Introduction
Cerebellar ataxia is a progressive neurological
disorder that is most frequently caused by inflammation or injury
to the cerebellum. As the cerebellum regulates movement and
muscular function, individuals with cerebellar ataxia often
experience a lack of coordination and struggle to accomplish
everyday chores. Ataxia with oculomotor apraxia type 1 (AOA1) is an
autosomal recessive disease. It presents early in life and its
symptoms include progressive cerebellar ataxia, oculomotor apraxia,
dysarthria, peripheral axonal neuropathy and hypoalbuminemia
(1).
Acquired ataxias may be transient or chronic and may
be triggered by environmental (trauma or toxin exposure) or medical
(infection, tumors or stroke) factors. Hereditary ataxias are
heterogeneous, with causal mutations documented in >50 genes and
inheritance patterns ranging from classical dominant to recessive,
mitochondrial to X-linked (2). Of
note, four genes have been implicated in the development of AOA
(3). AOA1 is prevalent in Japanese
and Portuguese populations (4).
Mutations in the APTX gene are primarily responsible for
AOA1(5). This gene is located on
chromosome 9p21. It encodes aprataxin and is involved in
mitochondrial DNA repair through transcription regulation (5). Pathogenic variants of the APTX
gene destabilize aprataxin and subsequently increase the effects of
single-strand breaks in DNA, even when there are no apparent gross
errors (6).
A total of 18 other mutations were identified as
pathogenic variants of APTX; these mutations were found in
39 families (5). In the present
case report, a patient with AOA1 is described, who had a novel
homozygous missense mutation, His251Tyr, due to c.751 C>T
substitution.
Case report
An eight-year-old female patient presented in 2021
at the pediatric neurology clinic of King Fahad Specialist Hospital
Dammam (Dammam, Saudi Arabia) with progressive ataxia and an
unsteady gait. The patient was the first child of healthy
consanguineous parents. The patient had three healthy brothers and
no family history of gait disturbances or neurological disorders.
The patient's mother's pregnancy with the patient was uneventful
and the patient was delivered at full term via normal vaginal
delivery. The patient's birth weight was within the normal range
and she was a healthy infant. The patient also achieved normal
developmental milestones until the age of 14 months. The patient
was able to sit without support at eight months of age and began
walking at 14 months of age. The patient then began to experience
imbalances during walking and recurrent falls. As the patient grew
older, the patient's family noticed that the patient's ataxic gait
was progressively worsening. The patient also subsequently
presented with dysarthria and ataxic handwriting. A brief mental
examination revealed that the patient performed normally at school
and that the patient's cognitive function was normal. Prior to
presentation, the patient was a healthy child with an unremarkable
medical and surgical history. The patient had no history of recent
infection, abnormal movements, visual or hearing difficulties, or
developmental regression.
General examination revealed mild telangiectasia.
The patient's growth parameters were within normal ranges, apart
from the patient's head circumference, which was 50 cm below the
25th percentile. Motor examination revealed normal muscle bulk,
tone and power bilaterally. The patient's deep tendon reflexes were
+1 bilaterally in both the upper and lower extremities.
Furthermore, the patient's Babinski reflex was bilaterally normal.
Sensory examination results were also normal. An ocular examination
revealed oculomotor apraxia. The patient had no nystagmus and the
rest of the cranial nerves were unremarkable. A cerebellar
examination revealed wide-based ataxic gait, head titubation,
dysarthria, scanning speech, intention tremor, dysmetria and
dysdiadochokinesia.
