mitofusin 1
and mitofusin 2 (Mfn-1 and Mfn-2, respectively; the functions of
these proteins are determined by their tissue-specific mRNA and
protein expression), which is promoted via disulfide-bond
formation, causing OMMs to come into proximity with each other and
to be tethered, subsequently leading to fusion (74-77).
Secondly, fusion of the IMM is then facilitated by OXPHOS, which
accelerates the proteolytic action of the dynamin-associated GTPase
optic atropy-1 (Opa-1), and this process cleaves long Opa-1 into a
short and soluble form. Only the long Opa-1 (L-Opa-1) form is a
component of the IMM, which facilitates fusion of the IMM (78,79).
NM23-H4 is a type of nucleotide diphosphate kinase that promotes
the GTP loading of Opa-1 in the presence of ATP (80). Opa-1 GTP loading/hydrolysis and
S-S-proteolytic processing are two necessary steps in the process
of IMM fusion (80). Opa-1 also
has a key role in various mitochondria-associated cellular
functions, such as participating in the respiratory chain and
apoptosis (81). Thirdly, under
conditions of metabolic stress, such as that which occurs during
membrane potential dissipation, the metallopeptidase Oma-1 (an
inner membrane ATP-independent protease) exerts its function via
inducing Opa-1 to ultimately degrade into the short form of Opa-1
(S-Opa-1), which leads to damaged mitochondria being selected for
mitophagy (82). Mitochondrial
fission occurs mainly during the S-, G2- and M-phases of the cell
cycle in order to ensure that the mitochondria are distributed
equally in the daughter cells (83). Mitochondrial fission is a complex
and multistep process that serves a crucial role in the regulatory
mechanisms of cellular proliferation, differentiation, apoptosis,
mitochondrial quality control and ROS production (84-87).
For example, mitochondrial fission has been demonstrated to
accelerate the segregation and autophagy of damaged mitochondria
under stress conditions, which effectively reduces the accumulation
of dysfunctional mitochondria and subsequently ameliorates the
cellular stress conditions (88).
The GTPase dynamin-related protein 1 (Drp1/DNM1L) is the primary
driving factor of mitochondrial fission (89). The site of mitochondrial fission is
marked by an initial constriction at the OMM that is generated by
endoplasmic reticulum (ER) and actin filaments, which helps the
recruitment of Drp1 at the fission site (90). In mammals, although Drp1 is unable
to bind to the phospholipid membrane directly due to the loss of a
pleckstrin homology (PH) domain, Drp1 is nevertheless able to exert
this function via adaptor proteins [mitochondrial fission factor
(Mff) and mitochondrial dynamics proteins 51 and 49 (MiD51 and
MiD49)] (91-93).
Mitochondrial scission mainly occurs at the site of ER contact,
suggesting that both phospholipids [predominantly phosphatidic acid
(PA) and cardiolipin (CL)] and calcium transfer are indispensable
during this scission process (94,95).
Dynamin 2 (Dnm-2; a GTPase) is recruited at the contact site, where
Drp1 recruits and induces the membrane constriction, finally
leading to mitochondrial fission (30). There also exists another mechanism
of IMM fission which is dependent on calcium instead of Drp1. When
inverted formin 2 (INF2) proteins are recruited at the contact
site, more INF-2-mediated calcium enters into the mitochondria,
which leads to a decrease in the membrane potential, the cleavage
of Opa-1, and the activation of Oma-1(96). S-Opa-1 is an important component of
the mitochondrial contact site and the intermembrane space bridging
(MIB) complex. It controls the OMM-IMM tethering mediated by Mic60,
promotes the release of IMM tethering and possible shrinkage, and
ultimately regulates the mitochondrial inner compartment (CoMIC)
(97). S-Opa-1 participates in the
procedure of cristae morphogenesis and the tethering of the OMM
along with other associated proteins, such as mitofilin
(Mic60/Immt), ChchD3, ChchD6 and Sam50 (a type of outer membrane
protein) (97,98). In addition, there exists other
mitochondrial-fission regulatory proteins, including the
leucine-rich repeat kinase 2 (LRRK2) and the small GTPase, Rab32
(99,100).
Mitophagy clears damaged or dysfunctional
mitochondria to control the mitochondrial quality, and the
dysregulation of mitophagy is associated with a number of different
diseases (101). There are both
canonical and non-canonical modes that mediate the signaling
pathway in mitophagy. Mitophagy induced by PTEN-induced putative
kinase 1 (PINK1) and Parkin is the most common mechanism, which is
a multistep process of degrading unhealthy mitochondria via the
activation of PINK1, Parkin and other recruited proteins (102-104).
In a normal cellular environment, PINK1 protein at the OMM is
constitutively cleaved and degraded by mitochondrial processing
protease (MPP) and presenilin-associated rhomboid-like (PARL)
protein. When the mitochondrial membrane potential is perturbed,
and subsequently the mitochondrial membrane is depolarized, PINK1
is stabilized at the OMM since both MPP and PARL are inhibited,
which could be considered as the signal of mitochondrial
dysfunction (105). PINK1 is
activated via its autophosphorylation, which further leads to the
phosphorylation of its substrates. PINK1 phosphorylates serine-65
of ubiquitin to activate and recruit Parkin, which serves to
amplify the PINK1-initiated signal. Subsequently, activated Parkin
induces the ubiquitinylation of mitochondrial fusion-associated
proteins such as Mfn-1 and Mfn-2, which serves to prevent them from
participating in the fusion process (106,107). The ubiquitinylation of Miro1
protein induced by Parkin weakens the protein ability of Miro1 to
bind with microtubules, and strengthens the ability of this protein
to bind with the PINK1-Parkin complex, which causes the isolation
of associated damaged mitochondria (108). Concurrently, the phosphorylation
of the ubiquitin chain mediated by PINK1 enhances the recruitment
and activation of Parkin. Microtubule-associated protein 1 light
chain 3 (LC3) on the autophagosome directly interacts with
polyubiquitinylated proteins recognized by cargo adaptors, leading
finally to the formation of a complex that is degraded by autophagy
(109).
In other canonical mechanisms, both Bcl-2 homology 3
(BH3)-only protein (Nix, also known as Bnip3L) and BCL-2/adenovirus
E1-interacting protein 3 (Bnip3) not only interact with LC3, but
also exert their function under the regulation of
hypoxia-associated factors, which serves an important role in
mitophagy (110,111). FUN14 domain-containing 1
(FUNDC1), an OMM protein, is also associated with mitophagy through
its interaction with LC3, Opa-1 and Drp1 (112,113).
In addition, there are a variety of non-canonical
mitophagy pathways. Depolarized mitochondrial CL is able to
directly interact with GABAA receptor-associated protein (Gabarap)
in the OMM, and the oxidation status of CL serves to regulate the
balance between cytoprotective mitophagy and other mitochondrial
death pathways (114). Prohibitin
2, a receptor present in the IMM, is able to promote
Parkin-mediated mitophagy through interacting with LC3(115). Furthermore, Beclin-1 regulator 1
(AMBRA1) and Bcl-2-like protein 13 (BCL-2L13), as mitophagy
receptors, have been shown to induce and promote mitophagy
(116,117). BCL-2L13 is also involved in
Parkin-independent mitophagy (118).
Considering all the evidence, it has been clearly
demonstrated that dysfunction of mitophagy leads to the
accumulation of damaged mitochondria, which induces oxidative
stress and various pathological states.
With the ETC (also called the respiratory chain), a
liberation of electrons results from the oxidation of NADH and
FADH2. The liberated electrons are passed along the
carrier complexes, and eventually transferred to an oxygen molecule
(119). ROS derived from
mitochondrial superoxide are mainly produced by Complex I/III of
the ETC in mitochondria (120).
Heightened ROS production is induced by infection, inflammation,
air pollution and oxidative stress, supporting the notion that ALI
may lead to the high levels of ROS that are observed (121). Low concentrations of ROS and
superoxide (such as peroxynitrite and hydrogen peroxide) are
considered to be important components of a normally functioning
cellular signaling pathway, whereas high levels of these molecules
are able to induce ETC damage under pathological conditions
(122). As one of the underlying
causes of ALI, sepsis also has an impact on the function of the
ETC. It has been demonstrated that the level of mitochondrial ROS
increases in numerous organs, which results in abnormalities of the
ETC in the same organs, as observed in cases of sepsis (123,124). It has been shown that the
concentrations of ETC proteins that are associated with or contain
iron-sulfur centers are reduced in sepsis, since Fenton reactions
induce the depletion of ETC protein constituents (125). When the mitochondrial ETC
components are damaged or even lost in cases of severe disease, the
decreased production of ATP may accelerate the pathological
processes of certain diseases (126-128).
In addition to the direct damage caused to ETC
proteins, an abnormal expression of ROS derived from mitochondria
may influence other cellular constituents, and the interactions
between ROS and these constituents subsequently alter their
function; this includes proteins, DNA, and lipid peroxidation
(129,130). Since mtDNA lacks protective
histones and the expression of bases modified by oxidation in mtDNA
is several times higher compared with that in nuclear DNA, mtDNA is
more easily impaired by ROS (131,132). It has been shown that superoxide
anions produced by mitochondria have a limited ability to directly
pass through the mitochondrial membranes, although they exit the
mitochondrion more easily by forming new molecular species and
reacting with cellular components in the cytoplasm (133,134). For example, under conditions in
which the generation of nitric oxide significantly increases,
nitric oxide may combine with superoxide forming peroxynitrite,
which is able to impair proteins and modify lipids (135). Moreover, ROS oxidize proteins and
alter their activity, promoting the release of proteases and
inhibiting the activation of antioxidant enzymes (136). In ALI, the overproduction of ROS
is widely derived from parenchymal cells, a high concentration of
oxygen and oxidant-generating enzymes, leading to the induction of
oxidative stress and cell damage (137,138).
