Introduction
Bardet-Biedl syndrome (BBS) a rare autosomal
recessive genetic disease with clinical manifestations that can
affect multiple systems throughout the body, was first reported by
Georges Bardet (1). The incidence
of BBS is nearly 1 in 125,000-160,000 in North America and Europe
(2). Beales et al (3) analyzed the clinical symptoms of 109
families with BBS and summarized and revised the diagnostic
criteria of the disease. The proposed diagnostic criteria for BBS
are the presence of either four primary features or three primary
and two secondary features, which differentiate this syndrome from
other phenotype overlapped syndromes.
The incidence rate of retinal degeneration/dystrophy
in patients with BBS is >90% (4), and it mostly occurs as night
blindness at the beginning of the disease (3). Obesity is the second primary feature
of BBS, with an incidence rate of 72-92% (3-5);
it often starts in childhood and gradually worsens with age, and
can further develop into type 2 diabetes (4). The average body mass index (BMI) of
adult men and adult women with BBS is 36.6 and 31.5
kg/m2 (normal ranges: 18-24 kg/m2),
respectively (5). Postaxial
polydactyly is the only symptom that can be observed at birth, with
both upper and lower extremities being simultaneously involved in
21% of patients, lower limb involvement occurring in 21% of the
patients and upper limb involvement occurring in 9% of the patients
(4,6). The incidence rate of hypogonadism in
BBS populations is 59-98% (6). The
indicators of hypogonadism range from late sexual maturity to
hypogenitalism in males (7). Most
individuals with this condition have a micropenis and/or low
testicular volume at birth, and 9% have cryptorchidism (3). Females with hypogonadism exhibit
features such as hypoplastic fallopian tubes, uterus and ovaries,
partial and complete vaginal atresia, a septate vagina, a duplex
uterus, hydrocolpos, hydrometrocolpos and a persistent urogenital
sinus (8). Renal abnormality is a
major cause of morbidity and mortality in patients with BBS, and
has an incidence rate ranging between 20 and 53% (3), including cystic tubular disease and
anatomical deformities. Intellectual disability occurs in 50-61% of
patients with BBS (7). and a
previous study has shown that the volume of the hippocampus is
decreased in patients with BBS (9).
Secondary BBS phenotypes include hepatic fibrosis,
diabetes mellitus, neurological impairment, speech deficits,
behavioral abnormalities, craniofacial dysmorphism, dental
irregularities, developmental delay, hypertension, cardiovascular
abnormalities, hearing loss and olfactory impairments (7).
In total, >500 cases of BBS have been reported
globally, of which only 80 cases have been accurately diagnosed in
China (10). To date, 21 BBS genes
have been identified and mapped on various chromosomes (11), and ~80% of the clinically examined
cases can be explained by the known identified BBS genes (6). BBS has both a high degree of genetic
heterogeneity and extensive clinical heterogeneity, and the
association between genotype and phenotype is not significant
(12). Novel interventions are
developing at a rapid pace including genetic therapeutics such as
gene therapy, exon skipping therapy, nonsense suppression therapy,
and gene editing. Other non-genetic therapies such as gene
repurposing, targeted therapies, and non-pharmacological
interventions are also ongoing. A major challenge in developing
genetic therapies for BBS is the generation of a long lasting
therapy. A successful example of this is the retinal gene therapy
(Luxturna), which has been developed for RPE65-associated Leber
congenital amaurosis (13).
The present report describes the case of a girl
(age, 7 years and 6 months) who was initially diagnosed with BBS
due to early breast development and obesity, and gradually
developed central precocious puberty (CPP) during follow-up.
Whole-exome gene sequencing revealed new heterozygous mutations in
the BBS type 10 (BBS10) gene, which, to the best of our
knowledge, have not yet been reported.
Case report
Patient
The patient was a girl (age, 7 years and 6 months),
with a height of 127.8 cm [+0.4 standard deviation score (SDS)
girls, i.e. 0.4 SDs above average for girls this age] (14), weight of 38.0 kg (+3.0 SDS girls
(14)] and a BMI of 23.3
kg/m2. The patient visited the Department of Children's
Health Care, Fifth People's Hospital of Foshan City (Foshan, China)
for the first time in July 2020 due to a rapid increase in body
weight from the age of 6 years and breast development for a month
prior to the visit. The patient was the only daughter in the family
and was born at full-term after spontaneous labor (measuring 49 cm
in length and 3.0 kg in weight, with a 43.5-cm head circumference
at birth).
