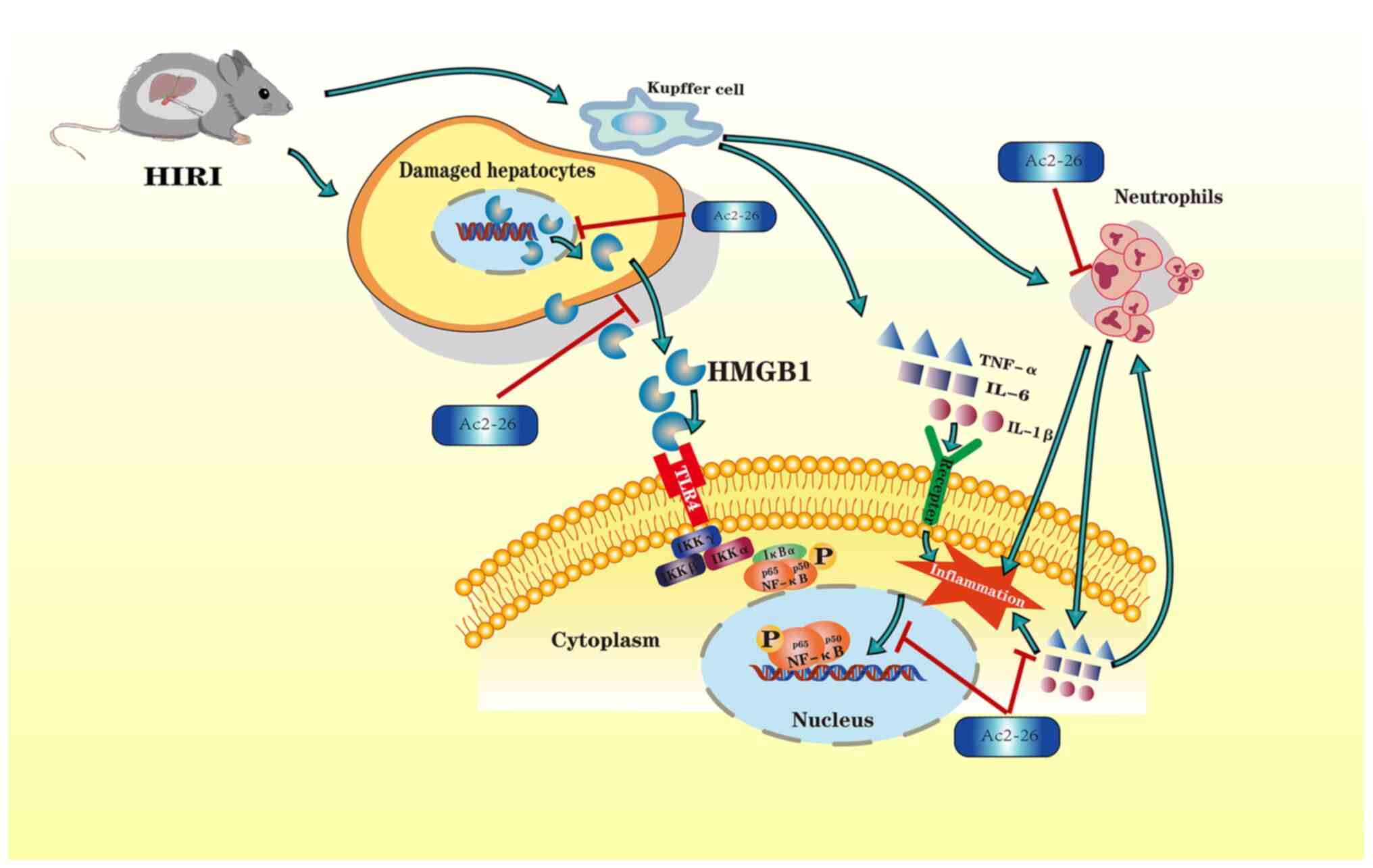
Introduction
As a common pathophysiological condition, hepatic
ischemia-reperfusion injury (HIRI) represents the primary cause of
liver injury during hepatolobectomy, liver transplantation and
hemorrhagic shock and may cause hepatocyte damage and even liver
failure (1). Although numerous
studies (1-4)
have been conducted on HIRI, the mechanism underlying this
condition remains largely unknown and there are currently no
effective measures of prevention or treatment measures. Therefore,
there is an urgent need to study the potential mechanism of HIRI
and explore new therapeutic targets.
During HIRI, local ischemia and hypoxia cause rapid
depletion of ATP and glycogen, leading to cell death and the
release of damage associated molecular patterns (DAMPs) from
damaged or dying cells (2,3,5).
High mobility group box 1 (HMGB1), one of the most studied DAMPs
involved in the ischemia-reperfusion (I/R) process, has recently
been found to be released from dying or damaged hepatocytes and
bind to toll-like receptor 4 (TLR4). Once bound to TLR4, HMGB1 can
induce an intracellular signaling cascade, which involves
activating NF-κB, which subsequently regulates the expression of
inflammatory cytokines, containing TNF-α, IL-1β and IL-6 (4,6).
Excessive inflammatory factors and chemokines can recruit
neutrophils to infiltrate and damage the hepatic parenchymal cells.
Furthermore, infiltrating neutrophils intensify hepatic injury by
exacerbating inflammatory cascades induced by neutrophil
extracellular traps (NETs). Subsequently, dead hepatocytes release
additional HMGB1, which forms a positive feedback loop, thus
augmenting hepatic injury (7).
Accordingly, a strategy for inhibiting the positive feedback loop
may be of therapeutic value for the prevention and treatment of
HIRI.
Annexin A1 (AnxA1), an important
glucocorticoid-regulating protein, inhibits the migration of
leukocytes to the inflammation site as well as inflammatory
response induced by pathogenic microbial infections and aseptic
inflammation (8). Ac2-26, the
N-terminal-derived peptide of AnxA1, serves an
anti-inflammatory role through multiple pathways (8,9). As
AnxA1 is easily inactivated in vitro, Ac2-26 is often used
as its substitute and is widely employed in experimental models of
inflammation. Increasing evidence (10-13)
suggests that Ac2-26 exhibits protective effects against the
I/R-induced inflammatory response and ameliorates organ
dysfunctions. However, whether Ac2-26 can alleviate HIRI by
inhibiting neutrophil infiltration through regulation of the
HMGB1/TLR4/NF-κB signaling pathway remains to be elucidated.
Due to the involvement of sterile inflammation in
the pathogenesis of HIRI and the effect of Ac2-26 on the
inflammatory cascades of IRI of various organs, it was hypothesized
that Ac2-26 reduced inflammatory responses induced by HIRI and the
present study was conducted with the aim of determining the effects
of Ac2-26 on the HMGB1/TLR4/NF-κB axis and neutrophil infiltration.
The present study could provide a potential therapeutic
intervention for sterile inflammation following HIRI.
Materials and methods
Chemicals
Ac2-26 was purchased from Shanghai Qiang Yao
Biotechnology Co., Ltd. Anti-GAPDH antibody (cat. no. 60004-1-Ig)
was obtained from ProteinTech Group, Inc. Anti-lymphocyte antigen 6
complex locus G6D (Ly6G) antibody (cat. no. 87048) was purchased
from Cell Signaling Technology, Inc. Anti-HMGB1 antibody (cat. no.
ab18256) was obtained from Abcam. Anti-TLR4 (cat. no. bs-20379R),
anti-NF-κB (cat. no. bs-3485R), anti-p-NF-κB (cat. no. bs-5512R)
and anti-IκBα (cat. no. bs-1287R) antibodies were obtained from
BIOSS. Anti-CD16/32 was purchased from Elabscience Biotechnology,
Inc. Peridinin chlorophyll protein complex-conjugated anti-CD45
antibody (cat. no. 103130) was from BioLegend, Inc. The
Phycoerythrin-conjugated anti-Ly6G antibody (cat. no. 551461) was
purchased from BD Bioscience. The BCA protein assay kit (cat. no.