Mitochondrial dysfunction in lung cells has an
important role in the pathological process of ALI (146). The main pathological feature of
ALI is the infiltration of inflammatory cells, such as macrophages
and neutrophils (147,148). Mitochondria are involved in the
modulation of immune cells via different mechanisms. It has been
demonstrated that mitochondrial ROS can stimulate the activation of
macrophage-surface Toll-like receptors (TLRs), enhancing their
anti-pathogenic ability (149).
Triggering receptor expressed on myeloid cells 1 (TREM-1) maintains
the integrity of mitochondria to prolong the survival of
macrophages which plays a key role in ALI (150). Moreover, macrophages can be
divided into two phenotypes, M1 (proinflammatory) and M2
(anti-inflammatory) respectively, which are associated with the
bioenergetic function of mitochondria and are an essential part in
the process of lung infection and inflammation (151,152). The mitochondrial ETC is involved
in the activation of lipopolysaccharide (LPS)-induced nuclear
factor-κB (NF-κB), suggesting that mitochondria alleviate the
degree of damage of ALI by regulating neutrophils (153).
LPS is known to damage alveolar epithelial cells and
is one of the main causes of ALI. Islam et al found that
mitochondria derived from bone marrow-derived stromal cells were
released in the microvesicles engulfed by the alveolar cells, which
increased the concentration of alveolar ATP and decreased the
mortality of animal models in LPS-induced ALI (154). In current research, the heme
oxygenase-1/carbon monoxide system has been revealed to alleviate
ALI induced by endotoxin via regulating the mitochondrial dynamic
equilibrium (155,156). In LPS-induced ALI rat models, the
expression of Mfn-1 is negatively regulated by HO-1 expression
possibly related to the PI3K/Akt signaling pathway, which can
improve the condition of oxidative stress by regulating
mitochondrial fusion (157). The
mitochondrial division inhibitor-1 (Mdivi-1) has the ability to
relieve the activation of mitogen-activated protein kinases
(MAPKs), oxidative stress and apoptosis induced by LPS and reduce
pro-inflammatory cytokine release, which inhibits the mitochondrial
fission and mitigates the degree of damage by ALI (158). In LPS-induced ALI, the severity
of inflammation and lung injury can be restrained by regulating the
Drp1-induced mitochondrial fission (159). Dexmedetomidine (DEX) affords lung
protection and mitigates the damage of ALI by keeping the dynamic
balance between mitochondrial fusion and fission via the
HIF-1a/HO-1 pathway (160).
Normal mitophagy maintains the homeostasis of cells by cleaving and
degrading damaged mitochondria, while excessive mitophagy may lead
to mitochondrial dysfunction, cell damage and death. Sestrin2
(Sesn2), a highly conserved protein, protects alveolar macrophages
and reduces the release of the Nod-like receptor protein 3 (NLRP3)
inflammasome by promoting mitophagy, which finally plays a
protective role in LPS-induced ALI (161). Transcription factor EB (TFEB)
negatively regulates mitophagy and decreases mitochondrial injury
to protect LPS-induced ALI (162,163). Overexpression of PPARγ
coactivator 1α (PGC-1α) may positively regulate the expression of
TFEB and then affect mitophagy, which in turn alleviates lung edema
and decreases inflammation in LPS-induced ALI (164). Zhao et al demonstrated
that oxyberberine inhibited the translation of Parkin1 from the
cytoplasm to mitochondria and Parkin-mediated mitophagy to ease the
degree of inflammation in LPS-induced ALI (165). Moreover, overexpression of Bcl-2
proteins also attenuated LPS-induced ALI via PINK1/Parkin-mediated
mitophagy (166).
The lungs are one of the organs most often affected
by sepsis, which usually leads to ALI. Damage-associated molecular
pattern (DAMP) is the general term for numerous endogenous risk
molecules, existing in the nucleus, mitochondria, or cytoplasm
(167,168), which are released in response to
cell death or stress (169,170). The released DAMPs are recognized
and bond to multiple receptors which include pattern recognition
receptors (PRRs), and then activate downstream pathways to trigger
the inflammatory response, aggravating the damage of the lungs
(171,172). mtDNA, a type of cellular toxicity
compound, acts as a DAMP and contains materials only found in
bacteria and induces cellular toxicity via two main mechanisms
(173,174). The first one is to activate and
interact with NLRP3 inflammasome, and the second one is to
recognize the bacteria-like mtDNA via the activation of TLR9
(175,176). For example, the release of mtDNA
depends on the level of TLR4, and mtDNA induces ALI together with
TLR9(177). In addition, mtDNAs,
as mitochondrial DAMPs, increase the permeability of lung
endothelial cells in sepsis-induced ALI (178). The balance between mitochondrial
fusion and fission is broken when massive ROS exist, which
accelerate the progression of sepsis and are an indirect cause of
ALI (179). Chen et al
found that PINK1/Parkin-mediated mitophagy played a protective role
in cecal ligation and puncture (CLP)-induced ALI (180). It has been proven that Nrf2
regulates mitophagy in lung cells and exerts a protective function
in sepsis (181). MAP kinase
kinase 3 (MKK3) promotes the activation of mitochondrial biogenesis
and mitophagy through the PINK1/Parkin pathway and PGC-1α/Nrf-1
axis, which in turn increase the number of healthy mitochondria and
protect against sepsis-induced ALI (182).
Reportedly, hydrochloric acid-induced ALI may result
in the direct damage of the alveolar epithelium and then induce
proinflammatory signaling (183,184). However, the pathophysiological
mechanisms of hydrochloric acid-induced ALI are still not clear.
Hough et al found the mitochondrial function of alveolar
cells impaired in hydrochloric acid-induced ALI (185). Acute PM2.5 exposure was related
to enhanced airway inflammation, immune cell infiltration, and the
release of proinflammatory cytokines and chemokines, inducing ALI
(186,187). The activation of the
TLR4/NF-κB/p38 MAPK and NLRP3/caspase-1 signaling pathways may
inhibit ALI and mitochondrial damage by regulating the expression
of the related mitochondrial fusion and fission proteins, such as
Opa-1, Drp1, and Mfn-2(188).
Ischemia/reperfusion injury (IRI) usually includes the release of
cytokines and inflammatory mediators, extensive oxidative stress,
and the induction of apoptosis, increasing the dysfunction and
damage of lungs (189).
Tanshinone IIA (TIIA) combined with cyclosporine A (CsA) attenuated
the apoptosis of the lung tissue by improving the mitochondrial
dynamics via the PI3K/Akt/Bad signaling pathway (190). Prolonged, high oxygen
concentration promoted the production of ROS and the level of
proapoptotic proteins, finally inducing ALI. Research has shown
that thyroid hormone T3 increases mitochondrial biogenesis and
mitophagy, thus providing effective protection in hyperoxia-induced
ALI (191).
In summary, the dysfunction of mitochondria plays a
crucial role in ALI. These findings suggest the potential of
mitochondrial biology and mitophagy as targets for the treatment
and intervention of ALI.