At 1 year and 3 months of age, the parents of the
patient noticed rapid weight increase, with a height of 76.0 cm [-1
SDS girls (14)], a weight of 13.0
kg [+3.1 SDS girls (14)] and a
BMI of 22.5 kg/m2 being reached. The patient experienced
retinal degeneration in both eyes. Sixth finger/toe deformities in
both hands and the left foot were treated with surgical operations
when she was 2 years old. At the first visit, motor functions and
speech development were delayed, and the patient had attention
problems and a poor academic performance (age, 7 years and 6
months). The father and mother were aged 44 and 42 years,
respectively, and both were healthy; the family history revealed a
non-consanguinous marriage and no notable genetic findings.
Clinical findings
At the first visit to the Department of Children's
Health Care, Fifth People's Hospital of Foshan City in July 2020,
the patient was 7 years and 6 months of age. A physical examination
indicated the following: Blood pressure, 92/60 mmHg; height, 127.8
cm [+0.4 SDS girls (14)]; weight,
38.0 kg [+3.0 SDS girls (14)];
and BMI, 23.3 kg/m2. The abdominal circumference was
73.5 cm. Facial features revealed a low hairline, crowding of the
teeth, malocclusion and no abnormal facial features or limb
malformation appearance. The patient had small hands and feet, with
surgical scars of 1.0-1.5 cm in length on the outside of the little
fingers of both hands and the little toe of the left foot. The
breasts were at Tanner stage 2(15) of development and female genitalia
were present. The vaginal opening was normal and located below the
urethral opening. The patient's father and mother were 167 cm [-0.9
SDS (14)] and 163 cm [+0.4 SDS
(14)] in height, respectively.
Both parents had normal sexual characteristics and both were Tanner
stage 5. After this visit, the patient was referred to other
hospitals. Therefore, no other examination and treatment was
provided.
The patient returned for another visit in January
2021, at 8 years and 2 months of age. The patient had experienced
breast development and intermittent pain for 3 months. The
following measurements were recorded: Height, 130.8 cm [+0.3 SDS
girls (14)]; weight, 40.5 kg
[+2.8 SDS girls (14)]; and BMI,
23.6 kg/m2. The abdominal circumference was 75.8 cm and
the patient was at Tanner breast stage 2.
Diagnostic assessment
In July 2020, at the time of the first visit, the
patient's intelligence test score (Chinese-Wechsler Intelligence
Scale for Children) was 75 (normal range: 85-115) (16). Routine blood and urine tests were
within the normal ranges. Her hormones and biochemical data were
normal, and they were in the state of Tanner Stages 2 female. And
her ovarian function was also normal (Table I).
 | Table IHormones and biochemical data in July
2020. |
Table I
Hormones and biochemical data in July
2020.
| Parameter | Result | Reference range |
|---|
| Triglyceride,
mmol/l | 1.70 | 0.56-1.70 |
| Total cholesterol,
mmol/l | 3.40 | 3.10-5.70 |
| Fasting blood
glucose, mmol/l | 5.40 | 3.90-6.10 |
| Fasting insulin,
mU/l | 15.40 | 3.00-25.00 |
| Blood
17-hydroxyprogesterone, ng/ml | 0.76 | 0.33-2.97 |
| α-fetoprotein,
IU/ml | 1.10 | 0-20.00 |
| β-human chorionic
gonadotropin, mIU/ml | 2.00 | 0-10.00 |
| Inhibin B, pg/ml | 11.20 | 0-43.91 |
| Anti-Müllerian
hormone, ng/ml | 2.20 | 0.05-10.40 |
| Testosterone,
nmol/l | 0.08 | <0.24-0.97 |
| Estradiol,
pmol/l | 99.80 | 37.00-88.00 |
| FSH, IU/l | 3.51 | 1.00-10.80 |
| LH, IU/l | 0.16 | 0.02-4.70 |
| Free
triiodothyronine, pmol/l | 5.80 | 3.50-6.60 |
| Free thyroxine,
pmol/l | 17.30 | 11.50-22.70 |
| Thyroid-stimulating
hormone, mIU/l | 3.87 | 0.64-6.27 |
Ultrasonography indicated that the patient's uterus
and ovaries were in the prepubertal stage, and there was no adrenal
and celiac ectopic hyperplastic disease. Additionally, the bone age
was 10 years (17), which was 2
years and 6 months advanced of that expected. Magnetic resonance
imaging of the pituitary gland was normal.