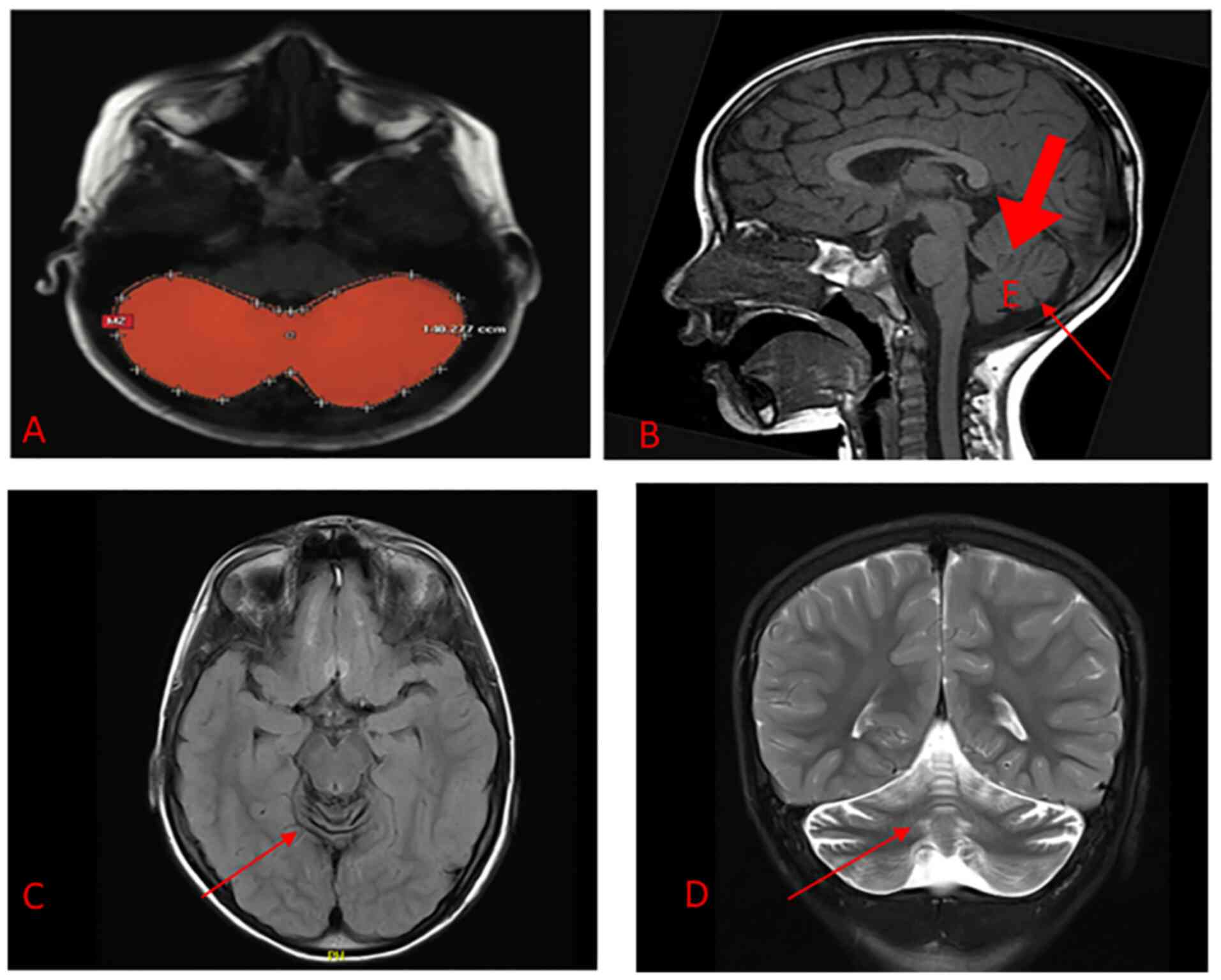
Brain magnetic resonance imaging (MRI) indicated
diffused cerebellar atrophy (Fig.
1). No other brain parenchymal abnormalities were detected.
Otherwise, the patient's MRI was unremarkable.
