Introduction
Hereditary spherocytosis (HS), a type of
erythrocytosis, is heterogeneous in terms of clinical
manifestations, biochemical data and genetics (1). It is a common hereditary hemolytic
anemia in Nordic populations and has been reported in other
backgrounds, but the global incidence rate is unknown (2). The main clinical features of HS are
anemia, jaundice, reticulocytosis, splenomegaly and spherical
erythrocytes, which may be observed in peripheral blood smears
(3). Patients with severe HS may
be of short stature and have a delayed puberty (4). The phenotypic severity of HS is
related to different pathogenic gene defects and may start at any
age, particularly in the neonatal stage. A phenotype characterized
by hyperbilirubinemia may lead to severe jaundice. In addition,
according to guidelines for the diagnosis and management of HS
(2011 update), HS may be suspected when the mean corpuscular
hemoglobin concentration (MCHC) in newborns exceeds 360 g/l
(5). However, according to the
reported Asian cases, an increase in MCHC is not common in neonatal
HS (3,6-8).
This may be due to differences in ethnic background or statistical
errors caused by lower numbers of neonatal cases. The present study
reported a case of neonatal HS in a Han Chinese pedigree. The
patient developed jaundice and hyperbilirubinemia after birth, but
MCHC remained within the reference range. Gene detection revealed a
novel frameshift mutation in spectrum, β, erythrocytic (SPTB). In
conclusion, the present case report further expands the variation
spectrum of SPTB and provides reliable clinical data for the study
of the mechanisms underlying HS.
Case report
Case presentation
The patient was a male neonate and the first child
of the mother. At the gestational age of 40+3 weeks, the
infant was born through natural vaginal birth. The birth weight was
3,420 g. The Apgar score was 10 points at both 1 and 5 mins
post-birth. In February 2022, 10 h after birth, the infant was
admitted to the Department of Neonatology of the Fourth Affiliated
Hospital of Anhui Medical University (Hefei, China) due to
‘abnormal skin yellowing’. Physical examination revealed lethargy,
yellow colour of the skin and mucous membranes of the whole body
and no splenomegaly. Routine blood examination indicated that
symptoms of anemia and jaundice progressively aggravated, mainly
due to the continuous decrease in red blood cell count (RBC) and
hemoglobin. Biochemical examination revealed an abnormal increase
in the reticulocyte count and reticulocyte ratio, as well as
aspartate aminotransferase, alanine aminotransferase and total
bilirubin levels (Table I).
Peripheral blood smear revealed that the size of mature RBCs varied
and that spherical RBCs were occasionally observed. Results of the
anti-globulin (Coombs') test and glucose-6-phosphate dehydrogenase
screening were negative. No abnormality was found on
B-ultrasonography of the head or abdomen. The patient was
hospitalized for 17 days without being treated with blood
transfusion. After phototherapy (12 h per day for 4 days), the skin
yellowing gradually improved. The main indication for discharge was
the stable level of total bilirubin <15 µg/l; furthermore,
symptoms associated with anemia, such as shortness of breath and
laborious feeding, were not observed. The parents of the patient
were unrelated (the father was 30 years old and the mother was 29
years old when the patient was hospitalized). The father had a
history of anemia and splenomegaly, and after splenectomy (at 15
years of age), his anemia symptoms resolved.
 | Table ILaboratory indicators of the neonatal
case of the present study. |
Table I
Laboratory indicators of the neonatal
case of the present study.