P0012) was purchased from Beyotime Institute of Biotechnology. The
radioimmunoprecipitation assay (RIPA; cat. no. R0020) and
phenylmethanesulfonyl fluoride (PMSF; cat. no. P0100) were
purchased from Beijing Solarbio Science & Technology Co., Ltd.
Horseradish peroxidase (HRP)-conjugated secondary anti-mouse
antibody (cat. no. ZB-2301) and DAB two-step kit (cat. no. PV-6001)
were obtained from OriGene Technologies, Inc. The FITC-conjugated
secondary antibodies (cat. no 805-095-180) came from Jackson
ImmunoResearch Laboratories, Inc. The alanine transaminase (ALT;
cat. no. C009-2-1), aspartate transaminase (AST; cat. no. C010-2-1)
and myeloperoxidase assay kits (MPO; cat. no. A044-1-1) were
manufactured from Nanjing Jiancheng Bioengineering Institute. The
HMGB1 enzyme-linked immunosorbent assay kit (cat. no. ST51011) was
purchased from IBL International. TRIzol® reagent (cat.
no. 15596026) was purchased from Thermo Fisher Scientific, Inc. The
ReverTra Ace qPCR RT kit (cat. no. FSQ-101) was obtained from
Toyobo Life Science. Lastly, the collagenase type Ⅳ (cat. no.
LS004186) was purchased from Worthington Biochemical
Corporation.
Animals and treatment
A total of 72 healthy male C57 black 6 (C57BL/6)
mice, aged 8 weeks with weights of 22-25 g and SPF grade, were
purchased from the Jinan Pengyue Experimental Animal Center. All
mice were treated humanely during the entire experimental process,
including housing under constant temperature and humidity (21-26˚C
and 50-70%, respectively), with a 12-h light/dark cycle and food
and water provided ad libitum.
A week later, before the operation, the animals were
randomly allocated into sham, I/R, I/R + Ac2-26 and Ac2-26 groups
(n=6). Mice in the I/R + Ac2-26 group were administered Ac2-26 at a
dose of 250 µg/kg via intraperitoneal injection 30 min before
ischemia, whereas those in the sham and Ac2-26 groups received the
same anesthetic and laparotomy, with the exception of surgery.
Briefly, mice were anesthetized via intraperitoneal injection of 1%
pentobarbital sodium (40 mg/kg) and the left and middle branches of
the hepatic pedicle were occluded by clamping them with a
noninvasive vascular clip for 45 min, followed by 24 h of
reperfusion. The liver color change from red to dark purple, which
indicated that the model was successfully established. Body
temperature was maintained with an appropriative heating pad at
37±0.4˚C upon surgical procedures. Animals were provided food and
water after fully awake. During the operation three mice succumbed
due to intraoperative bleeding (mortality rate of 4.2%). Animal's
health and behavior was monitored every half hour after surgery,
including respiratory rate and depth, mucous membrane coloration
and wounds.
After 24 h of reperfusion, blood samples were
collected from the retro-orbital vein under pentobarbital sodium
(40 mg/kg) until the mice lost consciousness. The blood samples
were centrifuged at room temperature at 3,000 x g for 10 min in an
Eppendorf centrifuge (5424R) to obtain the corresponding serum
samples. The serum and left lobe samples were collected and then
stored at -80˚C for further experiments. After blood sample
collection, all animals were sacrificed by cervical dislocation
under anesthesia (1% pentobarbital sodium 40 mg/kg). Animals were
sacrificed if any of the following conditions occurred: The animal
was unable to move and or respond to gentle stimuli, or had
difficulty in breathing, or experienced inability to eat or drink.
Animal mortality was verified by cardiac arrest, disappearance of
spontaneous breathing and no response to tail pinch.
All procedures were carried out in line with the US
National Institutes of Health Guide for the Care and Use of
Laboratory Animals 8th edition (14) and the guidelines of the Animal
Ethics Committee of Weifang Medical University (Weifang, China),
which approved the present study (approval no. 2020SDL187).
ALT and AST enzyme activity
determination
The serum levels of ALT and AST were detected by
commercial assay kits in line with the manufacturer's protocols.
Briefly, the serum samples (5 µl) were mixed with matrix solution
(20 µl) in the assay wells and kept at 37˚C for 30 min. Next, 20 µl
chromogenic reagent was added and kept at room temperature again
for 15 min. A wavelength of 510 nm was used to read plates with a
microplate reader (Multiskan FC; Thermo Fisher Scientific, Inc.).
The absolute OD value of ALT and AST was calculated by the
following formula:
Absolute OD value = the OD of the experimental group
- the OD of the control.
Immunohistochemistry and
immunofluorescent staining
Paraffin sections (5 µm) were dewaxed, hydrated,
incubated in a microwave with citrate buffer repair solution,
inactivated with 3% hydrogen peroxide and blocked with 1% goat
serum (cat. no. AR0009; Wuhan Boster Biological Technology, Ltd.)
for 1 h at 37˚C. Then the primary antibody against HMGB1 (1:150)
was used to incubate the sections overnight at 4˚C, followed by
incubation with a streptavidin-peroxidase complex. The peroxidase
conjugates were later visualized by the diaminobenzidine (DAB)
solution for 5 min and counterstained with hematoxylin for 20 sec
at room temperature. The slides were subsequently washed with PBS
between each step. The slides were then dehydrated, cleared, sealed
with neutral gum, observed under an optical microscope and images
captured (magnification, x200) for analysis.
For Ly6G immunofluorescence staining, 10-µm-thick
frozen sections were collected, fixed in 4% paraformaldehyde for 10
min and inactivated with 3% hydrogen peroxide for 10 min at room
temperature, repaired by microwave at 95-100˚C until cooled to room
temperature and blocked with goat serum of 1% for 1 h at 37˚C.
Next, sections were incubated overnight with an antibody against
Ly6G (1:200) at 4˚C and a FITC-conjugated secondary antibody
(1:150) for 45 min at 37˚C, then mounting medium with DAPI was used
to cover. Using a fluorescence microscope (Olympus FV500, Olympus
Corporation; magnification, x200), the slides were judged by an
investigator who was unaware of the corresponding study group.
Reverse transcription-quantitative
(RT-q)PCR
TRIzol® reagent was used to extract the
total RNA from hepatic tissue and the total RNA was reverse
transcribed with the ReverTra Ace® qPCR RT kit. In line
with the manufacturer's instructions, 0.6 µl primers and 1 µl cDNA
were used for reverse transcription polymerase. The thermocycling
conditions were as follows: Pre-denaturation at 95˚C for 3 min,
annealing at 60˚C for 45 sec and repetition for 35 cycles. The
primer sequences for HMGB1, TLR4 and GAPDH are shown in Table I.
GAPDH standardized the total mRNA for genes apiece and the relative
expression level was calculated using the reported
2-ΔΔCq method (15).
All the experiments were repeated ≥3 times for each condition
Western blot analysis
Western blotting was used to analyze the protein
expression level as previously reported (16). Tissue protein samples were prepared
from protein extraction reagents with hepatic tissues. In short, 80
mg of hepatic tissue was homogenized on ice in 1 ml of RIPA lysis
buffer with a protease inhibitor mixture and PMSF. The BCA method
was used to measure the protein concentrations. Next, a total of 20
µg protein was subjected to 10% SDS-PAGE and the protein was
transferred to a PVDF membrane. After blocking in TBS with 0.1%
Tween-20 (TBST) buffer with 5% skimmed milk for 2 h at room
temperature, the PVDF membranes were incubated with the primary
antibodies, such as anti-GAPDH (1:1,000), anti-HMGB1 (1:2,000),
anti-TLR4 (1:1,500), anti-NF-κB (1:500), anti-p-NF-κB (1:500) and
anti-IκBα (1:1,000) on a shaker at 4˚C overnight with agitation,
followed by incubation with a horseradish peroxidase-conjugated
secondary antibody lasted 1 h at room temperature. Band
densitometry was visualized with an ECL detection system (ChemiDoc™
Touch; Bio-Rad Laboratories, Inc.) and analyzed by ImageJ 1.8.0
software (National Institutes of Health), the relative protein
expression level were normalized to the ratio of GAPDH. Experiments
were repeated ≥3 times for each condition.