The gonadotropin-releasing hormone (GnRH)
stimulation test showed that the peaks of FSH and LH, which were
5.66 IU/l and 1.23 IU/l (Reference range: peak of LH>5.0IU/l and
LH/FSH>0.6), respectively, appeared at 60 min
post-administration. The ratio of LH to FSH was 0.22. Peripheral
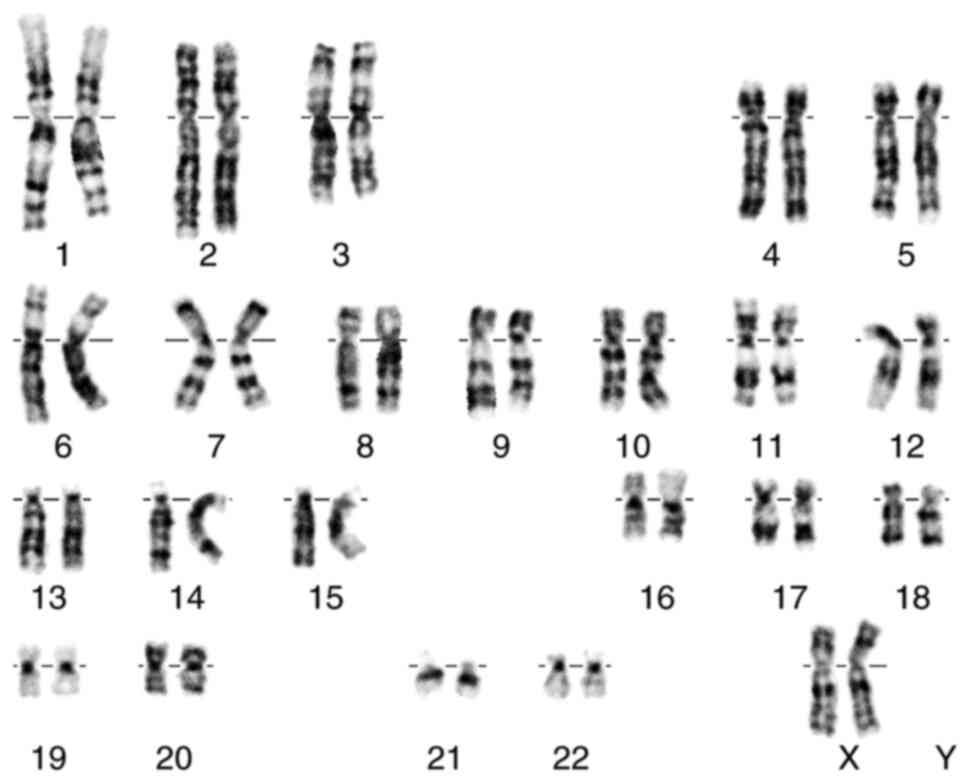
blood lymphocyte karyotype showed a result of 46, XX (Fig. 1).
According to all the aforementioned results, the
patient was initially diagnosed with BBS and pseudo-precocious
puberty. However, whole-exome gene sequencing was not performed, as
it was too expensive for the parents. After 3 months of follow-up,
the patient's breasts returned to Tanner stage B1.
In January 2021, at 8 years and 2 months old, the
patient returned for another visit. This time, her hormones and
biochemical data were still normal, and they were in the state of
Tanner Stages 2 female. And her ovarian and adrenal function were
also normal (Table II).
| Table IIHormones and biochemical data,
January 2021. |
Table II
Hormones and biochemical data,
January 2021.