| Parameter | Reference range | Day 1 | Day 2 | Day 5 |
|---|
| WBC,
x109/l | 3.50-9.50 | 21.50 | 16.03 | 16.16 |
| Neutrophils, % | 40.00-75.00 | 74.50 | 69.00 | 61.60 |
| Lymphocytes, % | 20.00-50.00 | 16.10 | 20.60 | 20.80 |
| RBC,
x1012/l | 6.00-7.00 | 4.43 | 4.31 | 3.65 |
| Hemoglobin, g/l | 170-200 | 147 | 142 | 120 |
| Hematocrit, % | 40.00-50.00 | 44.40 | 42.80 | 35.00 |
| Mean corpuscular
volume, fl | 82.0-100.0 | 100.1 | 99.2 | 95.9 |
| Mean corpuscular
hemoglobin, pg | 27.0-34.0 | 33.2 | 33.1 | 32.8 |
| Mean corpuscular
hemoglobin concentration, g/l | 316-354 | 332 | 333 | 342 |
| Reticulocytes, % | 0.50-1.50 | 7.91 | 8.98 | 5.50 |
| Reticulocyte count,
x1012/l | 0.024-0.084 | 0.350 | 0.387 | 0.201 |
| Total bilirubin,
µg/l | 5.0-21.0 | 206.1 | 218.9 | 222.6 |
| Direct bilirubin,
µg/l | 0-6.8 | 0 | 0 | 28.5 |
| Indirect bilirubin,
µg/l | 2.0-18.0 | 206.1 | 218.9 | 194.5 |
| Aspartate
aminotransferase, U/l | 0-34 | 131 | 81 | 73 |
| Alanine
aminotransferase, U/l | 0-49 | 18 | 13 | 16 |
| γ-Glutamyl
transpeptidase, U/l | 0-73 | 243 | 213 | 204 |
Genetic analysis
To further clarify the pathogenic factors of the
patient's clinical phenotype, ~3 ml of peripheral blood was
extracted from the patient and the patient's parents for
trio-whole-exome sequencing testing. The work described in this
case report was conducted in accordance with the Code of Ethics of
the World Medical Association (Declaration of Helsinki) for
experiments involving humans (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/).
The ethics committee of the Fourth Affiliated Hospital of Anhui
Medical University (Hefei, China) approved the study and the
publication of this report (PJ-YX2022-008). The parents of the
patient provided written informed consent regarding the publication
of the medical data and images of the case. In brief, the leukocyte
genome was extracted according to the instructions of a DNA
extraction kit (CoWin Biosciences). After constructing the exome
library, the Illumina Novaseq 6000 series sequencer (Illumina,
Inc.) was used for high-throughput sequencing. The sequencing data
were subjected to quality control, reference sequence comparison
and screening, normal population distribution frequency analysis
(dbSNP, www.ncbi.nlm.nih.gov/snp/; ExAC, www.exac.broadinstitute.org/; and 1000 Genomes,
www.1000genomes.org/), and pathogenicity
prediction analysis using various software (SIFT, www.sift.bii.a-star.edu.sg/; Polyphen-2,
www.genetics.bwh.harvard.edu/pph2/; and
MutationTaster, www.mutationtaster.org/).
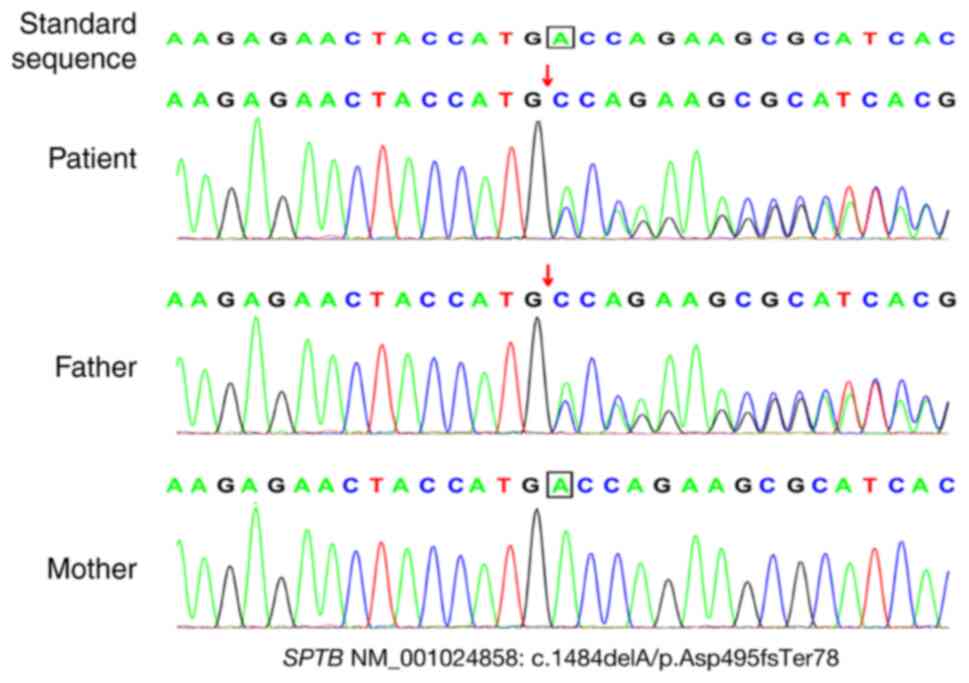
The results indicated that the patient harbored a
frameshift mutation (NM_001024858: c.1484dela/p.asp495fster78); the
patient's father carried a heterozygous mutation and the patient's
mother carried the wild-type gene. There were no pathogenic
mutations in other hemoglobin disease- or HS-related genes, such as
ANK1, SLC4A1, SPTA1, HBA1 and HBA2. The mutation was not included
in the public database for the normal control population. The
mutation was rated as pathogenic according to the American College
of Medical Genetics and Genomics guidelines and the rating evidence
was ‘pathogenic very strong 1 + pathogenic moderate 2 + possibly
pathogenic 4’ (9). Sanger
sequencing confirmed the presence of this mutation (Fig. 1). Finally, based on the clinical
manifestations and family history, the patient was diagnosed with
HS2 [Online Mendelian Inheritance in Man (OMIM) #616649].