Enzyme-linked immunosorbent assay
(ELISA)
The serum levels of HMGB1 were observed by an ELISA
kit (cat. no. ST51011; IBL International GmbH) according to the
manufacturer's instructions. A wavelength of 450 nm was used to
read the ELISA plates with a microplate reader (Thermo Fisher
Scientific, Inc.).
Flow cytometry
Non-parenchymal cells of the hepatic tissues were
isolated using the enzyme digestion method. Briefly, liver samples
were collected after in situ perfusion with HEPES solution
without calcium ions until there was no capillary flow out and then
perfused with 0.02% collagenase type Ⅳ, which had been preheated at
37˚C. After digestion in situ for 12 min, the left lobe
samples were harvested, cut into tiny pieces and digested into a
single-cell suspension using 0.02% collagenase type Ⅳ for 20 min at
37˚C. Next, DMEM/F12 medium with 10% fetal bovine serum was added
to the digested tissue to terminate the reaction. The sample was
then filtered through a 200-mesh nylon mesh and centrifuged at 4˚C
and 30 x g. The supernatant was aspirated and the collected cells
were washed twice, resuspended, counted and suspended to a density
of 1x106 cells/ml. Cells, except those in the control
group, were then blocked with an anti-CD16/CD32 (1:100) antibody
for 10 min to prevent non-specific binding and next stained with a
cocktail of fluorochrome-tagged anti-CD45 (1:1,800) and anti-Ly6G
(1:1,800) antibodies for 25 min in the dark at room temperature. A
BD FACSVerse flow cytometer (cat. no. 651155; BD Biosciences) was
used for the assay and FlowJo software 10.4.0 (FlowJo LLC) was used
for data analysis. The results were expressed as the percentage of
positive cells.
MPO activity assay
The activities of MPO in the liver tissues were
identified using assay kits from Nanjing Jiancheng Bioengineering
Institute according to the manufacturer's protocol. The optical
density at 450 nm was measured using a microplate reader. MPO
activity was calculated with following formula:
MPO activity (U/mg) = ΔOD/[11.3 x volume of hepatic
tissues (mg/reaction solution)].
Statistical analysis
The Shapiro-Wilk test was used to confirm the
normality of the data. The data that met the normal distribution
were expressed as the mean ± standard error of the mean. The Levene
test was used for homogeneity of variance, one-way ANOVA was used
to analyze the data, followed by Tukey's test as appropriate. Data
that did not show the normal distribution (P>0.05), such as MPO
(Fig. 5E), were represented by the