| Parameter | Result | Reference
range |
|---|
| Blood
17-hydroxyprogesterone, ng/ml | 0.28 | 0.33-2.97 |
| DHEA-S, µmol/l | 1.06 | 0.88-3.35 |
| α-fetoprotein,
IU/ml | 0.70 | 0-20.00 |
| β-human chorionic
gonadotropin, mIU | 3.10 | 0-10.00 |
| Inhibin B,
pg/ml | 7.20 | 0-43.91 |
| Anti-Müllerian
hormone, ng/ml | 4.40 | 0.05-10.40 |
| Testosterone,
nmol/l | 0.21 | <0.24-0.97 |
| Estradiol,
pmol/l | 81.30 | 37.00-88.00 |
| FSH, IU/l | 1.47 | 1.00-10.80 |
| LH, IU/l | 0.26 | 0.02-4.70 |
| Free
triiodothyronine, pmol/l | 5.30 | 3.50-6.60 |
| Free thyroxine,
pmol/l | 14.00 | 11.50-22.70 |
| Thyroid-stimulating
hormone, mIU/l | 5.23 | 0.64-6.27 |
Furthermore, the bone age was 11 years (17), which was almost 3 years advanced of
that expected. Ultrasonography revealed the following results:
Uterine volume, 26x10x17 mm; endometrial thickness, 4 mm; left
ovary volume, 21x13x15 mm or ~2.1 ml; and right ovary volume,
17x11x12 mm or ~1.2 ml. No early antral follicles were observed.
The GnRH stimulation test showed that the peaks of FSH and LH,
which were 9.29 and 6.31 IU/l, respectively, appeared 60 min after
administration. The ratio of LH to FSH was 0.68.
Whole-exome gene sequencing was performed by the
Guangzhou Daan Clinical Laboratory Center in Guangzhou, China).
Genomic DNA from peripheral blood leukocytes, derived from the
proband was extracted using a QIAamp DNA Blood Mini kit (cat. no.
51185; Qiagen, GmbH). Concentration was measure by Qubit 3.0. A
total of 1 µg each genomic DNA sample was fragmented by sonication
and purified to yield fragments of 200-300 bp. Paired-end adaptor
oligonucleotides from Illumina, Inc. were ligated to the shared
genomic DNA. A total of 500 nanograms of these tailed fragments
were then hybridized to the probe library of the Sure Select Human
All Exon V6 (Agilent; catalogue number: 51908864). The enrichment
libraries were sequenced on the Illumina Novaseq 6000 sequencer
(Illumina, San Diego, California) as 150-bp paired-end reads. Reads
were aligned to the human reference genome (GRCh37/hg19) with the
burrows-wheeler aligner (BWA) (18) and potential duplicate paired-end
reads were removed with Genome Analysis Toolkit (GATK) v.4.2.0.0
(https://github.com/broadinstitute/gatk/releases/tag/4.2.2.0).
GATK v.4.2.0.0 was used for base quality-score recalibration and
indel realignment as well as for single-nucleotide-variant and
indel discovery and genotyping with standard hard-filtering
parameters (19). Variants with
low quality were flagged and excluded from subsequent analyses.
Bamdst v.1.0.9 (github.com/shiquan/bamdst) was used to assess coverage
of the clean data of each sample with default parameters. All
variants identified in the affected individuals were annotated with
databases, including refGene (http://varianttools.sourceforge.net/Annotation/RefGene),
Avsnp150(https://www.ncbi.nlm.nih.gov/snp/), gnomAD211
(http://gnomad-sg.org/), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/),
dbnsfp41a (https://sites.google.com/site/jpopgen/dbNSFP),
Intervar (wintervar.wglab.org/) by snpeff5.0d
(sourceforge.net/projects/snpeff/) and annovar 2020 Jun
(annovar.openbioinformatics.org/en/latest/).
Candidate mutational events were then inspected with the
integrative genomics viewer (20).