Literature review
A total of 26 studies were reviewed, including 160
reported cases of HS in the Chinese population, of which 24 were
neonatal cases (Tables SI and
SII). The incidence of the
ankyrin 1 (ANK1) mutation in the neonatal group (15/24, 63%) was
higher than that in the non-neonatal group (58/136, 43%), while the
incidence of the SPTB mutation was similar in the two groups. In
addition, most of the mutations in the neonatal group were
loss-of-function (LOF) mutations. For instance, the incidence of an
LOF mutation in ANK1 was ~87% (13/15), while that in SPTB was 100%
(8/8) (Fig. 2). This may indicate
that patients with HS and LOF mutations are more likely to have a
neonatal onset.
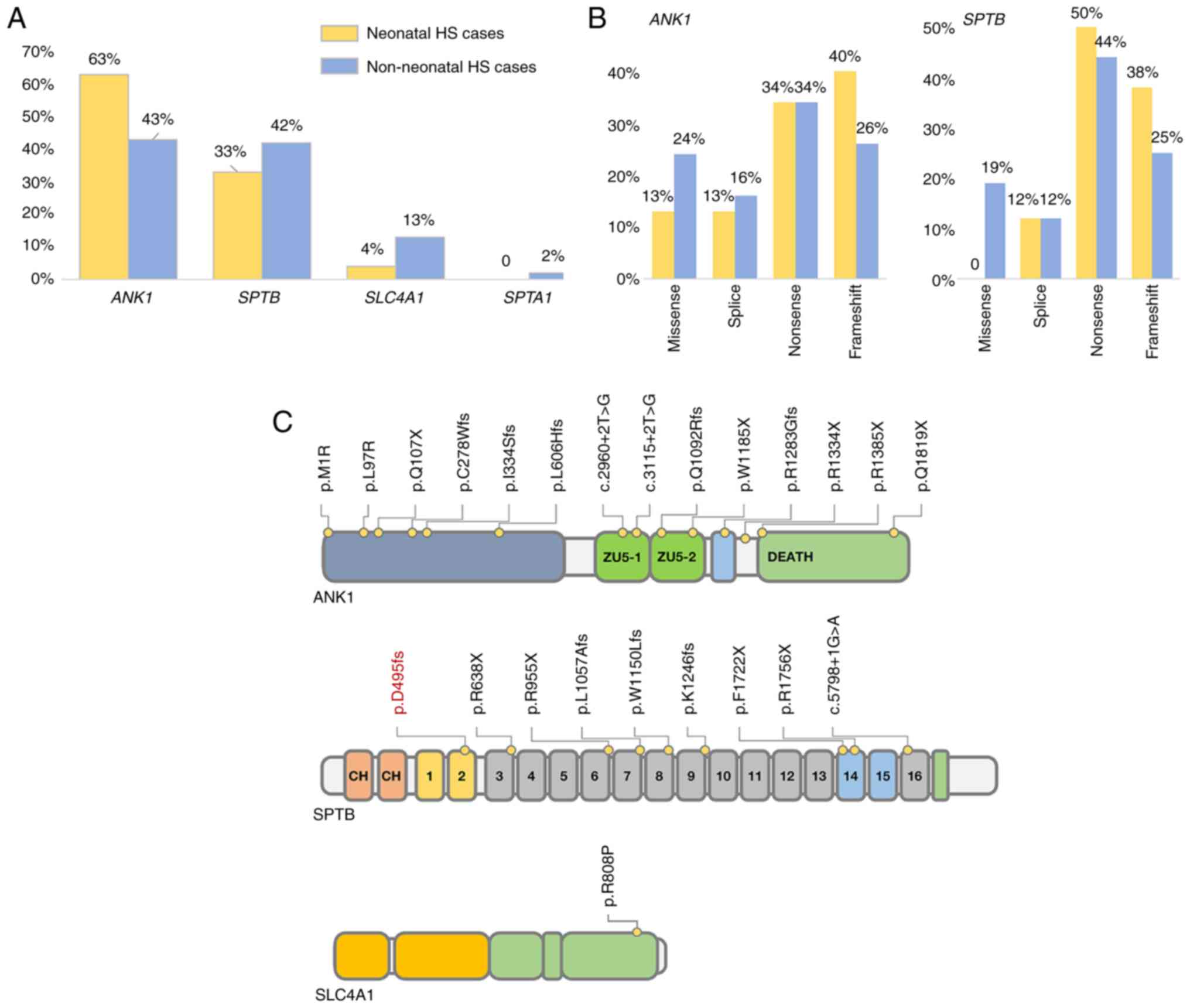 | Figure 2Genetic characteristics of the
neonatal cases in China. (A) The incidence of ANK1 and SPTB
mutations in neonatal and non-neonatal Chinese patients with HS.