Laboratory analyses were performed to measure
ammonia, lactate, albumin, lipid profile, immunoglobulins,
alpha-fetoprotein, peripheral smear, serum vitamin E, serum and
urine amino acids, serum and urine acylcarnitine, urine organic
acids and fatty acids; all were normal and so were the patient's
liver and renal function. The patient's audiological and
ophthalmological examinations were also unremarkable.
Next-generation sequencing of 214 genes was
performed using genomic DNA extracted from a blood sample using
Human All Exon V6 (SureSelectXT Target Enrichment System for
Illumina Paired-End Sequencing Library, version 1.3.1) (7). A variant of uncertain significance
was identified; c.751C>T p.(His251Tyr) was detected with
probable homozygosity in the APTX gene (chromosome 9). This
variant of the APTX gene may be responsible for early-onset
ataxia with oculomotor apraxia and hypoalbuminemia.
Discussion
Pathogenic variants of the APTX gene have
been associated with early-onset ataxia. The present study reported
on a rare case of an APTX gene mutation in an eight-year-old
female patient. The patient was diagnosed with hereditary
progressive ataxia. The identified variant, c.751C>T
p.(His251Tyr) with probable homozygosity in the APTX gene
(chromosome 9), was of uncertain significance. It is located in a
highly conserved residue. This variant was detected with high
confidence according to best practice guidelines (8,9). The
following discussion explores the previously published literature
on autosomal recessive cerebellar ataxias, with an emphasis on the
APTX gene.
Autosomal recessive cerebellar ataxia (ARCA) is a
set of disorders. ARCA subtypes related to AOA have recently been
identified and classified into two major types: AOA1 and
AOA2(10). Mutations in the
APTX gene may lead to ataxia, with oculomotor apraxia and
hypoalbuminemia (11,12). Aprataxin, a member of the histidine
triad family, is encoded by the APTX gene (10). It has been identified as the causal
gene in ataxia-oculomotor apraxia syndrome in a different group of
individuals with autosomal recessive inheritance.
Individuals of various ethnicities may have
mutations in the APTX gene (4,10,13,14)
(Table I). The most prevalent ARCA
mutation in Japan is c.689 690insT; in Portugal, it is c.837G>A
(15,16). Shimazaki et al (15) performed a sequencing analysis of
the APTX gene in six patients from four Japanese families.
Except for one patient with a sporadic mutation, all other patients
had inherited the condition in an autosomal recessive manner.
Furthermore, two patients had a novel homozygous missense mutation
(c.80A>G). In one case, a missense compound heterozygous
c.95C>T mutation led to the replacement of proline with leucine
at amino acid position 32. Tranchant et al (16) found two variants of the APTX
gene in three non-Portuguese and non-Japanese individuals. One of
these patients had a nonsense W279X mutation; the other two
patients were French siblings who carried a missense K197Q mutation
and a compound heterozygous nonsense W279X mutation. Sekijima et
al (11) reported 689 insT in
the APTX gene in a 14-year-old female with severe
generalized dystonia. Another study reported 14 patients with
APTX gene mutations in nine families, including five novel
variants in exons 5 and 6 (A198V, D267G, W279R and IVS5+1)
(13). To screen for APTX
mutations, Habeck et al (17) tested 165 patients with early-onset
ataxia in Germany. Another genetic study of 13 patients from three
unrelated Tunisian families with AOA1 identified mutations in the
APTX gene (18). APTX exon
7 deletions were detected in a family with normal clinical
presentation of AOA1 and no severe phenotypes. With AOA1, it is
difficult to establish an association between genotype and
phenotype, as symptoms vary among different families with the same
mutation (18).
 | Table IComparative studies including clinical
characteristics of previously reported cases. |
Table I
Comparative studies including clinical
characteristics of previously reported cases.