The resulting variants were excluded when the frequency was
>1/1,00 in genome aggregation database (gnomAD). Variants were
correlated with patient phenotypes and the results of clinical
investigations. All variants were classified in the American
College of Medical Genetics and Genomics(ACMG) standards and
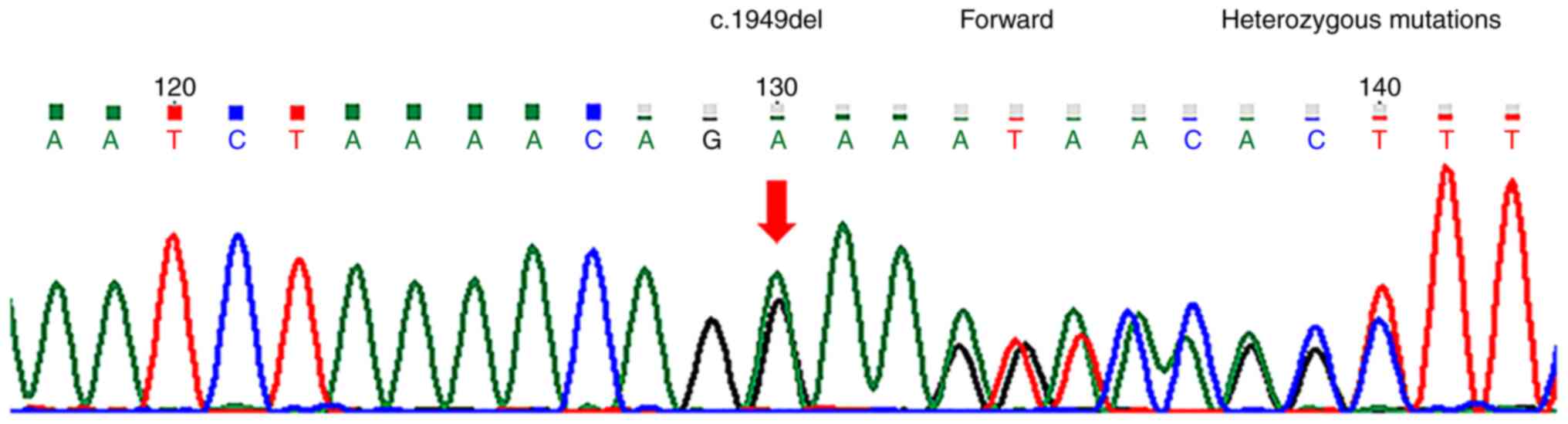
guidelines (21). chr12:76739816
(c.1949del) (Fig. 2) and
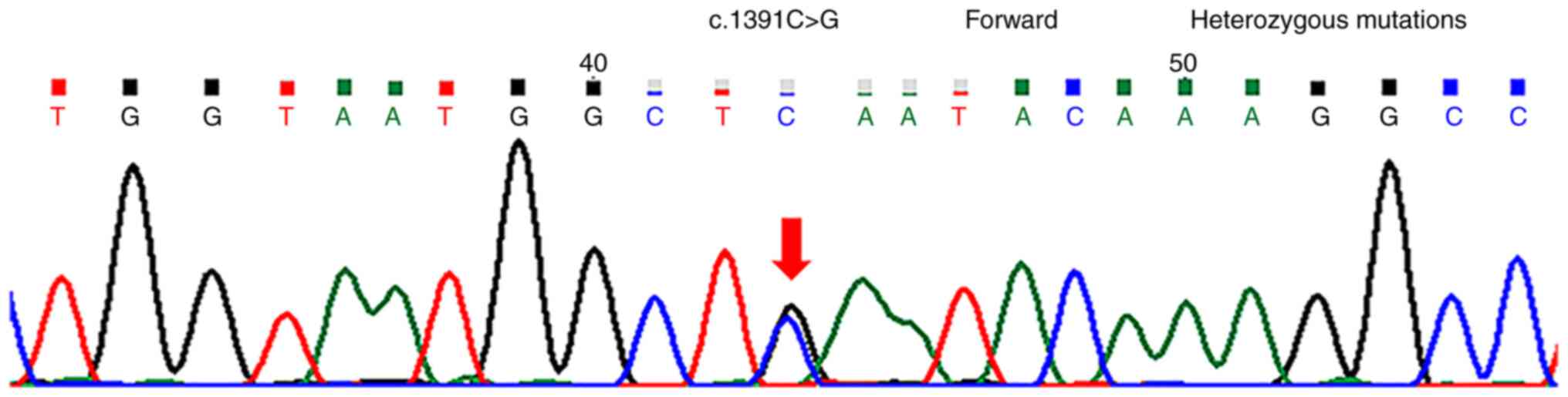
chr12:76740374 (c.1391C>G) (Fig.
3) heterozygous mutations associated with the patient's
clinical phenotype were located in the BBS10 gene. However,
no possible pathogenic variants in the 59 genes recommended by the
ACMG (21) were detected. The
final diagnosis was BBS10 and CPP.