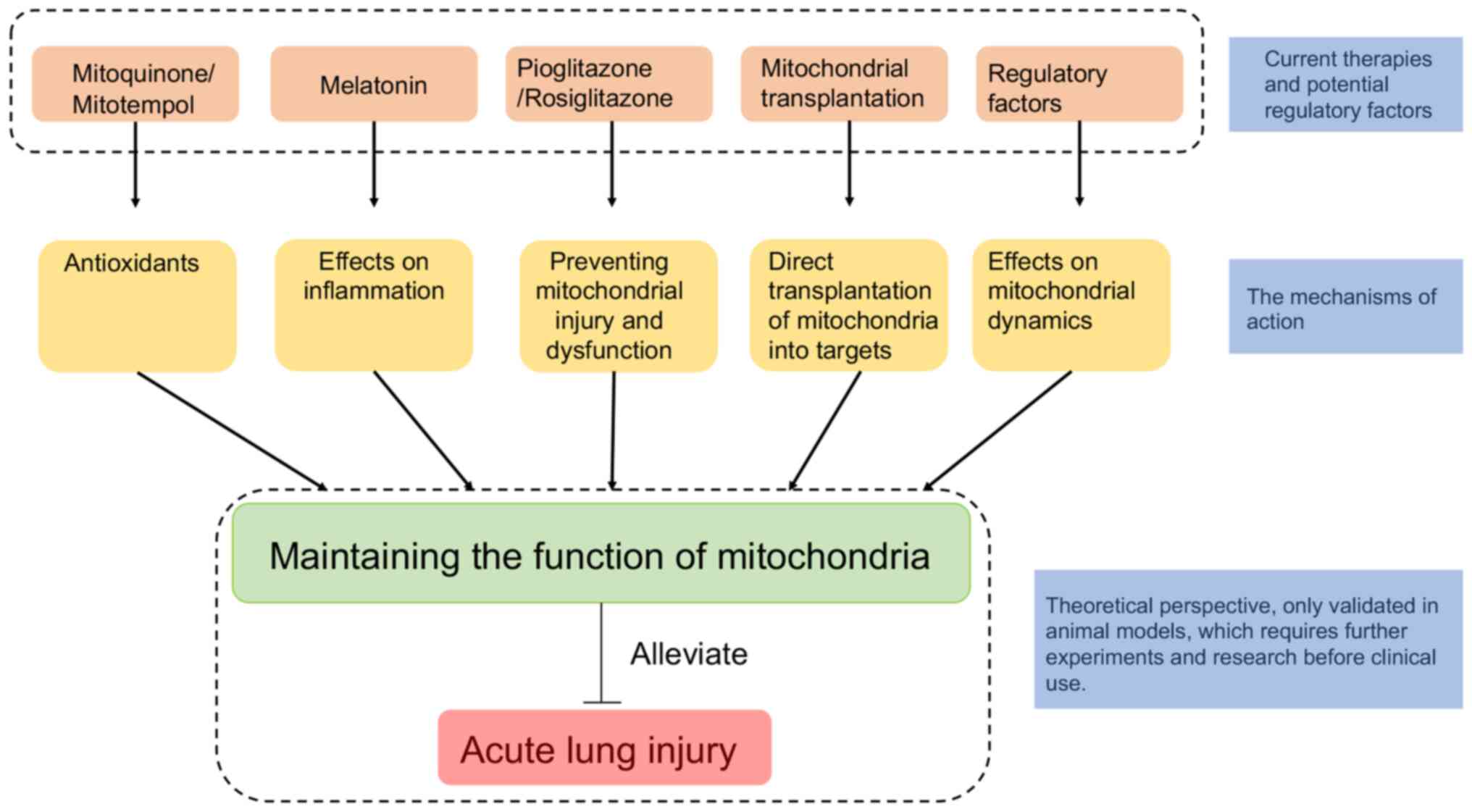
Mitochondria play an indispensable role in the
occurrence and development of ALI. In addition, in ALI models,
cells undergo abnormal mitochondrial biological processes or
mitophagy. Thus, factors associated with mitochondrial
pathophysiology may be the potential therapeutic targets for ALI
(Fig. 3) (192). Mitochondrial-targeted
antioxidants can protect against the mitochondrial dysfunction and
oxidative stress induced by mechanical ventilation, suggesting
improvement of the prognosis of ALI treated by mechanical
ventilation (193). Research has
also demonstrated that both pioglitazone and rosiglitazone
effectively induce mitochondrial biogenesis and prevent the related
cell dysfunction and damage (194). Mitochondrial transplantation, as
an important method to replace damaged mitochondria, can
significantly improve the condition of lungs and reduce the lung
tissue damage induced by ALI (195). Melatonin can effectively inhibit
superoxide and nitric oxide and protect against the mitochondrial
damage (196-198).
In addition to current therapies, there are also
certain factors that regulate mitochondrial dynamics, which may aid
in understanding the role of mitochondria in ALI and may be
translated into novel therapies in the future. Membrane lipid
composition and post-translation modification are two main factors
that regulate mitochondrial dynamics. CL and PA are two minor
constituents of phospholipids, but they are both involved in the
remodeling of the mitochondrial membrane. PA, a saturated lipid
synthesized in the endoplasmic reticulum (ER), is transferred from
the ER to the mitochondria and then converted into CL at the inner
mitochondrial membrane (199).
The respective microdomain formation of PA and CL determines
mitochondrial fusion or fission (200-202).
It has been shown that Drp1 can interact with these two
phospholipids to influence mitochondrial fusion or fission (200,
203-205). Drp1 is the core protein of mitochondrial dynamics, and
how post-translational modifications of Drp1 regulate mitochondrial
dynamics has been widely explored (206-208).
In addition to membrane lipid composition and post-translation
modification, there are also other proteins that modulate this
dynamic process, including ganglioside-induced differentiation
associated protein 1 (GDAP1) (209), mitochondrial fission process 1
(MTFP1/MTP18) (210), and
reactive oxygen species modulator 1 (ROMO1) (211).
To sum up, there is still a long way to go before
these therapies and regulatory factors can be formally used in the
clinic, because some of these potential treatments are still
speculative and others have only been verified in animal
models.
The present review summarized the structure,
function, pathophysiology of mitochondria and the role of
mitochondria in ALI, which could pave the way to provide novel
therapeutic methods to treat ALI.
ALI is a complex and severe pathological disease
with high morbidity and mortality in the ICU, which triggers the
sustaining inflammatory response, lung epithelial and endothelial
cell death, and alveolar barrier destruction (212). Mitochondria are considered to be
the powerhouse of cells, take part in metabolite biosynthesis and
produce ROS (213). They have
also been proven to be involved in necrosis, immunological
response, thermoregulation, and intracellular calcium regulation
(214,215). Generally, the dysfunction of
mitochondria usually occurs in the pathophysiological processes of
diseases.
Mitochondria play an important role in ALI.
Macrophages and neutrophils are essential effector cells that are
involved in ALI, and mitochondria regulate the polarization of
macrophages and the apoptosis and NETosis of neutrophils (216,217). In addition, mitochondrial
dynamics and mitophagy are associated with the outcome of ALI.
Collectively, mtDNA, as a DAMP, induces ALI, and ROS produced by
mitochondria affect the process and outcome of ALI.
However, there are some remaining issues that need
to be addressed. The research on mitochondria-related elements in
ALI is still in its infancy, and the changes in mitochondria and
regulatory factors are complex and interactive. Moreover, the
research concerning the role of mitochondria in ALI is based on
animal models, warranting more experiments, in order to be brought
into clinical practice.
Not applicable.
Funding: The present review was supported by the Mass Chemical
Injury First-Aid, Core Discipline Improvement Project, 3-year
(2020-2022) Action Plan of Shanghai Public Health System
Development (program no. GWV-10.1-XK26; budget no. 1304), the
Emergency and Critical Care Centre, Discipline Platform Improvement
Project, 3-year Talents Echelon Action Plan of Jinshan Hospital
(program no. XPT-2020-3; budget no. 1257), and the Emergency and
Critical Care, Class A, Core Medicine Discipline Improvement
Project of Jinshan District, 6th Season (program no. JSZK2019A01;
budget no. 1195).
Not applicable.
BZ and JS conceived and designed the review,
collected and analyzed the data, and co-wrote the manuscript. JS
reviewed the manuscript. Data authentication is not applicable.
Both authors (BZ and JS) read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Jiang Z, Zhang L and Shen J: MicroRNA:
Potential biomarker and target of therapy in acute lung injury. Hum
Exp Toxicol. 39:1429–1442. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mowery NT, Terzian WTH and Nelson AC:
Acute lung injury. Curr Probl Surg. 57(100777)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bernard G, Artigas A, Brigham K, Carlet J,
Falke K, Hudson L, Lamy M, Legall JR, Morris A and Spragg R: Report
of the American-European consensus conference on ARDS: Definitions,
mechanisms, relevant outcomes and clinical trial coordination. The
consensus committee. Intensive Care Med. 20:225–232.
1994.PubMed/NCBI View Article : Google Scholar
|
|
4
|
ARDS Definition Task Force. Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hudson LD, Milberg JA, Anardi D and
Maunder RJ: Clinical risks for development of the acute respiratory
distress syndrome. Am J Respir Crit Care Med. 151:293–301.
1995.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Looney MR, Su X, Van Ziffle JA, Lowell CA
and Matthay MA: Neutrophils and their Fc gamma receptors are
essential in a mouse model of transfusion-related acute lung
injury. J Clin Invest. 116:1615–1623. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Martin T, Hagimoto N, Nakamura M and
Matute-Bello G: Apoptosis and epithelial injury in the lungs. Proc
Am Thorac Soc. 2:214–220. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Weiser M, Pechet T, Williams J, Ma M,
Frenette PS, Moore FD, Kobzik L, Hines RO, Wagner DD, Carroll MC
and Hechtman HB: Experimental murine acid aspiration injury is
mediated by neutrophils and the alternative complement pathway. J
Appl Physiol (1985). 83:1090–1095. 1997.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Spadaro S, Park M, Turrini C, Tunstall T,
Thwaites R, Mauri T, Ragazzi R, Ruggeri P, Hansel TT, Caramori G
and Volta CA: Biomarkers for acute respiratory distress syndrome
and prospects for personalised medicine. J Inflamm (Lond).
16(1)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huang X, Xiu H, Zhang S and Zhang G: The
role of macrophages in the pathogenesis of ALI/ARDS. Mediators
Inflamm. 2018(1264913)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Han S and Mallampalli RK: The acute
respiratory distress syndrome: From mechanism to translation. J
Immunol. 194:855–860. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Meduri G, Kohler G, Headley S, Tolley E,
Stentz F and Postlethwaite A: Inflammatory cytokines in the BAL of
patients with ARDS. Persistent elevation over time predicts poor
outcome. Chest. 108:1303–1314. 1995.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Parsons PE, Eisner MD, Thompson BT,
Matthay MA, Ancukiewicz M, Bernard GR and Wheeler AP: NHLBI Acute
Respiratory Distress Syndrome Clinical Trials Network. Lower tidal
volume ventilation and plasma cytokine markers of inflammation in
patients with acute lung injury. Crit Care Med. 33:1–6, 230-232.