median and interquartile range and submitted to the Kruskal-Wallis
nonparametric test, followed by the Dunn's test to determine the
differences among the groups (P<0.05). SPSS 25.0 software (IBM
Corp.) was applied to calculate the statistical significance, while
GraphPad Prism 7 software (GraphPad Software, Inc.) was employed
for drawing graphs. P<0.05 was considered to indicate a
statistically significant difference.
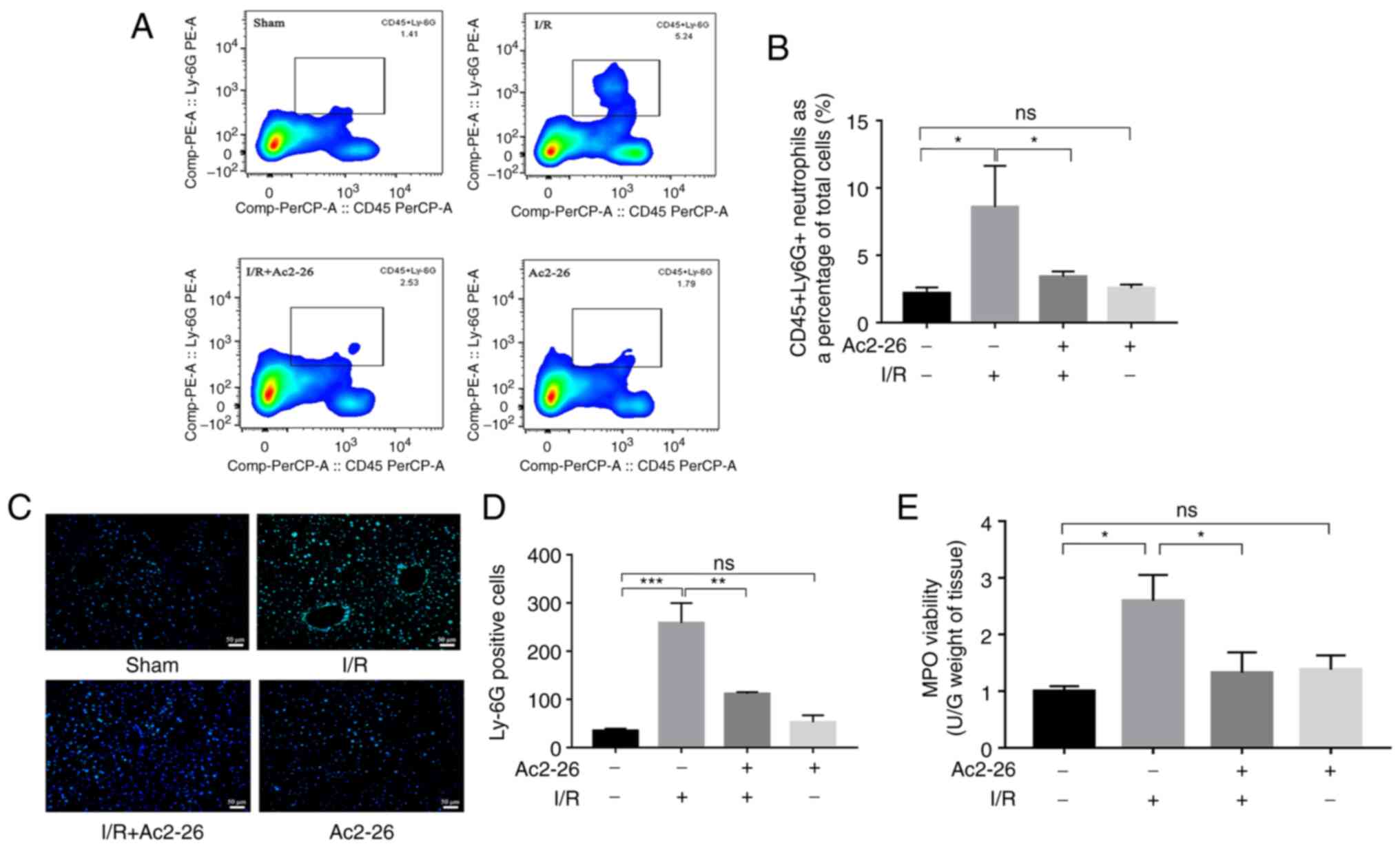 | Figure 5Ac2-26 ameliorates HIRI-induced
hepatic neutrophil infiltration. (A) The neutrophil infiltration of
liver tissues was examined by flow cytometry. (B) The proportion of
neutrophils in different groups of liver tissues was quantified by
flow cytometry. (C) Immunofluorescence staining showed the
localization of Ly6G positive reaction in hepatic tissue
(magnification, x200). (D) The number of Ly6G positive cells in
different groups. Data were analyzed as Tukey's test and expressed
as means ± standard error of the mean, n ≥3. *P<0.05,
**P<0.01 and ***P<0.001; ns, not
significant. (E) The MPO activity was measured with Colorimetric
method. Data was not shown a normal distribution, analyzed as
Kruskal-Wallis nonparametric test, followed by the Dunn's test, n
≥3, *P<0.05; ns, not significant; HIRI, hepatic
ischemia-reperfusion injury. |
Results
Ac2-26 preconditioning attenuates
hepatic injury induced by HIRI
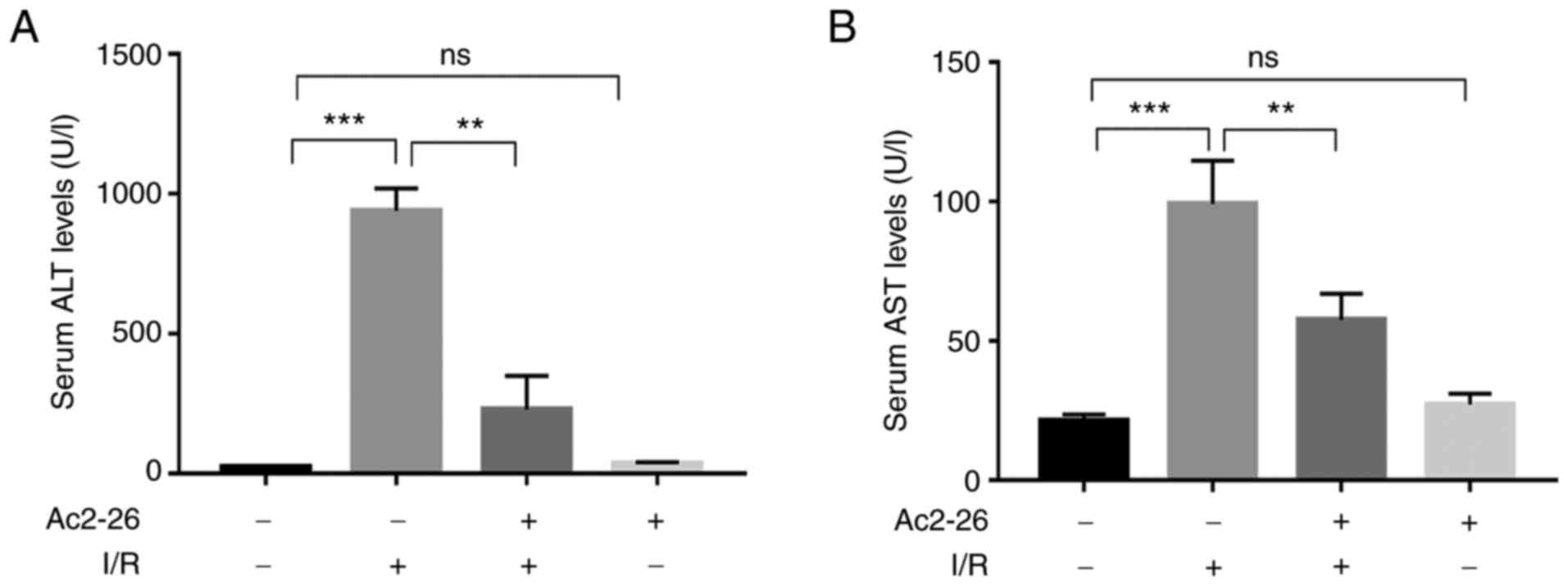
To evaluate whether Ac2-26 protects hepatocytes
against HIRI, the serum levels of ALT and AST, which are biomarkers
of hepatocellular damage, were assessed spectrophotometrically
using a microplate reader. The results showed that the serum levels
of ALT and AST in the Ac2-26 group were similar to those in the
sham group (P>0.05; Fig. 1A and
B), which indicated that Ac2-26
had no significant toxic effects on hepatocytes. As shown in
Fig. 1, compared with those in the
sham group, the serum levels of ALT and AST were markedly increased
in the I/R group (P<0.001). By contrast, the levels of these
indicators were significantly reduced (P<0.001) in the I/R +
Ac2-26 group compared with those in the I/R group (P<0.01).
Collectively, these data suggested that Ac2-26 pretreatment
ameliorated the I/R-induced hepatocyte dysfunction.
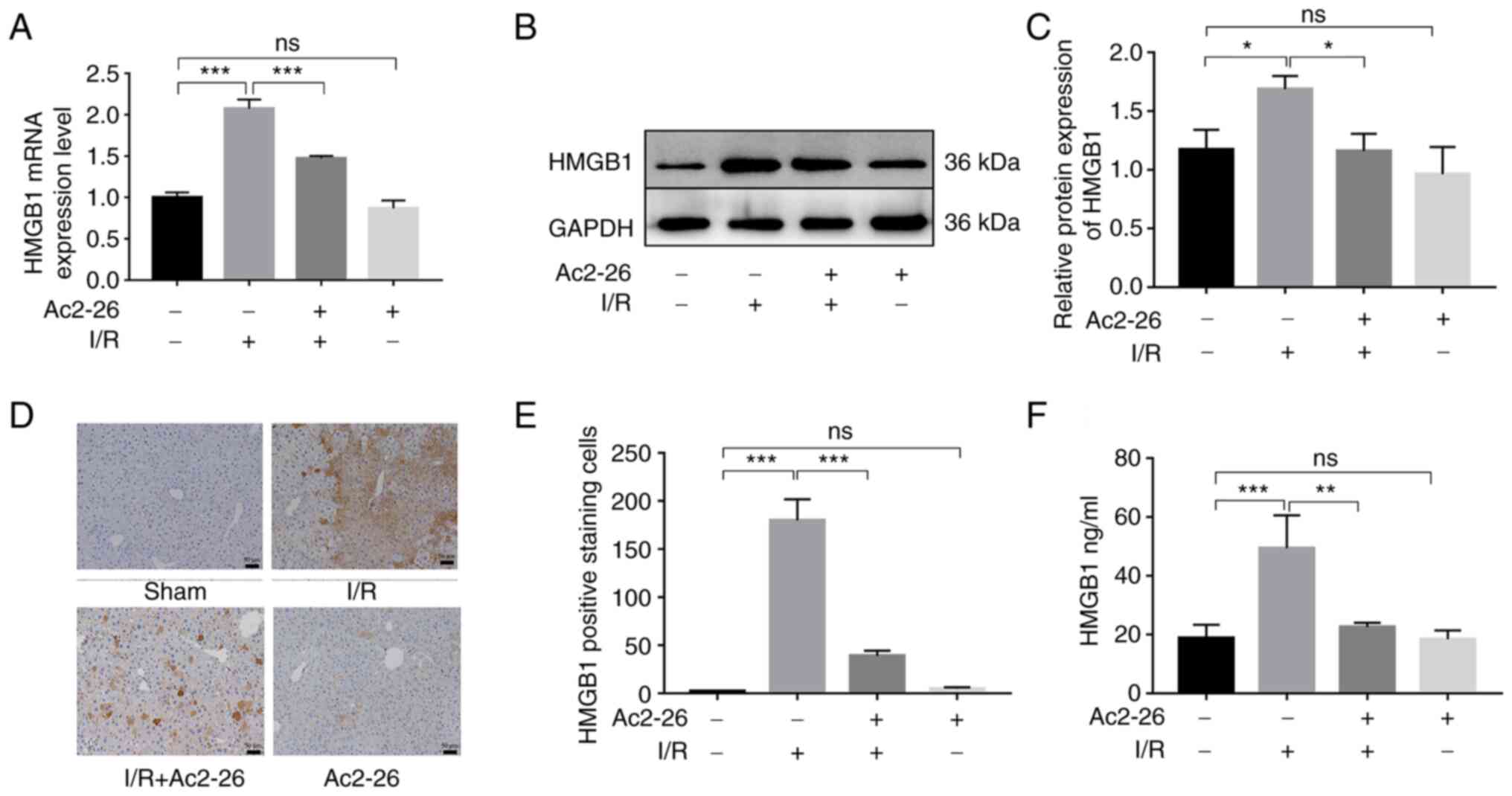
Ac2-26 preconditioning inhibits the
HMGB1/TLR4 signaling pathway in HIRI
HMGB1, as a crucial intermediary factor in the
pathophysiology of the inflammatory response induced by I/R,
induces hepatocyte injury upon HIRI by binding and activating TLR4.