Therapeutic intervention
To avoid premature depletion of gonadal function,
the patient's parents agreed to use GnRH analog (GnRHa) for CPP
treatment. The initial dose was 3.75 mg subcutaneous injection, and
the maintenance dose was subcutaneous injection 50-100 mg/kg every
4 weeks. The patient's height and sexual development will be fully
assessed every 3 months. This treatment plan will last until the
age of 11 or until the precocious puberty is well controlled.
Follow-up and outcomes
The patient showed good intervention compliance and
tolerance. So far, no unfavorable and unanticipated events have
been observed. After 3 months of treatment with GnRHa, height and
weight had increased to 132.0 cm [+0.2 SDS girls (14)] and 42.0 kg [+2.8 SDS girls
(14)], respectively. The Tanner
breast stage was now 1. The GnRH stimulation test showed that the
peaks of FSH and LH, which were 7.15 and 3.42 IU/l, respectively,
appeared 60 min after administration. The ratio of LH to FSH was
0.48. Ultrasonography showed that the uterus and ovaries had
reduced to their prepubertal size.
In May 2022, the patient was 9 years and 6 months
old, and after receiving 15 months of treatment with GnRHa, height
and weight measurements had increased to 138.6 cm [+0.26 SDS girls
(14)] and 46.8 kg [+2.4 SDS girls
(14)], respectively. The BMI was
24.4 kg/m2 and the abdominal circumference was 76.8 cm.
The patient was at Tanner breast stage 1. The GnRH stimulation test
showed that the peaks of FSH and LH, which were 9.66 and 3.15 IU/l,
respectively, appeared 90 min after administration. The ratio of LH
to FSH was 0.33. The level of anti-Müllerian hormone was 3.9 ng/ml
(reference range for a 0 to 10-year-old girl, 0.05-10.40 ng/ml).
Ultrasonography showed that the uterus and ovaries were still their
prepubertal size. The patient's bone age was 11 years and 6 months
(17), which was 2 years advanced
of that expected.
Discussion
The final diagnosis in the present case was BBS10
and CPP. This is the first ever encounter of a patient with BBS10
and CPP in the Fifth People's Hospital of Foshan City.
The first visit of the patient was due to a rapid
increase in body weight, but in fact, weight and BMI measurements
did not change significantly from the beginning of
pseudo-precocious puberty to CPP. A recent study showed that
early-onset obesity enhanced paraventricular nucleus expression of
serine palmitoyltransferase long chain base subunit 1 and advanced
the maturation of the ovarian noradrenergic system (22). Although the age of thelarche has
decreased from 1977 to 2013(23),
it is questionable whether this type of obesity is sufficient to
cause precocious puberty in BBS, which is characterized by
hypogonadism.
Patients with precocious puberty often have
secondary sex characteristic of mismatched gonadal development.
However, the Tanner stage of the breast, and the uterus and ovaries
of the present patient markedly lagged behind the development of
bone age. We hypothesized that this may have been associated with
the clinical features of BBS. Clinical manifestations included
retinal degeneration, obesity, postaxial polydactyly and
intellectual disability, which were in line with the
characteristics of BBS10, except for renal abnormality. No adrenal
gland diseases and germ cell tumors were found, and there was no
chronic steroid use. The final whole-exome gene sequencing revealed
that the c.1949del and c.1391C>G heterozygous mutations
associated with the patient's clinical phenotype were located in
the BBS10 gene, and were not included in the recommendations
of the ACMG guidelines of 59 genes. A literature search revealed no
clinical studies reporting these two mutations. Therefore, none of
these mutations of the previous case reports can explain the
occurrence of CPP.
To explain the cause of the CPP with BSS10, the
literature was searched using the key words ‘precocious puberty’,
‘gonad dysplasia’, ‘Turner syndrome’, ‘Klinefelter syndrome’,
‘Kallmann syndrome’ and ‘Prader-Willi syndrome’ in
Medline(https://www.medline.com/),
Pubmed(https://pubmed.ncbi.nlm.nih.gov/), Embase (https://www.embase.com/landing?status=grey) and CNKI
databases(https://www.cnki.net/) up to May 2021,
and a notable phenomenon was found: Regardless of hypergonadotropic
hypogonadism and hypogonadotropic hypogonadism, cases of precocious
puberty have been reported, which is somewhat unusual.