2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Greene KE, Wright JR, Steinberg KP,
Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T,
Kuroki Y, et al: Serial changes in surfactant-associated proteins
in lung and serum before and after onset of ARDS. Am J Respir Crit
Care Med. 160:1843–1850. 1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Terpstra ML, Aman J, van Nieuw Amerongen
GP and Groeneveld AB: Plasma biomarkers for acute respiratory
distress syndrome: A systematic review and meta-analysis*. Crit
Care Med. 42:691–700. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ware LB, Matthay MA, Parsons PE, Thompson
BT, Januzzi JL and Eisner MD: National Heart, Lung and Blood
Institute Acute Respiratory Distress Syndrome Clinical Trials
Network. Pathogenetic and prognostic significance of altered
coagulation and fibrinolysis in acute lung injury/acute respiratory
distress syndrome. Crit Care Med. 35:1821–1828. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: Epidemiology, patterns of care, and mortality for
patients with acute respiratory distress syndrome in intensive care
units in 50 countries. JAMA. 315:788–800. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Peter JV, John P, Graham PL, Moran JL,
George IA and Bersten A: Corticosteroids in the prevention and
treatment of acute respiratory distress syndrome (ARDS) in adults:
Meta-analysis. BMJ. 336:1006–1009. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
McAuley DF and Matthay MA: Is there a role
for beta-adrenoceptor agonists in the management of acute lung
injury and the acute respiratory distress syndrome? Treat Respir
Med. 4:297–307. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bernard GR, Luce JM, Sprung CL, Rinaldo
JE, Tate RM, Sibbald WJ, Kariman K, Higgins S, Bradley R, Metz CA,
et al: High-dose corticosteroids in patients with the adult
respiratory distress syndrome. N Engl J Med. 317:1565–1570.
1987.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Steinberg KP, Hudson LD, Goodman RB, Hough
CL, Lanken PN, Hyzy R, Thompson BT and Ancukiewicz M: National
Heart, Lung and Blood Institute Acute Respiratory Distress Syndrome
(ARDS) Clinical Trials Network. Efficacy and safety of
corticosteroids for persistent acute respiratory distress syndrome.
N Engl J Med. 354:1671–1684. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Doig C: Aerosolized surfactant in
sepsis-induced adult respiratory distress syndrome. JAMA.
273:1259–1260. 1995.PubMed/NCBI
|
|
24
|
Domenighetti G, Suter PM, Schaller MD,
Ritz R and Perret C: Treatment with N-acetylcysteine during acute
respiratory distress syndrome: A randomized, double-blind,
placebo-controlled clinical study. J Crit Care. 12:177–182.
1997.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Dellinger RP, Zimmerman JL, Taylor RW,
Straube RC, Hauser DL, Criner GJ, Davis K Jr, Hyers TM and
Papadakos P: Effects of inhaled nitric oxide in patients with acute
respiratory distress syndrome: Results of a randomized phase II
trial. Inhaled nitric oxide in ARDS study group. Crit Care Med.
26:15–23. 1998.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vincent JL, Brase R, Santman F, Suter PM,
McLuckie A, Dhainaut JF, Park Y and Karmel J: A multi-centre,
double-blind, placebo-controlled study of liposomal prostaglandin
E1 (TLC C-53) in patients with acute respiratory distress syndrome.
Intensive Care Med. 27:1578–1583. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Abraham E, Carmody A, Shenkar R and
Arcaroli J: Neutrophils as early immunologic effectors in
hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol
Lung Cell Mol Physiol. 279:L1137–L1145. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ketoconazole for early treatment of acute
lung injury and acute respiratory distress syndrome: A randomized
controlled trial. The ARDS network. JAMA. 283:1995–2002.
2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Randomized placebo-controlled trial of
lisofylline for early treatment of acute lung injury and acute
respiratory distress syndrome. Crit Care Med. 30:1–6.
2002.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Perkins GD, Gates S, Park D, Gao F, Knox
C, Holloway B, McAuley DF, Ryan J, Marzouk J, Cooke MW, et al: The
beta agonist lung injury trial prevention. A randomized controlled
trial. Am J Respir Crit Care Med. 189:674–683. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Morris PE, Papadakos P, Russell JA,
Wunderink R, Schuster DP, Truwit JD, Vincent JL and Bernard GR: A
double-blind placebo-controlled study to evaluate the safety and
efficacy of L-2-oxothiazolidine-4-carboxylic acid in the treatment
of patients with acute respiratory distress syndrome. Crit Care
Med. 36:782–788. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu KD, Levitt J, Zhuo H, Kallet RH, Brady
S, Steingrub J, Tidswell M, Siegel MD, Soto G, Peterson MW, et al:
Randomized clinical trial of activated protein C for the treatment
of acute lung injury. Am J Respir Crit Care Med. 178:618–623.
2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Murphy MB, Moncivais K and Caplan AI:
Mesenchymal stem cells: Environmentally responsive therapeutics for
regenerative medicine. Exp Mol Med. 45(e54)2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rajasekaran S, Pattarayan D, Rajaguru P,
Sudhakar Gandhi P and Thimmulappa RK: MicroRNA regulation of acute
lung injury and acute respiratory distress syndrome. J Cell
Physiol. 231:2097–2106. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chakraborty C, Sharma AR, Sharma G, Doss
CGP and Lee SS: Therapeutic miRNA and siRNA: Moving from bench to
clinic as next generation medicine. Mol Ther Nucleic Acids.
8:132–143. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Johnson ER and Matthay MA: Acute lung
injury: Epidemiology, pathogenesis, and treatment. J Aerosol Med
Pulm Drug Deliv. 23:243–252. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Porporato PE, Filigheddu N, Pedro JMB,
Kroemer G and Galluzzi L: Mitochondrial metabolism and cancer. Cell
Res. 28:265–280. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bar-Ziv R, Bolas T and Dillin A: Systemic
effects of mitochondrial stress. EMBO Rep.
21(e50094)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Anderson AJ, Jackson TD, Stroud DA and
Stojanovski D: Mitochondria-hubs for regulating cellular
biochemistry: Emerging concepts and networks. Open Biol.
9(190126)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rowlands DJ: Mitochondria dysfunction: A
novel therapeutic target in pathological lung remodeling or
bystander? Pharmacol Ther. 166:96–105. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wu H, Wei H, Sehgal SA, Liu L and Chen Q:
Mitophagy receptors sense stress signals and couple mitochondrial
dynamic machinery for mitochondrial quality control. Free Radic
Biol Med. 100:199–209. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gottlieb RA and Stotland A: MitoTimer: A
novel protein for monitoring mitochondrial turnover in the heart. J
Mol Med (Berl). 93:271–278. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Collins TJ, Berridge MJ, Lipp P and
Bootman MD: Mitochondria are morphologically and functionally
heterogeneous within cells. EMBO J. 21:1616–1627. 2002.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Rongvaux A: Innate immunity and tolerance
toward mitochondria. Mitochondrion. 41:14–20. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
de-Lima-Júnior JC, Souza GF, Moura-Assis
A, Gaspar RS, Gaspar JM, Rocha AL, Ferrucci DL, Lima TI, Victório
SC, Bonfante ILP, et al: Abnormal brown adipose tissue
mitochondrial structure and function in IL10 deficiency.
EBioMedicine. 39:436–447. 2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Vögtle FN, Burkhart JM, Gonczarowska-Jorge
H, Kücükköse C, Taskin AA, Kopczynski D, Ahrends R, Mossmann D,
Sickmann A, Zahedi RP and Meisinger C: Landscape of
submitochondrial protein distribution. Nat Commun.
8(290)2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ralto KM and Parikh SM: Mitochondria in
acute kidney injury. Semin Nephrol. 36:8–16. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Abate M, Festa A, Falco M, Lombardi A,
Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia
M and Misso G: Mitochondria as playmakers of apoptosis, autophagy
and senescence. Semin Cell Dev Biol. 98:139–153. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Cogliati S, Enriquez JA and Scorrano L:
Mitochondrial cristae: Where beauty meets functionality. Trends
Biochem Sci. 41:261–273. 2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Rath S, Sharma R, Gupta R, Ast T, Chan C,
Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al:
MitoCarta3.0: An updated mitochondrial proteome now with
sub-organelle localization and pathway annotations. Nucleic Acids
Res. 49:D1541–D1547. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Smith AC and Robinson AJ: MitoMiner v3.1,
an update on the mitochondrial proteomics database. Nucleic Acids
Res. 44 (D1):D1258–D1261. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Rhee HW, Zou P, Udeshi ND, Martell JD,
Mootha VK, Carr SA and Ting AY: Proteomic mapping of mitochondria
in living cells via spatially restricted enzymatic tagging.
Science. 339:1328–1331. 2013.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hartl FU and Neupert W: Protein sorting to
mitochondria: Evolutionary conservations of folding and assembly.
Science. 247:930–938. 1990.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Wiedemann N and Pfanner N: Mitochondrial
machineries for protein import and assembly. Annu Rev Biochem.
86:685–714. 2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Boczonadi V, Ricci G and Horvath R:
Mitochondrial DNA transcription and translation: Clinical
syndromes. Essays Biochem. 62:321–340. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Gilkerson RW, Selker JM and Capaldi RW:
The cristal membrane of mitochondria is the principal site of
oxidative phosphorylation. FEBS Lett. 546:355–358. 2003.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Youle RJ: Mitochondria-striking a balance
between host and endosymbiont. Science.
365(eaaw9855)2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Aw WC, Towarnicki SG, Melvin RG, Youngson
NA, Garvin MR, Hu Y, Nielsen S, Thomas T, Pickford R, Bustamante S,
et al: Genotype to phenotype: Diet-by-mitochondrial DNA haplotype
interactions drive metabolic flexibility and organismal fitness.