To explore the action of Ac2-26 on the HMGB1/TLR4 signaling pathway
in HIRI, the expression levels of HMGB1 were first detected using
RT-qPCR and western blotting. The expression of HMGB1 was
significantly increased in the I/R group compared with that in the
sham group (RT-qPCR: P<0.001; western blotting: P<0.05). As
expected, Ac2-26 pretreatment remarkably reduced the expression of
HMGB1 in liver tissues induced by HIRI (RT-qPCR: P<0.001;
western blotting: P<0.05) (Fig.
2A-C).
Meanwhile, immunohistochemical staining showed that
there was almost no HMGB1-positive reaction in the hepatocytes of
the sham group. However, the hepatocytes of the I/R group showed
increased levels of HMGB1 within the cytoplasm. By contrast,
pretreatment with Ac2-26 clearly reduced the expression of HMGB1
(P<0.001; Fig. 2D and E). Further, the serum level of HMGB1 was
increased in the I/R group (P<0.001), but in the Ac2-26-treated
group, it was significantly reduced (P<0.01) (Fig. 2F). These results demonstrated that
Ac2-26 inhibited the overexpression and release of HMGB1 caused by
HIRI.
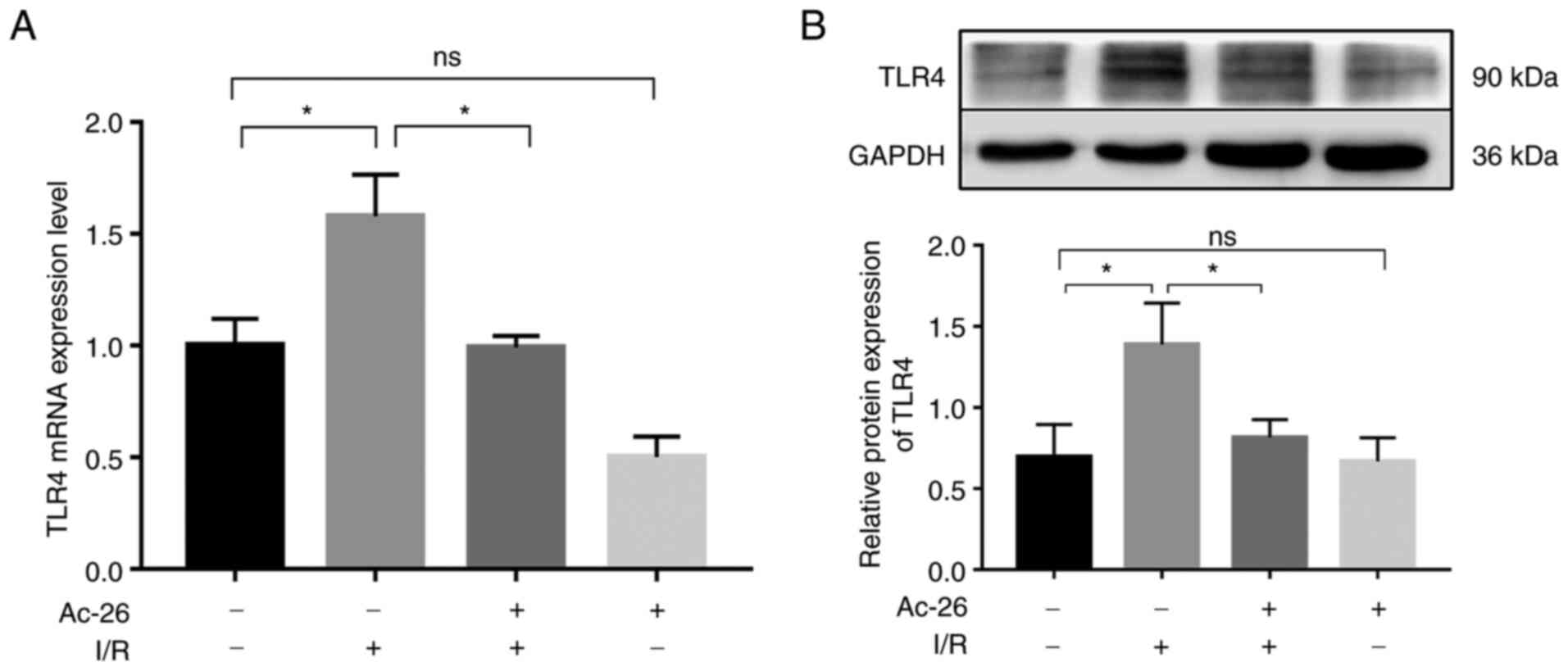
As the main receptor of HMGB1, TLR4 is responsible
for the HMGB1-induced inflammatory response in various disease
models. Subsequently, to explore whether the protective effect of
Ac2-26 was related to the HMGB1/TLR4 pathway, the expression of
TLR4 was determined via RT-qPCR. As illustrated in Fig. 3A, the mRNA levels of TLR4
were remarkably higher in the I/R group than those in the sham
group (P<0.05) and this elevation was notably suppressed by
Ac2-26 pretreatment (P<0.05; Fig.
3A). The aforementioned changes in the levels of TLR4 were
further supported by western blot assay, as shown in Fig. 3B (P<0.05). Collectively, these
findings suggested that Ac2-26 had a protective effect on
I/R-induced liver injury by targeting the HMGB1/TLR4 signaling
pathway.
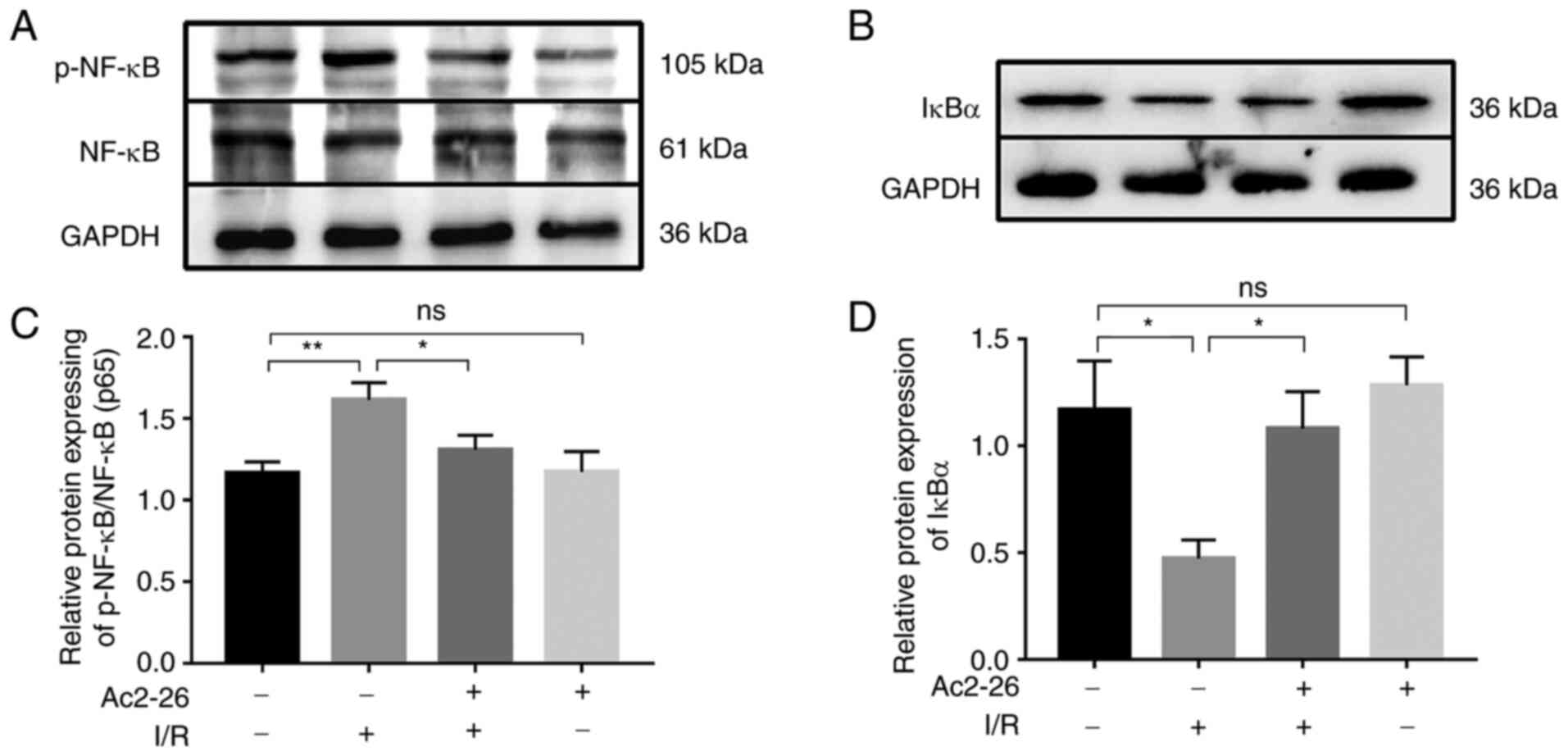
Ac2-26 exerts protective effects
against HIRI by inhibiting the TLR4/NF-κB signaling pathway
Since TLR4 is the main receptor of HMGB1 and the
TLR4/NF-κB signaling axis contributes to the activity of the HMGB1
signaling pathway in various disease models, the expression of the
NF-κB p65 subunit and its inhibitor IκBα was detected by western
blot analysis. As shown in Fig. 4A
and C, the expression of p-NF-κB
and p-NF-κB/NF-κB ratio were markedly increased (P<0.05) in the
I/R group compared with that in the sham group and this effect was
significantly attenuated (P<0.05) by Ac2-26 pretreatment
(Fig. 4A and C).