U-shaped gonadotrophin levels are present from birth
to puberty in normal males, while the same pattern, but at markedly
higher levels, is present in anorchid boys, indicating that the
gonads serve a role in the negative feedback of gonadotrophins in
childhood (24). In addition,
patients with Turner syndrome and triple X syndrome show premature
activation of the GnRH pulse generator, even without signs of
puberty (25). Both of these
chromosomal aneuploidies have increased gonadotropin levels as
compensation for the restricted ovarian function, to the extent
that they manifest as CPP, but eventually progress to premature
ovarian failure. Gonadal dysplasia may reduce the negative feedback
of gonadotrophins, resulting in the earlier activation of the
hypothalamic-pituitary-gonadal axis (25). These theories seem to reasonably
explain the occurrence of precocious puberty cases in
hypergonadotropic hypogonadism.
Another question is with regard to the manner in
which precocious puberty then occurs in hypogonadotropic
hypogonadism. No pathogenic allelic variants of genes known to
cause monogenic CPP (KISS1 receptor, KiSS-1 metastasis suppressor,
makorin ring finger protein 3 and δ like non-canonical Notch ligand
1) (26-28)
were found in the present case. Perhaps hints can be taken from
other biological studies; for example, adult Drosophila
accelerate their mating behavior to defend against the threat of
certain parasitic wasps (29). It
is unclear whether CPP occurring in BBS-10 would be a prelude of
gonad dysplasia (30) or a
self-protection mechanism of human reproductive function.
Central precocious puberty occurred in case of
hypogonadotropic hypogonadis. Therefore, further clinical data and
molecular biological evidence is required to confirm the etiology
and mechanism of the present case.
Acknowledgements
The authors would like to thank Ms Liyu Yuan
(Institute of Fashion Technology of Guangdong Polytechnic, Foshan
China) for editing the figures associated with the manuscript.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HL, JH and IL were responsible for the study
investigation. HL, JH, IL, RH and XS conceived and designed the
study. HL wrote the original manuscript. HL, RH and XS reviewed and
edited the manuscript. All authors read and approved the final
manuscript. HL, JH and RH confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The experimental protocol was established according
to the ethical guidelines of the Helsinki Declaration and was
approved by the Human Ethics Committee of the Fifth People's
Hospital of Foshan City (Foshan, China; approval no.
2021060704).
Patient consent for publication
Written informed consent for publication of the case
report was obtained from parents.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bardet G: On congenital obesity syndrome
with polydactyly and retinitis pigmentosa (a contribution to the
study of clinical forms of hypophyseal obesity). Obes Res.
3:387–99. 1920.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ajmal M, Khan MI, Neveling K, Tayyab A,
Jaffar S, Sadeque A, Ayub H, Abbasi NM, Riaz M, Micheal S, et al:
Exome sequencing identifies a novel and a recurrent BBS1 mutation
in Pakistani families with Bardet-Biedl syndrome. Mol Vis.
19:644–653. 2013.PubMed/NCBI
|
|
3
|
Beales PL, Elcioglu N, Woolf AS, Parker D
and Flinter FA: New criteria for improved diagnosis of Bardet-Biedl
syndrome: Results of a population survey. J Med Genet. 36:437–446.
1999.PubMed/NCBI
|
|
4
|
Forsythe E and Beales PL: Bardet-Biedl
syndrome. Eur J Hum Genet. 21:8–13. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Moore SJ, Green JS, Fan Y, Bhogal AK,
Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JC,
et al: Clinical and genetic epidemiology of Bardet-Biedl syndrome
in Newfoundland: A 22-year prospective, population-based, cohort
study. Am J Med Genet A. 132A:352–360. 2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
M'hamdi O, Ouertani I and
Chaabouni-Bouhamed H: Update on the genetics of bardet-biedl
syndrome. Mol Syndromol. 5:51–56. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Khan SA, Muhammad N, Khan MA, Kamal A,
Rehman ZU and Khan S: Genetics of human Bardet-Biedl syndrome,an
updates. Clin Genet. 90:3–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Uğuralp S, Demircan M, Cetin S and Siğirci
A: Bardet-Biedl syndrome associated with vaginal atresia: A case
report. Turk J Pediatr. 45:273–275. 2003.PubMed/NCBI
|
|
9
|
Baker K, Northam GB, Chong WK, Banks T,
Beales P and Baldeweg T: Neocortical and hippocampal volume loss in
a human ciliopathy: A quantitative MRI study in Bardet-Biedl
syndrome. Am J Med Genet A. 155A:1–8. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shen T, Yan XM and Xiao CJ: Current status
and implication of research on Bardet-Biedl syndrome. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 30:570–573. 2013.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
11
|
Heon E, Kim G, Qin S, Garrison JE, Tavares
E, Vincent A, Nuangchamnong N, Scott CA, Slusarski DC and Sheffield
VC: Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum
Mol Genet. 25:2283–2294. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Daniels AB, Sandberg MA, Chen J,
Weigel-DiFranco C, Fielding Hejtmancic J and Berson EL:
Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch
Ophthalmol. 130:901–907. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Forsythe E, Kenny J, Bacchelli C and
Beales PL: Managing Bardet-Biedl syndrome-now and in the future.