PLoS Genet. 14(e1007735)2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Gorman GS, Schaefer AM, Ng Y, Gomez N,
Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery
PF, et al: Prevalence of nuclear and mitochondrial DNA mutations
related to adult mitochondrial disease. Ann Neurol. 77:753–759.
2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Melvin RG and Ballard JW: Intraspecific
variation in survival and mitochondrial oxidative phosphorylation
in wild-caught Drosophila simulans. Aging Cell. 5:225–233.
2006.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Princepe D and De Aguiar MAM: Modeling
Mito-nuclear compatibility and its role in species identification.
Syst Biol. 70:133–144. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Telschow A, Gadau J, Werren J and
Kobayashi Y: Genetic incompatibilities between mitochondria and
nuclear genes: Effect on gene flow and speciation. Front Genet.
10(62)2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Roth KG, Mambetsariev I, Kulkarni P and
Salgia R: The mitochondrion as an emerging therapeutic target in
cancer. Trends Mol Med. 26:119–134. 2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Liu X, Kim CN, Yang J, Jemmerson R and
Wang X: Induction of apoptotic program in cell-free extracts:
Requirement for dATP and cytochrome c. Cell. 86:147–157.
1996.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Bossy-Wetzel E, Newmeyer DD and Green DR:
Mitochondrial cytochrome c release in apoptosis occurs upstream of
DEVD-specific caspase activation and independently of mitochondrial
transmembrane depolarization. EMBO J. 17:37–49. 1998.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Hine C, Harputlugil E, Zhang Y,
Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Treviño-Villarreal JH,
Mejia P, Ozaki CK, et al: Endogenous hydrogen sulfide production is
essential for dietary restriction benefits. Cell. 160:132–144.
2015.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Eisner V, Picard M and Hajnóczky G:
Mitochondrial dynamics in adaptive and maladaptive cellular stress
responses. Nat Cell Biol. 20:755–765. 2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Mitra K, Wunder C, Roysam B, Lin G and
Lippincott-Schwartz J: A hyperfused mitochondrial state achieved at
G1-S regulates cyclin E buildup and entry into S phase. Proc Natl
Acad Sci USA. 106:11960–11965. 2009.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Rambold A, Kostelecky B, Elia N and
Lippincott-Schwartz J: Tubular network formation protects
mitochondria from autophagosomal degradation during nutrient
starvation. Proc Natl Acad Sci USA. 108:10190–10195.
2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Aiello A, Cristofaro M, Carrozza F,
Verdone F and Carile L: Lymphocyte subpopulations and the soluble
interleukin-2 receptor in Hashimoto's thyroiditis and subacute
thyroiditis. Clin Ter. 133:401–404. 1990.PubMed/NCBI(In Italian).
|
|
73
|
Leduc-Gaudet JP, Hussain SNA, Barreiro E
and Gouspillou G: Mitochondrial dynamics and mitophagy in skeletal
muscle health and aging. Int J Mol Sci. 22(8179)2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Santel A, Frank S, Gaume B, Herrler M,
Youle RJ and Fuller MT: Mitofusin-1 protein is a generally
expressed mediator of mitochondrial fusion in mammalian cells. J
Cell Sci. 116:2763–2774. 2003.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Eura Y, Ishihara N, Yokota S and Mihara K:
Two mitofusin proteins, mammalian homologues of FZO, with distinct
functions are both required for mitochondrial fusion. J Biochem.
134:333–344. 2003.PubMed/NCBI View Article : Google Scholar
|
|
76
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Detmer SA and Chan DC: Complementation
between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects
caused by CMT2A disease mutations. J Cell Biol. 176:405–414.
2007.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Ehses S, Raschke I, Mancuso G, Bernacchia
A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI and
Langer T: Regulation of OPA1 processing and mitochondrial fusion by
m-AAA protease isoenzymes and OMA1. J Cell Biol. 187:1023–1036.
2009.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Mishra P, Carelli V, Manfredi G and Chan
DC: Proteolytic cleavage of Opa1 stimulates mitochondrial inner
membrane fusion and couples fusion to oxidative phosphorylation.
Cell Metab. 19:630–641. 2014.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Schlattner U, Tokarska-Schlattner M,
Ramirez S, Tyurina YY, Amoscato AA, Mohammadyani D, Huang Z, Jiang
J, Yanamala N, Seffouh A, et al: Dual function of mitochondrial
Nm23-H4 protein in phosphotransfer and intermembrane lipid
transfer: A cardiolipin-dependent switch. J Biol Chem. 288:111–121.
2013.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Griparic L, van der Wel NN, Orozco IJ,
Peters PJ and van der Bliek AM: Loss of the intermembrane space
protein Mgm1/OPA1 induces swelling and localized constrictions
along the lengths of mitochondria. J Biol Chem. 279:18792–18798.
2004.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Pernas L and Scorrano L: Mito-morphosis:
Mitochondrial fusion, fission, and cristae remodeling as key
mediators of cellular function. Annu Rev Physiol. 78:505–531.
2016.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Margineantu DH, Gregory Cox W, Sundell L,
Sherwood SW, Beechem JM and Capaldi RA: Cell cycle dependent
morphology changes and associated mitochondrial DNA redistribution
in mitochondria of human cell lines. Mitochondrion. 1:425–435.
2002.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Mitra K: Mitochondrial fission-fusion as
an emerging key regulator of cell proliferation and
differentiation. Bioessays. 35:955–964. 2013.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Diebold L and Chandel NS: Mitochondrial
ROS regulation of proliferating cells. Free Radic Biol Med.
100:86–93. 2016.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Jheng HF, Tsai PJ, Guo S, Kuo LH, Chang
CS, Su IJ, Chang CR and Tsai YS: Mitochondrial fission contributes
to mitochondrial dysfunction and insulin resistance in skeletal
muscle. Mol Cell Biol. 32:309–319. 2012.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Deng X, Liu J, Liu L, Sun X, Huang J and
Dong J: Drp1-mediated mitochondrial fission contributes to
baicalein-induced apoptosis and autophagy in lung cancer via
activation of AMPK signaling pathway. Int J Biol Sci. 16:1403–1416.
2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhang H, Yan Q, Wang X, Chen X, Chen Y, Du
J and Chen L: The role of mitochondria in liver
ischemia-reperfusion injury: From aspects of mitochondrial
oxidative stress, mitochondrial fission, mitochondrial membrane
permeable transport pore formation, mitophagy, and
mitochondria-related protective measures. Oxid Med Cell Longev.
2021(6670579)2021.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Kraus F and Ryan MY: The constriction and
scission machineries involved in mitochondrial fission. J Cell Sci.
130:2953–2960. 2017.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Gandre-Babbe S and van der Bliek AM: The
novel tail-anchored membrane protein Mff controls mitochondrial and
peroxisomal fission in mammalian cells. Mol Biol Cell.
19:2402–2412. 2008.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Palmer CS, Osellame LD, Laine D,
Koutsopoulos OS, Frazier AE and Ryan MT: MiD49 and MiD51, new
components of the mitochondrial fission machinery. EMBO Rep.
12:565–573. 2011.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Losón OC, Song Z, Chen H and Chan DC:
Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in
mitochondrial fission. Mol Biol Cell. 24:659–667. 2013.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Chakrabarti R, Ji WK, Stan RV, de Juan
Sanz J, Ryan TA and Higgs HN: INF2-mediated actin polymerization at
the ER stimulates mitochondrial calcium uptake, inner membrane
constriction, and division. J Cell Biol. 217:251–268.
2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Kameoka S, Adachi Y, Okamoto K, Iijima M
and Sesaki H: Phosphatidic acid and cardiolipin coordinate
mitochondrial dynamics. Trends Cell Biol. 28:67–76. 2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Lee H and Yoon Y: Transient contraction of
mitochondria induces depolarization through the inner membrane
dynamin OPA1 protein. J Biol Chem. 289:11862–11872. 2014.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon
C, Kim H, Kim K, Sesaki H, Rhyu IJ, et al: Constriction of the
mitochondrial inner compartment is a priming event for
mitochondrial division. Nat Commun. 8(15754)2017.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Ding C, Wu Z, Huang L, Wang Y, Xue J and
Chen S, Deng Z, Wang L, Song Z and Chen S: Mitofilin and CHCHD6
physically interact with Sam50 to sustain cristae structure. Sci
Rep. 5(16064)2015.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Niu J, Yu M, Wang C and Xu Z: Leucine-rich
repeat kinase 2 disturbs mitochondrial dynamics via dynamin-like
protein. J Neurochem. 122:650–658. 2012.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Haile Y, Deng X, Ortiz-Sandoval C, Tahbaz
N, Janowicz A, Lu JQ, Kerr BJ, Gutowski NJ, Holley JE, Eggleton P,
et al: Rab32 connects ER stress to mitochondrial defects in
multiple sclerosis. J Neuroinflammation. 14(19)2017.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Mohsin M, Tabassum G, Ahmad S, Ali S and
Ali Syed M: The role of mitophagy in pulmonary sepsis.
Mitochondrion. 59:63–75. 2021.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Matsuda N, Sato S, Shiba K, Okatsu K,
Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al:
PINK1 stabilized by mitochondrial depolarization recruits Parkin to
damaged mitochondria and activates latent Parkin for mitophagy. J
Cell Biol. 189:211–221. 2010.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Bingol B and Sheng M: Mechanisms of
mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med.