(B) Comparison of the frequency of varying loss-of-function
mutations within ANK1 and SPTB in neonatal and non-neonatal
patients with HS. (C) The reported mutation distribution of Chinese
neonatal cases of HS; the position of the mutation investigated in
this case is indicated in red. HS, hereditary spherocytosis; ANK1,
ankyrin 1; SPTB, spectrum, β, erythrocytic; SLC4A1, solute carrier
family 4, member 1; SPTA1, spectrum, α, erythrocytic 1. |
Discussion
HS is characterized by anemia, jaundice, progressive
splenomegaly and reticulocytosis. Its clinical diagnosis is
challenging, as the disease phenotype is highly variable. For
instance, in patients with a mild phenotype and slight hemolysis,
the condition is frequently ignored; however, when infection,
fatigue and other specific factors aggravate hemolysis, subjects
may develop symptoms similar to those of acute hemolytic anemia
(10). The onset of HS in neonates
is usually serious and the condition mainly manifests as anemia and
hyperbilirubinemia. In general, neonatal cases with serious
hemolysis and anemia symptoms require transfusion of suspended
RBCs. However, with increasing neonatal age, the hematopoietic
function of the bone marrow and the compensatory ability of the
liver improve, which may alleviate the symptoms of anemia. To date,
five pathological genes related to HS have been identified: ANK1
(OMIM #612641), SPTB (OMIM #182870), spectrum, α, erythrocytic 1
(OMIM #182860), solute carrier family 4, member 1 (SLC4A1; OMIM
#109270) and erythrocyte membrane protein band 4.2 (OMIM #177070),
among which ANK1 and SPTB defects are the main pathogenic factors
of HS (3,11). In the present study, a patient with
HS from a Han Chinese family was analyzed and it was indicated that
the patient exhibited symptoms of jaundice, hyperbilirubinemia and
anemia after natural delivery. Genetic testing revealed a novel
frameshift mutation, p.Asp495fsTer78 in SPTB, which may lead to LOF
gene mutation. Finally, the patient was diagnosed with HS2. In
addition, the father of the patient carried the mutation and
exhibited symptoms of anemia and splenomegaly during adolescence;
however, the anemia symptoms subsided after splenectomy. The
phenotypic variability of HS was further demonstrated by phenotypic
differences among different family members.
SPTB is located on q23.3 of chromosome 14, which
encodes the spectrin family β-subunit, and α-spectrin forms a
tetramer α2β2 structure, which is an important part in forming the
erythrocyte membrane skeleton network. When the function of the
SPTB protein is altered, the cohesion of the membrane skeleton
decreases, resulting in spherical changes in erythrocytes (2,3). The
reported Chinese patients with HS-SPTB mainly harbored LOF
mutations, such as nonsense and frameshift mutations (3,7,11).
Wang et al (7) reported
that the phenotypic severity of patients with HS had no significant
correlation with pathogenic genes (ANK1, SPTB and SLC4A1) or
mutation types. However, the retrospective analysis of the reported
cases of HS in the Chinese population performed as part of the
present study suggests that patients with neonatal onset more
frequently harbor LOF mutations of pathogenic genes. However,
whether a correlation exists between the mutation types and the age
of onset of HS remains to be determined and requires further
investigation through the analysis of more cases, particularly
newborns, to establish the statistical significance of the
results.