| First author,
year | Number of
patients | Region | Age of onset | Clinical
features | Genes | (Refs.) |
|---|
| Moreira, 2001 | 15 families | Portugal and
Japan | 2-5 years | Early-onset ataxia,
cerebellar atrophy, ocula-motor apraxia and hypoalbuminemia | Exon 5:
c.617C>T; c.689dupT (689insT), (689-690insT); Exon 6:
c.837G>A | (4) |
| Date, 2001 | 7 families | Japan | First or second
decade of life | Progressive ataxia,
areflexia, sensory loss and hypoalbuminemia | Exon 5:
c.617C>T; c.689dupT (689insT), (689-690insT);Exon 6:
c.788T>G; c.841delT | (10) |
| Castellotti,
2011 | 13 | Italy | 3-7 years | Ataxia, dysarthria,
nystagmus, areflexia, sensory neuropathy | Exon 3: c.477delC
Exon 4: c.C541T Exon 7: c.C916T | (14) |
| Shimazaki,
2002 | 5 | Japan | 3-12 years | Cerebellar ataxia,
peripheral neuropathy, oculomotor apraxia, external
ophthalmoplegia, choreiform movements, facial grimacing, mental
deterioration, cerebellar atrophy, hypoalbuminemia,
hypercholesterolemia | Exon 5:
c.602A>G; c.617C>T; c.689dupT (689insT), (689-690insT) | (15) |
| Tranchant,
2003 | 3 | France and
Italy | 2 years and 15-22
years | 1st Italian
patient: Progressive ataxia, areflexia, oculomotor apraxia,
hypoalbuminemia and hypercholesterolemia; 2 French siblings: Later
onset of ataxia, areflexia, dysarthria and dystonia | Exon 5: c.589A>C
Exon 6: c.837G>A | (16) |
| Le Ber, 2003 | 14 | France (7
families), Italy (1 family) and Algeria (1 family) | 2-18 years | Gait ataxia,
chorea, dystonia, dysarthria, areflexia, axonal sensorimotor
neuropathy | Exon 5:
c.593C>T; c.770+1G>A Exon 6: c.835T>C; c.837G>A | (13) |
| Habeck, 2004 | 3 | Germany | 4-6 years | Cerebellar ataxia,
dysarthria, oculomotor apraxia, areflexia and hypotonia | Exon 6:
c.837G>A | (17) |
| Amouri, 2004 | 3 unrelated
families | Tunisia | Mean of 5
years | Cerebellar ataxia,
dysarthria, ocular apraxia, distal axonal neuropathy, cerebellar
atrophy, hypoalbuminemia, hypercholesterolemia | Exon 7:
c.875-1G>A; total deletion of gene | (18) |
Recently, the APTX mutation c.484-2A>T was
reported for the first time in a patient with Charcot-Marie-Tooth
disease (19). Pedroso et
al (20) described the case of
a female patient with slow progressive gait impairment.
Neurological tests revealed ocular motor apraxia and myoclonic
jerks. AOA1 was verified by a homozygous mutation in the
APTX gene (c.[837G>A];[837G>A]). In 2020, Ababneh
et al (21) discovered a
recurrent homozygous nonsense mutation (c.837G>A, p.W279*) in
the APTX gene in three patients with AOA1. In a study of
Palestinian and Israeli Arab families, WES identified a homozygous
mutation, c.837G>A: p.(Trp279*), in a patient with speech and
oculomotor apraxias and cerebellar ataxia (22).
Renaud et al (23) identified a p.Trp279 mutation in 53
patients with AOA1. In a consanguineous Iranian family with
hereditary AOA1 (five affected and six unaffected individuals), WES
revealed a novel homozygous stop-gain APTX gene mutation
(c.739A>T; p.Lys247*) (3).
Hirano et al (24)
identified a homozygous two-base deletion in the middle of exon 3
of the APTX gene. Karimzadeh et al (25) reported a homozygous frameshift
mutation, c.418_418 del, in the APTX gene. Complete
homozygous deletion of APTX (62 kb) has also been reported
in a patient with AOA1(26).