In addition, the expression of IκBα was notably
reduced (P<0.05) following I/R, but this effect was reversed by
Ac2-26 treatment (Fig. 4B and
D). These data suggested that
Ac2-26 preconditioning may suppress the inflammatory response
induced by HIRI by targeting the TLR4/NF-κB signaling pathway.
Ac2-26 ameliorates the HIRI-induced
hepatic neutrophil infiltration
Evidence from multiple experimental models and
clinical studies has shown that HIRI results in evident hepatic
neutrophil infiltration and activation and the proportion of
neutrophils infiltrating the hepatic tissue is associated with the
severity of the liver I/R injury (17). Based on the important role of
neutrophils in the inflammatory response, the effect of Ac2-26 on
neutrophil infiltration induced by HIRI was examined. First, the
proportion of neutrophils in the hepatic tissues of different
animal groups was measured with flow cytometry. As shown in
Fig. 5A and B, the proportion of neutrophils in the
I/R group was dramatically increased (P<0.05), whereas Ac2-26
pretreatment significantly reduced it (P<0.05; Fig. 5A). The levels of hepatic neutrophil
infiltration were further assessed using Ly6G immunofluorescence
staining, since Ly6G is a marker of neutrophils. As shown in
Fig. 5C and D, the number of Ly6G-positive cells, a
marker for neutrophils, was significantly increased (P<0.001) in
the I/R group compared with that in the sham group, which was
significantly reversed (P<0.01) by Ac2-26 administration
(Fig. 5B).
MPO is an enzyme that is mainly stored in the
azurophilic granules of neutrophils and is released from activated
neutrophils during IRI. MPO activity was measured as another
indicator of neutrophil aggregation and subsequent proteolytic
inflammation. In agreement with the results of Ly6G staining, MPO
activity was significantly increased (P<0.05) in the liver
tissue of the I/R group compared to that in the liver tissue of the
sham group, whereas pretreatment with Ac2-26 significantly
(P<0.05) antagonized these changes (Fig. 5E). In conclusion, the above
findings demonstrated that neutrophils served a critical role in
HIRI and Ac2-26 administration could inhibit the HIRI-induced
neutrophil infiltration, thereby reducing proteolytic
inflammation.
Discussion
HIRI is the major cause of liver dysfunction
following liver surgery and is associated with increased morbidity
and mortality (1). Thus, HIRI is a
choking point limiting the development of liver surgery. Despite
previous studies focused on characterizing the pathogenesis of
HIRI, there are currently no effective methods to prevent or
control it. Growing evidence supports that sterile inflammation
serves a crucial role in HIRI. Therefore, identifying effective
targets to protect hepatocytes against HIRI has become increasingly
important.
As a calcium-dependent phospholipid-binding protein,
AnxA1 is reported to inhibit leukocyte migration to the site of
inflammation and aseptic inflammation (8). Ac2-26, which is the pharmacologically
active center of AnxA1, was synthesized by Cirino et al
(18) as early as 1993 and it has
been shown to effectively inhibit the activation of the
transcription factor NF-κB and the subsequent production of
proinflammatory cytokines, such as TNF-α and IL-1β (19). Although previous studies (20,21)
have confirmed that Ac2-26 exhibits anti-inflammatory effects and
stability in the blood circulatory system, the inability to
quantify it in serum or plasma is a limitation of the present
study. Several studies have demonstrated that Ac2-26 exhibits
protective effects against ischemia-reperfusion injury (IRI) of the
heart (10), brain (22), kidney (13) and intestine (11) by inhibiting the expression of
inflammatory factors such as TNF-α, IL-1β and MPO. Huang et
al (5) found that Ac2-26
exerts anti-inflammatory and pro-repair effects by reducing
neutrophil accumulation and the production of proinflammatory
cytokines. However, the effects of Ac2-26 on HIRI have not been
reported to date. The present study aimed to determine whether
Ac2-26 could alleviate HIRI by inhibiting the HMGB1-mediated
inflammatory responses and neutrophil infiltration.
Previous studies have suggested that the HMGB1/TLR4
signaling pathway is involved in the regulation of IRI in various
parenchymal organs. Accumulating evidence suggests that the
expression of HMGB1 and TLR4 is enhanced in IRI rodent models. Wang
et al (23) confirmed that
HMGB1/TLR4 promotes the release of inflammatory factors by
activating NF-κB in a slight intestinal IRI. Furthermore, a study
by Xue et al (24) reveals
that HMGB1, which is released from necrotic cardiomyocytes in
response to ischemia, can activate TLR4, which can subsequently
trigger downstream signal transduction pathways and cause an
inflammatory cascade. Similarly, Tsaroucha et al (25) demonstrate that silibinin exhibits
protective effects against HIRI by inhibiting the release of HMGB1
from dying hepatic cells. The application of a HMGB1-neutralizing
antibody has been shown to reduce cytokine expression and preserve
hepatic function in a mouse model of HIRI (7). Furthermore, HMGB1 has been found to
be released by dying hepatic cells in response to ischemia and
activate proinflammatory signaling pathways by interacting with
TLR4(26). Inhibition of TLR4 also
provides significant protection against hepatic damage following
HIRI (27,28). However, exogenous HMGB1 or
neutralizing antibodies against HMGB1 have no effect on I/R damage
in TLR4 defective mice (7). The
aforementioned experimental results indicate that HMGB1 acts as a
key mediator of hepatic damage following IRI via interactions with
TLR4 and that activation of the HMGB1/TLR4 signaling pathway is
closely associated with the pathogenesis of HIRI. Accordingly, a
treatment targeting HMGB1/TLR4 signaling may be an effective
strategy to ameliorate hepatic damage by limiting neuroinflammatory
processes during IRI. Similarly to those results, the present study
showed that HIRI induced a significant increase in HMGB1 and TLR4
expression, as well as serum HMGB1 levels. Furthermore, it was
found that Ac2-26 pretreatment considerably inhibited the changes
in HMGB1 and TLR4 expression, which suggested that the protective
effect of Ac2-26 on hepatocytes partly depended on the inhibition
of the HMGB1/TLR4 signaling pathway.