As newborn HS cases usually have characteristics of
non-specific diseases, such as jaundice and hyperbilirubinemia,
clinical diagnosis is challenging. Furthermore, there are currently
no diagnostic guidelines for newborns or infants with HS. Existing
guidelines indicate that when the MCHC index of newborns is
>36.0 g/dl, the sensitivity and specificity of identifying HS
are as high as 82 and 98%, respectively (12). However, this index may not be
applicable to the Chinese population. In the present study,
previously reported data of 24 newborn cases of HS in the Chinese
population were reviewed and 16 cases without detailed clinical
data were excluded. Only one of the remaining eight newborn cases
of HS had an MCHC index greater than the reference range (13). Of note, different researchers have
used a variety of hematological indicators to upgrade the
algorithm, which may improve the sensitivity and accuracy of HS
identification. For instance, Tao et al (14) indicated through a cohort study that
the sensitivity and identification of HS may be markedly improved
when the mean sphered cell volume (MSCV) is less than the mean
corpuscular volume (MCV), and Arora et al (15) determined that MCV-MSCV >10 fl
may be used as the threshold range for diagnosing HS. However,
neonatal cohort studies and statistical evidence for these new
indicators for screening HS are lacking. In addition, in the
present study, no MSCV index was found in reported neonatal cases,
which may be related to the low availability of professional
equipment for detecting this index. At present, only the Beckman
Coulter blood analyzer is able to perform MSCV analysis (16).
Although key suggestive indicators for the screening
and diagnosis of neonatal HS are still lacking, guidelines have
listed the eosin-5'-maleimide binding test (EMABT) as one of the
diagnostic methods for HS and confirmed its high sensitivity and
specificity in numerous cohorts (17,18).
However, EMABT may have limitations in neonatal patients. There are
no obvious spherical RBCs in the peripheral blood images of certain
neonatal patients, which interferes with clinical judgment
(6,19). In addition, detection not only
relies on the analysis of patients' fresh blood samples, but also
the reproducibility of the results, which is affected by the
stability and concentration of dyes. Therefore, laboratories need
to have high standard requirements for detection timeliness and
standardized management (20). In
recent years, sequencing technology has been widely used in clinics
due to its convenience, accuracy and high-throughput application,
and it has an important role in HS diagnosis, differential
diagnosis and genetic counseling. Since more than half of neonatal
cases have a family history, the parents of newborns with
hyperbilirubinemia and anemia symptoms should be asked for detailed
family history information, which has a key role in
early-intervention gene detection (21).
In summary, the present study reported a case of
neonatal HS. The patient had hyperbilirubinemia and anemia
symptoms, but there were no abnormalities in MCHC or other
indicators. Gene detection suggested a frameshift mutation in SPTB.
A literature review suggested that Chinese neonatal cases are
mainly caused by ANK1 defects, which have a higher incidence of LOF
mutations than those in non-neonatal patients. In short, as
neonatal HS has non-specific indicators, which challenges accurate
diagnosis, it should be endeavored to actively improve the
application value of gene detection technology in the diagnosis of
HS.
Supplementary Material
Genetic information of reported
neonatal cases in china (including main laboratory data).
Genetic information of reported
non-neonatal cases in China.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CX and NW designed the experiments. YW, DW and XZ
collected and evaluated the clinical data. CX drafted the
manuscript. CX and NW checked and approved the authenticity of the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was carried out in accordance with the
code of Ethics of the World Medical Association (Declaration of
Helsinki) for experiments involving humans. This study was reviewed
and approved by the ethics committee of the Fourth Affiliated
Hospital of Anhui Medical University (Hefei, China; approval no.
PJ-YX2022-008).
Patient consent for publication
The parents of the patient provided written informed
consent regarding the publication of the medical data and images of
the case.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Delaunay J: The molecular basis of
hereditary red cell membrane disorders. Blood Rev. 21:1–20.
2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Perrotta S, Gallagher PG and Mohandas N:
Hereditary spherocytosis. Lancet. 372:1411–1426. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang R, Yang S, Xu M, Huang J, Liu H, Gu W
and Zhang X: Exome sequencing confirms molecular diagnoses in 38
Chinese families with hereditary spherocytosis. Sci China Life Sci.
61:947–953. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang D and Lai P: Global retardation and
hereditary spherocytosis associated with a novel deletion of
chromosome 8p11.21 encompassing KAT6A and ANK1. Eur J Med Genet.