100:210–222. 2016.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Sharma A, Ahmad S, Ahmad T, Ali S and Syed
MA: Mitochondrial dynamics and mitophagy in lung disorders. Life
Sci. 284(119876)2021.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Chen Y and Dorn GW II:
PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling
damaged mitochondria. Science. 340:471–475. 2013.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Glauser L, Sonnay S, Stafa K and Moore DJ:
Parkin promotes the ubiquitination and degradation of the
mitochondrial fusion factor mitofusin 1. J Neurochem. 118:636–645.
2011.PubMed/NCBI View Article : Google Scholar
|
|
108
|
López-Doménech G, Covill-Cooke C,
Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N and Kittler
JT: Miro proteins coordinate microtubule- and actin-dependent
mitochondrial transport and distribution. EMBO J. 37:321–336.
2018.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Real P, Benito A, Cuevas J, Berciano MT,
de Juan A, Coffer P, Gomez-Roman J, Lafarga M, Lopez-Vega JM and
Fernandez-Luna JL: Blockade of epidermal growth factor receptors
chemosensitizes breast cancer cells through up-regulation of
Bnip3L. Cancer Res. 65:8151–8157. 2005.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Hanna RA, Quinsay MN, Orogo AM, Giang K,
Rikka S and Gustafsson ÅB: Microtubule-associated protein 1 light
chain 3 (LC3) interacts with Bnip3 protein to selectively remove
endoplasmic reticulum and mitochondria via autophagy. J Biol Chem.
287:19094–19104. 2012.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Liu L, Feng D, Chen G, Chen M, Zheng Q,
Song P, Ma Q, Zhu C, Wang R, Qi W, et al: Mitochondrial
outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in
mammalian cells. Nat Cell Biol. 14:177–185. 2012.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Sekine S, Kanamaru Y, Koike M, Nishihara
A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y,
Ishihara N, et al: Rhomboid protease PARL mediates the
mitochondrial membrane potential loss-induced cleavage of PGAM5. J
Biol Chem. 287:34635–34645. 2012.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Kagan VE, Jiang J, Huang Z, Tyurina YY,
Desbourdes C, Cottet-Rousselle C, Dar HH, Verma M, Tyurin VA,
Kapralov AA, et al: NDPK-D (NM23-H4)-mediated externalization of
cardiolipin enables elimination of depolarized mitochondria by
mitophagy. Cell Death Differ. 23:1140–1151. 2016.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Wei Y, Chiang WC, Sumpter R Jr, Mishra P
and Levine B: Prohibitin 2 is an inner mitochondrial membrane
mitophagy receptor. Cell. 168:224–238.e10. 2017.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Di Rita A, Peschiaroli A, D Acunzo P,
Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M, Melino G,
Papaleo E, et al: HUWE1 E3 ligase promotes PINK1/PARKIN-independent
mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun.
9(3755)2018.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Ju L, Chen S, Alimujiang M, Bai N, Yan H,
Fang Q, Han J, Ma X, Yang Y and Jia W: A novel role for Bcl2l13 in
promoting beige adipocyte biogenesis. Biochem Biophys Res Commun.
506:485–491. 2018.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Murakawa T, Yamaguchi O, Hashimoto A,
Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H,
et al: Bcl-2-like protein 13 is a mammalian Atg32 homologue that
mediates mitophagy and mitochondrial fragmentation. Nat Commun.
6(7527)2015.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Benard G and Rossignol R: Ultrastructure
of the mitochondrion and its bearing on function and bioenergetics.
Antioxid Redox Signal. 10:1313–1342. 2008.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Goncalves RL, Quinlan CL, Perevoshchikova
IV, Hey-Mogensen M and Brand MD: Sites of superoxide and hydrogen
peroxide production by muscle mitochondria assessed ex vivo under
conditions mimicking rest and exercise. J Biol Chem. 290:209–227.
2015.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Zuo L and Wijegunawardana D: Redox role of
ROS and inflammation in pulmonary diseases. Adv Exp Med Biol.
1304:187–204. 2021.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Moncada S and Erusalimsky JD: Does nitric
oxide modulate mitochondrial energy generation and apoptosis? Nat
Rev Mol Cell Biol. 3:214–220. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
123
|
Gellerich FN, Trumbeckaite S, Opalka JR,
Gellerich JF, Chen Y, Neuhof C, Redl H, Werdan K and Zierz S:
Mitochondrial dysfunction in sepsis: Evidence from bacteraemic
baboons and endotoxaemic rabbits. Biosci Rep. 22:99–113.
2002.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Adrie C, Bachelet M, Vayssier-Taussat M,
Russo-Marie F, Bouchaert I, Adib-Conquy M, Cavaillon JM, Pinsky MR,
Dhainaut JF and Polla BS: Mitochondrial membrane potential and
apoptosis peripheral blood monocytes in severe human sepsis. Am J
Respir Crit Care Med. 164:389–395. 2001.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Callahan LA and Supinski GS: Sepsis
induces diaphragm electron transport chain dysfunction and protein
depletion. Am J Respir Crit Care Med. 172:861–868. 2005.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Ayala JC, Grismaldo A, Aristizabal-Pachon
AF, Mikhaylenko EV, Nikolenko VN, Mikhaleva LM, Somasundaram SG,
Kirkland CE, Aliev G and Morales L: Mitochondrial dysfunction in
intensive care unit patients. Curr Pharm Des.
27(3074)2021.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Fakhruddin S, Alanazi W and Jackson KE:
Diabetes-induced reactive oxygen species: Mechanism of their
generation and role in renal injury. J Diabetes Res.
2017(8379327)2017.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Stepien KM, Heaton R, Rankin S, Murphy A,
Bentley J, Sexton D and Hargreaves IP: Evidence of oxidative stress
and secondary mitochondrial dysfunction in metabolic and
non-metabolic disorders. J Clin Med. 6(71)2017.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Arulkumaran N, Deutschman CS, Pinsky MR,
Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P and Kellum
JA: ADQI XIV Workgroup. Mitochondrial function in sepsis. Shock.
45:271–281. 2016.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Boulos M, Astiz ME, Barua RS and Osman M:
Impaired mitochondrial function induced by serum from septic shock
patients is attenuated by inhibition of nitric oxide synthase and
poly(ADP-ribose) synthase. Crit Care Med. 31:353–358.
2003.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Biophys Res Commun. 460:72–81. 2015.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Sharma P and Sampath H: Mitochondrial DNA
integrity: Role in health and disease. Cells. 8(100)2019.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Shadel GS and Horvath TL: Mitochondrial
ROS signaling in organismal homeostasis. Cell. 163:560–569.
2015.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Poderoso JL: The formation of
peroxynitrite in the applied physiology of mitochondrial nitric
oxide. Arch Biochem Biophys. 484:214–220. 2009.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Sies H: Oxidative stress: A concept in
redox biology and medicine. Redox Biol. 4:180–183. 2015.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Chow CW, Herrera Abreu MT, Suzuki T and
Downey GP: Oxidative stress and acute lung injury. Am J Respir Cell
Mol Biol. 29:427–431. 2003.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Puri G and Naura AS: Critical role of
mitochondrial oxidative stress in acid aspiration induced ALI in
mice. Toxicol Mech Methods. 30:266–274. 2020.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Lopez-Crisosto C, Pennanen C,
Vasquez-Trincado C, Morales PE, Bravo-Sagua R, Quest AFG, Chiong M
and Lavandero S: Sarcoplasmic reticulum-mitochondria communication
in cardiovascular pathophysiology. Nat Rev Cardiol. 14:342–360.
2017.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Kwong JQ, Huo J, Bround MJ, Boyer JG,
Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K, et
al: The mitochondrial calcium uniporter underlies metabolic fuel
preference in skeletal muscle. JCI Insight.
3(e121689)2018.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Sommakia S, Houlihan PR, Deane SS, Simcox
JA, Torres NS, Jeong MY, Winge DR, Villanueva CJ and Chaudhuri D:
Mitochondrial cardiomyopathies feature increased uptake and
diminished efflux of mitochondrial calcium. J Mol Cell Cardiol.
113:22–32. 2017.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Denton RM: Regulation of mitochondrial
dehydrogenases by calcium ions. Biochim Biophys Acta.
1787:1309–1316. 2009.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Dey S, DeMazumder D, Sidor A, Foster DB
and O'Rourke B: Mitochondrial ROS drive sudden cardiac death and
chronic proteome remodeling in heart failure. Circ Res.
123:356–371. 2018.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Halestrap AP, Woodfield KY and Connern CP:
Oxidative stress, thiol reagents, and membrane potential modulate
the mitochondrial permeability transition by affecting nucleotide
binding to the adenine nucleotide translocase. J Biol Chem.
272:3346–3354. 1997.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Kiefmann M, Tank S, Keller P, Börnchen C,
Rinnenthal JL, Tritt MO, Schulte-Uentrop L, Olotu C, Goetz AE and
Kiefmann R: IDH3 mediates apoptosis of alveolar epithelial cells
type 2 due to mitochondrial Ca2+ uptake during
hypocapnia. Cell Death Dis. 8(e3005)2017.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Mu G, Deng Y, Lu Z, Li X and Chen Y:
miR-20b suppresses mitochondrial dysfunction-mediated apoptosis to
alleviate hyperoxia-induced acute lung injury by directly targeting
MFN1 and MFN2. Acta Biochim Biophys Sin (Shanghai). 53:220–228.