Once bound to TLR4, HMGB1 can activate TLR4, which
subsequently modulates the expression of inflammatory mediators and
triggers the downstream signaling pathways, including NF-κB. During
HIRI, NF-κB is not only the initiator of the inflammatory cascade
but also the central link of the inflammatory response (6). This indicates that the
HMGB1/TLR4/NF-κB signaling pathway serves an essential role in
regulation of the inflammatory response induced by IRI and may
represent an important therapeutic target. Studies have shown that
aucubin and berberine protect against inflammatory responses and
liver injury by regulating HMGB1/TLR4/NF-κB signaling (29,30).
Previous studies have shown that Ac2-26 exhibits
beneficial effects in the treatment of mucosal inflammation, wound
healing and I/R injury models (11,21,31).
The positive feedback between proinflammatory cytokines and NF-κB
signaling is blocked by Ac2-26 pretreatment, which alleviates the
inflammation (32). A substantial
increase in NF-κB activity along with a decrease in IκBα was
observed in the present study and Ac2-26 pretreatment was
sufficient to downregulate NF-κB via the expected upregulation of
IκBα, which served an anti-inflammatory role. This observation
supported the results of previous studies (21), which demonstrate that Ac2-26
treatment can significantly improve the perioperative resolution of
inflammation during I/R injury. This effect was associated with the
Ac2-26-mediated inhibition of NF-κB signaling and the normalization
of pro-inflammatory signaling molecules.
Similarly, infiltrating neutrophils can lead to
hepatic injury and amplify inflammation following IRI by releasing
proteases and oxidants that directly injure endothelial cells and
hepatocytes (33).
Pro-inflammatory signals coming from the damaged tissue can
activate neutrophils (2).
Furthermore, activated neutrophils can recruit additional
neutrophils to the liver by releasing inflammatory factors and
chemokines, thereby forming a positive feedback loop (34-36).
Early in 1991, Jaeschke et al (37) found that during the process of
HIRI, neutrophil infiltration was first observed during ischemia,
which was accelerated during reperfusion. Hoffman et al
(38) found that the degree of
neutrophil infiltration is proportional to the severity of
myocardial IRI. In myocardial IRI, anti-neutrophil interventions
could reduce the extent of myocardial infarction and myocardial
cell damage by 50%, which indicates that neutrophil infiltration
served an essential role in IRI (24). Furthermore, the presence and
accumulation of neutrophils in re-perfused tissues coincides with
the extent of tissue injury (2).
Activated neutrophils may cause considerable cell death and tissue
injury by releasing reactive oxygen species, proteases such as
elastase and collagenase, inflammatory mediators, or by forming
NETs (5,39,40).
Among them, inflammatory factors, such as TNF-α, IL-6 and IL-1β,
which are released by activated neutrophils, can further amplify
the recruitment of neutrophils to the damaged site (41). As a result, a vicious positive
feedback loop is underway, which may expand the inflammatory
response and aggravate liver injury. Anti-neutrophil interventions
have been reported to be able to ameliorate the I/R-induced damage
to hepatic cells (40,42).
To investigate whether Ac2-26 could reduce
neutrophil infiltration, non-parenchymal cells were collected from
liver tissues and the proportions of neutrophils were measured via
flow cytometry. The results showed that pretreatment with Ac2-26
significantly reduced the proportion of neutrophils in liver
tissues following HIRI. To further verify the changes in neutrophil
proportion during the I/R process, the Ly6G expression and MPO
activity in hepatic tissues were detected via immunofluorescence
staining and colorimetry, respectively. The results demonstrated
that Ac2-26 markedly reduced the HIRI-induced increase in
Ly6G-positive neutrophil infiltration as well as MPO activity.
These results suggested that Ac2-26 could inhibit HIRI-induced
neutrophil infiltration, thereby reducing the inflammatory
response.
In summary, the results of the present study
demonstrated that the HMGB1/TLR4/NF-κB/neutrophil axis may
participate in the development of HIRI. In order to illustrate the
molecular mechanisms of hepatoprotection by Ac2-26, the attendant
changes in the expression of HMGB1, TLR4 and NF-κB, as well as
neutrophil proportion, were investigated. The results demonstrated
that Ac2-26 could ameliorate the hepatic damage associated with
HIRI via the HMGB1/TLR4/NF-κB signaling pathway while inhibiting
neutrophil infiltration. A schematic illustration of the proposed
molecular mechanisms responsible for Ac2-26 hepatoprotection is
given in Fig. 6. The findings of
the present study may help to identify an effective targeted
therapy for the treatment of HIRI and improve the prognosis of
patients. In the future, ‘anti-neutrophil’ interventions or HMGB1
antagonists should be further investigated. However, the present
study has certain limitations, including the fact that the level of
Ac2-26 in serum and liver samples upon systemic administration was
not determined. Future research should be conducted in order to
explore the pharmacological mechanisms and metabolism of
Ac2-26.
Acknowledgements
The authors are grateful to Professor Jiyu Ju of
Weifang Medical University, for his help in the process of flow
cytometry analysis.
Funding
Funding: The present study was supported by the development
Project of Medical and Health Science and Technology in Shandong
Province (grant no. 202001020642) and the Key R&D Program of
Shandong Province (grant no. 2019GSF107056).
Availability of data and materials
The data used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CB, ZJ and HJ carried out the experimental work and
wrote the manuscript. SY and ML analyzed the data. CB and WL
confirm the authenticity of all the raw data. FC, YS and JL
analyzed the data, edited the manuscript and prepared the figures.
JJ and WL conceived and designed the experiments, edited the
manuscript and prepared the figures. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All procedures were conducted in accordance with the
animal ethics committee of the university. Weifang Medical
University Medical Ethics Committee provided full approval for this
research (approval no. 2020SDL187).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saidi RF and Kenari SK: Liver
ischemia/reperfusion injury: An overview. J Invest Surg. 6:366–379.
2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
van Golen RF, Reiniers MJ, Olthof PB, van
Gulik TM and Heger M: Sterile inflammation in hepatic
ischemia/reperfusion injury: Present concepts and potential
therapeutics. J Gastroenterol Hepatol. 3:394–400. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Quesnelle KM, Bystrom PV and
Toledo-Pereyra LH: Molecular responses to ischemia and reperfusion
in the liver. Arch Toxicol. 5:651–657. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wang Y, Liu ZS, Zhang SL, Diao QX and Ge
YJ: Effect and mechanism of portal blood stasis removal on
intestinal endotoxemia and hepatic ischemia reperfusion injury.
Transplant Proc. 9:2752–2756. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Huang H, Tohme S, Al-Khafaji AB, Tai S,
Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y and Tsung A:
Damage-associated molecular pattern-activated neutrophil
extracellular trap exacerbates sterile inflammatory liver injury.
Hepatology. 2:600–614. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xu L, Ge F, Hu Y, Yu Y, Guo K and Miao C:
Sevoflurane postconditioning attenuates hepatic
ischemia-reperfusion injury by limiting HMGB1/TLR4/NF-kappaB
pathway via modulating microRNA-142 in vivo and in vitro. Front
Pharmacol. 12(646307)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tsung A, Sahai R, Tanaka H, Nakao A, Fink
MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA and Billiar TR:
The nuclear factor HMGB1 mediates hepatic injury after murine liver
ischemia-reperfusion. J Exp Med. 7:1135–1143. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Perretti M and D'Acquisto F: Annexin A1
and glucocorticoids as effectors of the resolution of inflammation.
Nat Rev Immunol. 1:62–70. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Yang YH, Morand E and Leech M: Annexin A1:
Potential for glucocorticoid sparing in RA. Nat Rev Rheumatol.
10:595–603. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
La M, D'Amico M, Bandiera S, Di Filippo C,
Oliani SM, Gavins FN, Flower RJ and Perretti M: Annexin 1 peptides
protect against experimental myocardial ischemia-reperfusion:
Analysis of their mechanism of action. FASEB J. 12:2247–2256.