63(104082)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bolton-Maggs PH, Langer JC, Iolascon A,
Tittensor P and King MJ: General Haematology Task Force of the
British Committee for Standards in Haematology. Guidelines for the
diagnosis and management of hereditary spherocytosis-2011 update.
Br J Haematol. 156:37–49. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xie L, Xing Z, Li C, Liu SX and Wen FQ:
Identification of a De Novo c.1000delA ANK1 mutation associated to
hereditary spherocytosis in a neonate with Coombs-negative
hemolytic jaundice-case reports and review of the literature. BMC
Med Genomics. 14(77)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang D, Song L, Shen L, Zhang K, Lv Y, Gao
M, Ma J, Wan Y, Gai Z and Liu Y: Mutational characteristics of
causative genes in chinese hereditary spherocytosis patients: A
report on fourteen cases and a review of the literature. Front
Pharmacol. 12(644352)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wu C, Xiong T, Xu Z, Zhan C, Chen F, Ye Y,
Wang H and Yang Y: Preliminary study on the clinical and genetic
characteristics of hereditary spherocytosis in 15 Chinese children.
Front Genet. 12(652376)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sun Q, Xie Y, Wu P, Li S, Hua Y, Lu X and
Zhao W: Targeted next-generation sequencing identified a novel ANK1
mutation associated with hereditary spherocytosis in a Chinese
family. Hematology. 24:583–587. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Qin L, Nie Y, Zhang H, Chen L, Zhang D,
Lin Y and Ru K: Identification of new mutations in patients with
hereditary spherocytosis by next-generation sequencing. J Hum
Genet. 65:427–434. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Christensen RD and Henry E: Hereditary
spherocytosis in neonates with hyperbilirubinemia. Pediatrics.
125:120–125. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu Y, Zheng J, Song L, Fang Y, Sun C, Li
N, Liu G and Shu J: A novel SPTB gene mutation in neonatal
hereditary spherocytosis: A case report. Exp Ther Med.
20:3253–3259. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tao YF, Deng ZF, Liao L, Qiu YL, Chen WQ
and Lin FQ: Comparison and evaluation of three screening tests of
hereditary spherocytosis in Chinese patients. Ann Hematol.
94:747–751. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Arora RD, Dass J, Maydeo S, Arya V, Kotwal
J and Bhargava M: Utility of mean sphered cell volume and mean
reticulocyte volume for the diagnosis of hereditary spherocytosis.
Hematology. 23:413–416. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liao L, Xu Y, Wei H, Qiu Y, Chen W, Huang
J, Tao Y, Deng X, Deng Z, Tao H and Lin F: Blood cell parameters
for screening and diagnosis of hereditary spherocytosis. J Clin Lab
Anal. 33(e22844)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bianchi P, Fermo E, Vercellati C, Marcello
AP, Porretti L, Cortelezzi A, Barcellini W and Zanella A:
Diagnostic power of laboratory tests for hereditary spherocytosis:
A comparison study in 150 patients grouped according to molecular
and clinical characteristics. Haematologica. 97:516–523.
2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Crisp RL, Solari L, Vota D, García E,
Miguez G, Chamorro ME, Schvartzman GA, Alfonso G, Gammella D,
Caldarola S, et al: A prospective study to assess the predictive
value for hereditary spherocytosis using five laboratory tests
(cryohemolysis test, eosin-5'-maleimide flow cytometry, osmotic
fragility test, autohemolysis test, and SDS-PAGE) on 50 hereditary
spherocytosis families in Argentina. Ann Hematol. 90:625–634.
2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Christensen RD, Lambert DK, Henry E,
Eggert LD, Yaish HM, Reading NS and Prchal JT: Unexplained extreme
hyperbilirubinemia among neonates in a multihospital healthcare
system. Blood Cells Mol Dis. 50:105–109. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Da Costa L, Suner L, Galimand J, Bonnel A,
Pascreau T, Couque N, Fenneteau O and Mohandas N: Society of
Hematology and Pediatric Immunology (SHIP) group; French Society of
Hematology (SFH). Diagnostic tool for red blood cell membrane
disorders: Assessment of a new generation ektacytometer. Blood
Cells Mol Dis. 56:9–22. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Christensen RD, Yaish HM and Gallagher PG:
A pediatrician's practical guide to diagnosing and treating
hereditary spherocytosis in neonates. Pediatrics. 135:1107–1114.
2015.PubMed/NCBI View Article : Google Scholar
|