2021.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Szturmowicz M and Demkow U: Neutrophil
extracellular traps (NETs) in severe SARS-CoV-2 lung disease. Int J
Mol Sci. 22(8854)2021.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Lugg ST, Scott A, Parekh D, Naidu B and
Thickett DR: Cigarette smoke exposure and alveolar macrophages:
Mechanisms for lung disease. Thorax. 77:94–101. 2022.PubMed/NCBI View Article : Google Scholar
|
|
149
|
West AP, Brodsky IE, Rahner C, Woo DK,
Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS and
Ghosh S: TLR signalling augments macrophage bactericidal activity
through mitochondrial ROS. Nature. 472:476–480. 2011.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Yuan Z, Syed MA, Panchal D, Joo M, Colonna
M, Brantly M and Sadikot RT: Triggering receptor expressed on
myeloid cells 1 (TREM-1)-mediated Bcl-2 induction prolongs
macrophage survival. J Biol Chem. 289:15118–15129. 2014.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Guillén-Gómez E, Silva I, Serra N,
Caballero F, Leal J, Breda A, San Martín R, Pastor-Anglada M,
Ballarín JA, Guirado L and Díaz-Encarnación MM: From inflammation
to the onset of fibrosis through A2A receptors in
kidneys from deceased donors. Int J Mol Sci.
21(8826)2020.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Pearce EL, Poffenberger MC, Chang CH and
Jones RG: Fueling immunity: Insights into metabolism and lymphocyte
function. Science. 342(1242454)2013.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Zmijewski JW, Lorne E, Zhao X, Tsuruta Y,
Sha Y, Liu G, Siegal GP and Abraham E: Mitochondrial respiratory
complex I regulates neutrophil activation and severity of lung
injury. Am J Respir Crit Care Med. 178:168–179. 2008.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Islam MN, Das SR, Emin MT, Wei M, Sun L,
Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S and
Bhattacharya J: Mitochondrial transfer from bone-marrow-derived
stromal cells to pulmonary alveoli protects against acute lung
injury. Nat Med. 18:759–765. 2012.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Yu J, Shi J, Wang D, Dong S, Zhang Y, Wang
M, Gong L, Fu Q and Liu D: Heme oxygenase-1/carbon
monoxide-regulated mitochondrial dynamic equilibrium contributes to
the attenuation of endotoxin-induced acute lung injury in rats and
in lipopolysaccharide-activated macrophages. Anesthesiology.
125:1190–1201. 2016.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Shi J, Yu J, Zhang Y, Wu L, Dong S, Wu L,
Wu L, Du S, Zhang Y and Ma D: PI3K/Akt pathway-mediated HO-1
induction regulates mitochondrial quality control and attenuates
endotoxin-induced acute lung injury. Lab Invest. 99:1795–1809.
2019.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Yu J, Wang Y, Li Z, Dong S, Wang D, Gong
L, Shi J, Zhang Y, Liu D and Mu R: Effect of heme oxygenase-1 on
mitofusin-1 protein in LPS-induced ALI/ARDS in rats. Sci Rep.
6(36530)2016.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Deng S, Zhang L, Mo Y, Huang Y, Li W, Peng
Q, Huang L and Ai Y: Mdivi-1 attenuates lipopolysaccharide-induced
acute lung injury by inhibiting MAPKs, oxidative stress and
apoptosis. Pulm Pharmacol Ther. 62(101918)2020.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Jiang C, Zhang J, Xie H, Guan H, Li R,
Chen C, Dong H, Zhou Y and Zhang W: Baicalein suppresses
lipopolysaccharide-induced acute lung injury by regulating
Drp1-dependent mitochondrial fission of macrophages. Biomed
Pharmacother. 145(112408)2022.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Shi J, Yu T, Song K, Du S, He S, Hu X, Li
X, Li H, Dong S, Zhang Y, et al: Dexmedetomidine ameliorates
endotoxin-induced acute lung injury in vivo and in vitro by
preserving mitochondrial dynamic equilibrium through the
HIF-1a/HO-1 signaling pathway. Redox Biol.
41(101954)2021.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S
and Wang J: Sestrin 2 protects against LPS-induced acute lung
injury by inducing mitophagy in alveolar macrophages. Life Sci.
267(118941)2021.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Liu W, Li CC, Lu X, Bo LY and Jin FG:
Overexpression of transcription factor EB regulates mitochondrial
autophagy to protect lipopolysaccharide-induced acute lung injury.
Chin Med J (Engl). 132:1298–1304. 2019.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Luo X, Liu R, Zhang Z, Chen Z, He J and
Liu Y: Mitochondrial division inhibitor 1 attenuates mitophagy in a
rat model of acute lung injury. Biomed Res Int.
2019(2193706)2019.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Liu W, Li Y, Bo L, Li C and Jin F:
Positive regulation of TFEB and mitophagy by PGC-1α to alleviate
LPS-induced acute lung injury in rats. Biochem Biophys Res Commun.
577:1–5. 2021.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Zhao R, Wang B, Wang D, Wu B, Ji P and Tan
D: Oxyberberine prevented lipopolysaccharide-induced acute lung
injury through inhibition of mitophagy. Oxid Med Cell Longev.
2021(6675264)2021.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Zhang Z, Chen Z, Liu R, Liang Q, Peng Z,
Yin S, Tang J, Gong T and Liu Y: Bcl-2 proteins regulate mitophagy
in lipopolysaccharide-induced acute lung injury via PINK1/Parkin
signaling pathway. Oxid Med Cell Longev.
2020(6579696)2020.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Patel S: Danger-associated molecular
patterns (DAMPs): The derivatives and triggers of inflammation.
Curr Allergy Asthma Rep. 18(63)2018.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Frevert C, Felgenhauer J, Wygrecka M,
Nastase M and Schaefer L: Danger-associated molecular patterns
derived from the extracellular matrix provide temporal control of
innate immunity. J Histochem Cytochem. 66:213–227. 2018.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Vénéreau E, Ceriotti C and Bianchi ME:
DAMPs from cell death to new life. Front Immunol.
6(422)2015.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Bianchi ME: DAMPs, PAMPs and alarmins: All
we need to know about danger. J Leukoc Biol. 81:1–5.
2007.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Zedler S and Faist E: The impact of
endogenous triggers on trauma-associated inflammation. Curr Opin
Crit Care. 12:595–601. 2006.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Vourc'h M, Roquilly A and Asehnoune K:
Trauma-induced damage-associated molecular patterns-mediated remote
organ injury and immunosuppression in the acutely Ill patient.
Front Immunol. 9(1330)2018.PubMed/NCBI View Article : Google Scholar
|
|
173
|
West AP and Shadel GS: Mitochondrial DNA
in innate immune responses and inflammatory pathology. Nat Rev
Immunol. 17:363–375. 2017.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Lu B, Kwan K, Levine YA, Olofsson PS, Yang
H, Li J, Joshi S, Wang H, Andersson U, Chavan SS and Tracey KJ: α7
Nicotinic acetylcholine receptor signaling inhibits inflammasome
activation by preventing mitochondrial DNA release. Mol Med.
20:350–358. 2014.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Oka T, Hikoso S, Yamaguchi O, Taneike M,
Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et
al: Mitochondrial DNA that escapes from autophagy causes
inflammation and heart failure. Nature. 485:251–255.
2012.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Zhang L, Deng S, Zhao S, Ai Y, Zhang L,
Pan P, Su X, Tan H and Wu D: Intra-peritoneal administration of
mitochondrial DNA provokes acute lung injury and systemic
inflammation via Toll-like receptor 9. Int J Mol Sci.
17(1425)2016.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Sun S, Sursal T, Adibnia Y, Zhao C, Zheng
Y, Li H, Otterbein LE, Hauser CJ and Itagaki K: Mitochondrial DAMPs
increase endothelial permeability through neutrophil dependent and
independent pathways. PLoS One. 8(e59989)2013.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Gonzalez AS, Elguero ME, Finocchietto P,
Holod S, Romorini L, Miriuka SG, Peralta JG, Poderoso JJ and
Carreras MC: Abnormal mitochondrial fusion-fission balance
contributes to the progression of experimental sepsis. Free Radic
Res. 48:769–783. 2014.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Chen H, Lin H, Dong B, Wang Y, Yu Y and
Xie K: Hydrogen alleviates cell damage and acute lung injury in
sepsis via PINK1/Parkin-mediated mitophagy. Inflamm Res.
70:915–930. 2021.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Chang AL, Ulrich A, Suliman HB and
Piantadosi CA: Redox regulation of mitophagy in the lung during
murine staphylococcus aureus sepsis. Free Radic Biol Med.
78:179–189. 2015.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Mannam P, Shinn AS, Srivastava A, Neamu
RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Dela Cruz CS, Ahasic
AM, et al: MKK3 regulates mitochondrial biogenesis and mitophagy in
sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol.
306:L604–L619. 2014.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Westphalen K, Monma E, Islam MN and
Bhattacharya J: Acid contact in the rodent pulmonary alveolus
causes proinflammatory signaling by membrane pore formation. Am J
Physiol Lung Cell Mol Physiol. 303:L107–L116. 2012.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Kuebler WM, Parthasarathi K, Wang PM and
Bhattacharya J: A novel signaling mechanism between gas and blood
compartments of the lung. J Clin Invest. 105:905–913.
2000.PubMed/NCBI View Article : Google Scholar
|
|
185
|
Hough RF, Islam MN, Gusarova GA, Jin G,
Das S and Bhattacharya J: Endothelial mitochondria determine rapid
barrier failure in chemical lung injury. JCI Insight.