2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guido BC, Zanatelli M, Tavares-De-Lima W,
Oliani SM and Damazo AS: Annexin-A1 peptide down-regulates the
leukocyte recruitment and up-regulates interleukin-10 release into
lung after intestinal ischemia-reperfusion in mice. J Inflamm
(Lond). 1(10)2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Perretti M and Flower RJ: Annexin 1 and
the biology of the neutrophil. J Leukoc Biol. 1:25–29.
2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Facio FJ, Sena AA, Araujo LP, Mendes GE,
Castro I, Luz MA, Yu L, Oliani SM and Burdmann EA: Annexin 1
mimetic peptide protects against renal ischemia/reperfusion injury
in rats. J Mol Med (Berl). 1:51–63. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Care NRCU, Animals AUOL: Guide for the
care and use of laboratory animals. Washington (DC): National
Academies Press (US), 2011.
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 4:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang J, Yu S, Li J, Li H, Jiang H, Xiao P,
Pan Y, Zheng J, Yu L and Jiang J: Protective role of
N-acetyl-l-tryptophan against hepatic ischemia-reperfusion injury
via the RIP2/caspase-1/IL-1β signaling pathway. Pharm Biol.
1:385–391. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Datta G, Fuller BJ and Davidson BR:
Molecular mechanisms of liver ischemia reperfusion injury: Insights
from transgenic knockout models. World J Gastroenterol.
11:1683–1698. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cirino G, Cicala C, Sorrentino L,
Ciliberto G, Arpaia G, Perretti M and Flower RJ: Anti-inflammatory
actions of an N-terminal peptide from human lipocortin 1. Br J
Pharmacol. 3:573–574. 1993.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Perretti M and Flower RJ: Modulation of
IL-1-induced neutrophil migration by dexamethasone and lipocortin
1. J Immunol. 3:992–999. 1993.PubMed/NCBI
|
|
20
|
Perretti M, Ahluwalia A, Harris JG,
Goulding NJ and Flower RJ: Lipocortin-1 fragments inhibit
neutrophil accumulation and neutrophil-dependent edema in the
mouse. A qualitative comparison with an anti-CD11b monoclonal
antibody. J Immunol. 8:4306–4314. 1993.PubMed/NCBI
|
|
21
|
Kamaly N, Fredman G, Subramanian M, Gadde
S, Pesic A, Cheung L, Fayad ZA, Langer R, Tabas I and Farokhzad OC:
Development and in vivo efficacy of targeted polymeric
inflammation-resolving nanoparticles. Proc Natl Acad Sci USA.
16:6506–6511. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gavins FN, Dalli J, Flower RJ, Granger DN
and Perretti M: Activation of the annexin 1 counter-regulatory
circuit affords protection in the mouse brain microcirculation.
FASEB J. 8:1751–1758. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang J, He GZ, Wang YK, Zhu QK, Chen W and
Guo T: TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse
intestinal ischemia/reperfusion injury. World J Gastroenterol.
27:8314–8325. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xue J, Ge H, Lin Z, Wang H, Lin W, Liu Y,
Wu G, Xia J and Zhao Q: The role of dendritic cells regulated by
HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion
injury. J Cell Mol Med. 4:2849–2862. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tsaroucha AK, Valsami G, Kostomitsopoulos
N, Lambropoulou M, Anagnostopoulos C, Christodoulou E, Falidas E,
Betsou A, Pitiakoudis M and Simopoulos CE: Silibinin effect on
Fas/FasL, HMGB1, and CD45 expressions in a rat model subjected to
liver ischemia-reperfusion injury. J Invest Surg. 6:491–502.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sugihara M, Sadamori H, Nishibori M, Sato
Y, Tazawa H, Shinoura S, Umeda Y, Yoshida R, Nobuoka D, Utsumi M,
et al: Anti-high mobility group box 1 monoclonal antibody improves
ischemia/reperfusion injury and mode of liver regeneration after
partial hepatectomy. Am J Surg. 1:179–188. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mcdonald KA, Huang H, Tohme S, Loughran P,
Ferrero K, Billiar T and Tsung A: Toll-like receptor 4 (TLR4)
antagonist eritoran tetrasodium attenuates liver ischemia and
reperfusion injury through inhibition of high-mobility group box
protein B1 (HMGB1) signaling. Mol Med. 20:639–648. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nace GW, Huang H, Klune JR, Eid RE,
Rosborough BR, Korff S, Li S, Shapiro RA, Stolz DB, Sodhi CP, et
al: Cellular-specific role of toll-like receptor 4 in hepatic
ischemia-reperfusion injury in mice. Hepatology. 1:374–387.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhu JR, Lu HD, Guo C, Fang WR, Zhao HD,
Zhou JS, Wang F, Zhao YL, Li YM, Zhang YD, et al: Berberine
attenuates ischemia-reperfusion injury through inhibiting HMGB1
release and NF-kappaB nuclear translocation. Acta Pharmacol Sin.
11:1706–1715. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang S, Feng Z, Gao W, Duan Y, Fan G,
Geng X, Wu B, Li K, Liu K and Peng C: Aucubin attenuates liver
ischemia-reperfusion injury by inhibiting the HMGB1/TLR-4/NF-kappaB
signaling pathway, oxidative stress, and apoptosis. Front
Pharmacol. 11(544124)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Leoni G, Neumann PA, Kamaly N, Quiros M,
Nishio H, Jones HR, Sumagin R, Hilgarth RS, Alam A, Fredman G, et
al: Annexin A1-containing extracellular vesicles and polymeric
nanoparticles promote epithelial wound repair. J Clin Invest.
3:1215–1227. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Reischl S, Lee JH, Miltschitzky J,
Vieregge V, Walter RL, Twardy V, Kasajima A, Friess H, Kamaly N and
Neumann PA: Ac2-26-nanoparticles induce resolution of intestinal
inflammation and anastomotic healing via inhibition of NF-κB
signaling in a model of perioperative colitis. Inflamm Bowel Dis.
9:1379–1393. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Tsung A, Klune JR, Zhang X, Jeyabalan G,
Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR and Billiar TR:
HMGB1 release induced by liver ischemia involves Toll-like receptor
4 dependent reactive oxygen species production and calcium-mediated
signaling. J Exp Med. 12:2913–2923. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Baxter GF: The neutrophil as a mediator of
myocardial ischemia-reperfusion injury: Time to move on. Basic Res
Cardiol. 4:268–275. 2002.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Vinten-Johansen J: Involvement of
neutrophils in the pathogenesis of lethal myocardial reperfusion
injury. Cardiovasc Res. 3:481–497. 2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yago T, Petrich BG, Zhang N, Liu Z, Shao
B, Ginsberg MH and Mcever RP: Blocking neutrophil integrin
activation prevents ischemia-reperfusion injury. J Exp Med.
8:1267–1281. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jaeschke H, Farhood A and Smith CW:
Neutrophil-induced liver cell injury in endotoxin shock is a
CD11b/CD18-dependent mechanism. Am J Physiol. 261 (6 Pt
1):G1051–G1056. 1991.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hoffman JJ, Gilbert TB, Poston RS and
Silldorff EP: Myocardial reperfusion injury: Etiology, mechanisms,
and therapies. J Extra Corpor Technol. 4:391–411. 2004.PubMed/NCBI
|
|
39
|
Saiman Y and Friedman SL: The role of
chemokines in acute liver injury. Front Physiol.
3(213)2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Monson KM, Dowlatshahi S and Crockett ET:
CXC-chemokine regulation and neutrophil trafficking in hepatic
ischemia-reperfusion injury in P-selectin/ICAM-1 deficient mice. J
Inflamm (Lond). 4(11)2007.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Perry BC, Soltys D, Toledo AH and
Toledo-Pereyra LH: Tumor necrosis factor-α in liver
ischemia/reperfusion injury. J Invest Surg. 4:178–188.
2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Uchida Y, Freitas MC, Zhao D, Busuttil RW
and Kupiec-Weglinski JW: The inhibition of neutrophil elastase
ameliorates mouse liver damage due to ischemia and reperfusion.
Liver Transpl. 8:939–947. 2009.PubMed/NCBI View Article : Google Scholar
|