Front Pediatr. 6(23)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li H, Ji CY, Zong XN and Zhang YQ: Body
mass index growth curves for Chinese children and adolescents aged
0 to 18 years. Zhonghua Er Ke Za Zhi. 47:493–498. 2009.PubMed/NCBI(In Chinese).
|
|
15
|
Emmanuel M and Bokor BR: Tanner Stages.
In: StatPearls [Internet]. Treasure Island (FL): StatPearls
Publishing; 2022 Jan.
|
|
16
|
Dai X, Gong Y, Tang Q, Cai T, Zhou S,
Cheng Z, Yao S, Guo B and Xie Y: Chinese intelligence scale for
young children: Item analysis, reliability, and National norms.
Chin J Clin Psychol. 6:1–7. 1998.
|
|
17
|
William Walter Greulich, S.Idell Pyle
(eds): Radiographic atlas of skeletal development of hand and
wrist. Vol 1. 2nd edition. Stanford University Press, Stanford,
California, pp 161-165, 2017.
|
|
18
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al: The variant call format and VCFtools. Bioinformatics.
27:2156–2158. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the Association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Heras V, Castellano JM, Fernandois D,
Velasco I, Rodríguez-Vazquez E, Roa J, Vazquez MJ, Ruiz-Pino F,
Rubio M, Pineda R, et al: Central ceramide signaling mediates
obesity-induced precocious puberty. Cell Metab. 32:951–966.e8.
2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Eckert-Lind C, Busch AS, Petersen JH, Biro
FM, Butler G, Bräuner EV and Juul A: Worldwide secular trends in
age at pubertal onset assessed by breast development among girls: A
systematic review and meta-analysis. JAMA Pediatr.
174(e195881)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Grinspon RP, Ropelato MG, Bedecarrás P,
Loreti N, Ballerini MG, Gottlieb S, Campo SM and Rey RA:
Gonadotrophin secretion pattern in anorchid boys from birth to
pubertal age pathophysiological aspects and diagnostic usefulness.
Clin Endocrinol (Oxf). 76:698–705. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Stagi S, di Tommaso M, Scalini P, Lapi E,
Losi S, Bencini E, Masoni F, Dosa L, Becciani S and de Martino M:
Triple X syndrome and puberty: Focus on the
hypothalamus-hypophysis-gonad axis. Fertil Steril. 105:1547–1553.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Abreu AP, Dauber A, Macedo DB, Noel SD,
Brito VN, Gill JC, Cukier P, Thompson IR, Navarro VM, Gagliardi PC,
et al: Central precocious puberty caused by mutations in the
imprinted gene MKRN3. N Engl J Med. 368:2467–2475. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Leka-Emiri S, Chrousos GP and
Kanaka-Gantenbein C: The mystery of puberty initiation: Genetics
and epigenetics of idiopathic central precocious puberty (ICPP). J
Endocrinol Invest. 40:789–802. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Roberts SA and Kaiser UB: GENETICS IN
ENDOCRINOLOGY: Genetic etiologies of central precocious puberty and
the role of imprinted genes. Eur J Endocrinol. 183:R107–R117.
2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ebrahim SAM, Talross GJS and Carlson JR:
Sight of parasitoid wasps accelerates sexual behavior and
upregulates a micropeptide gene in Drosophila. Nat Commun.
12(2453)2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li L and Gong C: Central precocious
puberty as a prelude of gonad dysplasia. Pediatr Investig. 3:50–54.
2019.PubMed/NCBI View Article : Google Scholar
|