4(e124329)2019.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Ogino K, Nagaoka K, Okuda T, Oka A, Kubo
M, Eguchi E and Fujikura Y: PM2.5-induced airway inflammation and
hyperresponsiveness in NC/Nga mice. Environ Toxicol. 32:1047–1054.
2017.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Wei T and Tang M: Biological effects of
airborne fine particulate matter (PM2.5) exposure on
pulmonary immune system. Environ Toxicol Pharmacol. 60:195–201.
2018.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Xu M, Li F, Wang M, Zhang H, Xu L, Adcock
IM, Chung KF and Zhang Y: Protective effects of VGX-1027 in
PM2.5-induced airway inflammation and bronchial
hyperresponsiveness. Eur J Pharmacol. 842:373–383. 2019.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Ischemia/reperfusion. Compr Physiol. 7:113–170.
2016.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Tai H, Jiang X, Song N, Xiao HH, Li Y,
Cheng MJ, Yin XM, Chen YR, Yang GL, Jiang XY, et al: Tanshinone IIA
combined with cyclosporine a alleviates lung apoptosis induced by
renal ischemia-reperfusion in obese rats. Front Med (Lausanne).
8(617393)2021.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Zhang Y, Yu G, Kaminski N and Lee PJ:
PINK1 mediates the protective effects of thyroid hormone T3 in
hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol.
320:L1118–L1125. 2021.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Supinski GS, Schroder EA and Callahan LA:
Mitochondria and critical illness. Chest. 157:310–322.
2020.PubMed/NCBI View Article : Google Scholar
|
|
193
|
Powers SK, Hudson MB, Nelson WB, Talbert
EE, Min K, Szeto HH, Kavazis AN and Smuder AJ:
Mitochondria-targeted antioxidants protect against mechanical
ventilation-induced diaphragm weakness. Crit Care Med.
39:1749–1759. 2011.PubMed/NCBI View Article : Google Scholar
|
|
194
|
Miglio G, Rosa AC, Rattazzi L, Collino M,
Lombardi G and Fantozzi R: PPARgamma stimulation promotes
mitochondrial biogenesis and prevents glucose deprivation-induced
neuronal cell loss. Neurochem Int. 55:496–504. 2009.PubMed/NCBI View Article : Google Scholar
|
|
195
|
Moskowitzova K, Orfany A, Liu K,
Ramirez-Barbieri G, Thedsanamoorthy JK, Yao R, Guariento A,
Doulamis IP, Blitzer D, Shin B, et al: Mitochondrial
transplantation enhances murine lung viability and recovery after
ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol.
318:L78–L88. 2020.PubMed/NCBI View Article : Google Scholar
|
|
196
|
Ramachandran A and Jaeschke H:
Acetaminophen toxicity: Novel insights into mechanisms and future
perspectives. Gene Expr. 18:19–30. 2018.PubMed/NCBI View Article : Google Scholar
|
|
197
|
Tan DX, Manchester LC, Qin L and Reiter
RJ: Melatonin: A mitochondrial targeting molecule involving
mitochondrial protection and dynamics. Int J Mol Sci.
17(2124)2016.PubMed/NCBI View Article : Google Scholar
|
|
198
|
Srinivasan V, Pandi-Perumal SR, Spence DW,
Kato H and Cardinali DP: Melatonin in septic shock: Some recent
concepts. J Crit Care. 25:656.e1–e6. 2010.PubMed/NCBI View Article : Google Scholar
|
|
199
|
Vance JE: Phospholipid synthesis and
transport in mammalian cells. Traffic. 16:1–18. 2015.PubMed/NCBI View Article : Google Scholar
|
|
200
|
Adachi Y, Itoh K, Yamada T, Cerveny KL,
Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M and
Sesaki H: Coincident phosphatidic acid interaction restrains Drp1
in mitochondrial division. Mol Cell. 63:1034–1043. 2016.PubMed/NCBI View Article : Google Scholar
|
|
201
|
Macdonald PJ, Francy CA, Stepanyants N,
Lehman L, Baglio A, Mears JA, Qi X and Ramachandran R: Distinct
splice variants of dynamin-related protein 1 differentially utilize
mitochondrial fission factor as an effector of cooperative GTPase
activity. J Biol Chem. 291:493–507. 2016.PubMed/NCBI View Article : Google Scholar
|
|
202
|
Ban T, Heymann JA, Song Z, Hinshaw JE and
Chan DC: OPA1 disease alleles causing dominant optic atrophy have
defects in cardiolipin-stimulated GTP hydrolysis and membrane
tubulation. Hum Mol Genet. 19:2113–2122. 2010.PubMed/NCBI View Article : Google Scholar
|
|
203
|
Ugarte-Uribe B, Müller HM, Otsuki M,
Nickel W and García-Sáez AJ: Dynamin-related protein 1 (Drp1)
promotes structural intermediates of membrane division. J Biol
Chem. 289:30645–30656. 2014.PubMed/NCBI View Article : Google Scholar
|
|
204
|
Bustillo-Zabalbeitia I, Montessuit S,
Raemy E, Basañez G, Terrones O and Martinou J: Specific interaction
with cardiolipin triggers functional activation of dynamin-related
protein 1. PLoS One. 9(e102738)2014.PubMed/NCBI View Article : Google Scholar
|
|
205
|
Adachi Y, Iijima M and Sesaki H: An
unstructured loop that is critical for interactions of the stalk
domain of Drp1 with saturated phosphatidic acid. Small GTPases.
9:472–479. 2018.PubMed/NCBI View Article : Google Scholar
|
|
206
|
Qi X, Disatnik MH, Shen N, Sobel RA and
Mochly-Rosen D: Aberrant mitochondrial fission in neurons induced
by protein kinase C{delta} under oxidative stress conditions in
vivo. Mol Biol Cell. 22:256–265. 2011.PubMed/NCBI View Article : Google Scholar
|
|
207
|
Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS,
Ko SH and Han HJ: Aβ-Induced Drp1 phosphorylation through Akt
activation promotes excessive mitochondrial fission leading to
neuronal apoptosis. Biochim Biophys Acta. 1863:2820–2834.
2016.PubMed/NCBI View Article : Google Scholar
|
|
208
|
Xu S, Wang P, Zhang H, Gong G, Gutierrez
Cortes N, Zhu W, Yoon Y, Tian R and Wang W: CaMKII induces
permeability transition through Drp1 phosphorylation during chronic
β-AR stimulation. Nat Commun. 7(13189)2016.PubMed/NCBI View Article : Google Scholar
|
|
209
|
Niemann A, Ruegg M, La Padula V, Schenone
A and Suter U: Ganglioside-induced differentiation associated
protein 1 is a regulator of the mitochondrial network: New
implications for charcot-marie-tooth disease. J Cell Biol.
170:1067–1078. 2005.PubMed/NCBI View Article : Google Scholar
|
|
210
|
Tondera D, Santel A, Schwarzer R, Dames S,
Giese K, Klippel A and Kaufmann J: Knockdown of MTP18, a novel
phosphatidylinositol 3-kinase-dependent protein, affects
mitochondrial morphology and induces apoptosis. J Biol Chem.
279:31544–31555. 2004.PubMed/NCBI View Article : Google Scholar
|
|
211
|
Norton M, Ng AC, Baird S, Dumoulin A,
Shutt T, Mah N, Andrade-Navarro MA, McBride HM and Screaton RA:
ROMO1 is an essential redox-dependent regulator of mitochondrial
dynamics. Sci Signal. 7(ra10)2014.PubMed/NCBI View Article : Google Scholar
|
|
212
|
Zoulikha M, Xiao Q, Boafo GF, Sallam MA,
Chen Z and He W: Pulmonary delivery of siRNA against acute lung
injury/acute respiratory distress syndrome. Acta Pharm Sin B.
12:600–620. 2022.PubMed/NCBI View Article : Google Scholar
|
|
213
|
Singer M: The role of mitochondrial
dysfunction in sepsis-induced multi-organ failure. Virulence.
5:66–72. 2014.PubMed/NCBI View Article : Google Scholar
|
|
214
|
Hsu YC, Wu YT, Yu TH and Wei YH:
Mitochondria in mesenchymal stem cell biology and cell therapy:
From cellular differentiation to mitochondrial transfer. Semin Cell
Dev Biol. 52:119–131. 2016.PubMed/NCBI View Article : Google Scholar
|
|
215
|
Zhang H, Feng YW and Yao YM: Potential
therapy strategy: Targeting mitochondrial dysfunction in sepsis.
Mil Med Res. 5(41)2018.PubMed/NCBI View Article : Google Scholar
|
|
216
|
Morrison TJ, Jackson MV, Cunningham EK,
Kissenpfennig A, McAuley DF, O'Kane CM and Krasnodembskaya AD:
Mesenchymal stromal cells modulate macrophages in clinically
relevant lung injury models by extracellular vesicle mitochondrial
transfer. Am J Respir Crit Care Med. 196:1275–1286. 2017.PubMed/NCBI View Article : Google Scholar
|
|
217
|
Willson JA, Arienti S, Sadiku P, Reyes L,
Coelho P, Morrison T, Rinaldi G, Dockrell DH, Whyte MKB and
Walmsley SR: Neutrophil HIF-1α stabilization is augmented by
mitochondrial ROS produced via the glycerol 3-phosphate shuttle.
Blood. 139:281–286. 2022.PubMed/NCBI View Article : Google Scholar
|