Deletion of exon 6 of APTX in an 18-year-old female was
reported by Paucar et al (27). A new homozygous deletion in c.643
and an A>T single nucleotide polymorphism in c.641 in exon 6
were discovered in a seven-year-old pediatric patient (28). Another study described a patient
with a homozygous deletion of APTX and behavioral
abnormalities (29). Castellotti
et al (14) screened a
large cohort and found variants of the APTX gene in 13
ataxic individuals (6%), 11 of whom were homozygous for the
mutations p.W279X, p.W279R and p.P206L. They also observed three
new mutations: c.477delC, c.C541T and c.C916T. A unique homozygous
missense variant of APTX was discovered in a 34-year-old
female patient born to consanguineous parents (30).
Although the above-mentioned studies provide
valuable information about the likely function of APTX
mutations in early-onset ataxia, genotype-phenotype correlations
have not yet been clearly confirmed (18,31).
In essence, while further research is required to establish the
effect on the protein, a homozygous missense mutation, His251Tyr,
caused by a c.751 C>T substitution in APTX, is probably
deleterious for AOA1 in an autosomal recessive manner (32,33).
Yokoseki et al (34)
provided a comprehensive overview of this issue, finding that
patients with the p.Pro206Leu or p.Val263Gly mutations had less
gait disruption than those with the c.689 690insT mutations and
that patients with the p.Pro206Leu or p.Val263Gly mutations had
less gait disruption than those with the c.689 690insT mutations.
In that study, patients with the c.689 690insT mutation exhibited a
higher cumulative rate of inability to walk without assistance than
those with the other mutations. Patients with p.Pro206Leu or
p.Val263Gly mutations were found to have reduced ocular motor
apraxia and no cognitive impairment, whereas patients with
early-onset ataxia and hypoalbuminemia and the c.689 690insT
mutation had more severe phenotypes. Those with the p.Pro206Leu or
p.Val263Gly mutations presented with less ocular motor apraxia and
no cognitive impairment, whereas patients with the c.689 690insT
mutation had more severe symptoms, including early-onset ataxia and
hypoalbuminemia (34).
Before drawing broad conclusions from the present
study, it is necessary to recognize its limitations. The fact that
the present study is a case study on a single patient means that
various features of genotype-phenotype association may arise in
other contexts. In addition, the present analysis is based on only
exome sequencing data, which has constraints on its own. Future
research should extend this area by using other methods for copy
number variation detection, such as XHMM, CANOES and CLAMMS
(35,36). Utilizing Sanger sequencing to
validate the existence and homozygous condition of the variation
will further reaffirm the conclusion of this report. In addition,
it was not assessed whether the mutation was inherited, since no
genetic testing was performed on any of the other family members
because the parents refused further testing.
In conclusion, the present study reported on the
identification of a variant of uncertain significance, c.751C>T
p.(His251Tyr), in an eight-year-old female patient with hereditary
progressive ataxia. Based on a detailed review of the literature,
it was concluded that in patients with autosomal recessive or
solitary instances of cerebellar ataxia that worsen over time,
after Friedreich's ataxia has been ruled out, genetic testing
should be used to check for APTX mutations. At first, there
are usually no signs of oculomotor apraxia or other functional
problems. However, choreic movements are likely caused by AOA1. As
the AOA1 phenotype initially appears similar to other types of
choreic disorders, early-onset choreic patients who do not have
significant mutations in the IT15 or JPH3 genes should also be
checked for APTX mutations. Early detection of hyperkinetic
movement disorders in patients with AOA1 is important to ensure the
right treatment is provided. Finally, patients with early-onset
ataxia and mixed-movement disorders should be genetically tested
for a number of diseases, including AOA1.
With breakthroughs in genetic testing, the
identification of children with this disease will become easier in
the future. Current treatments for AOA1 focus on rehabilitation
therapy; there is currently no specific treatment for AOA1.
Although the present results are encouraging, more research is
required to establish the etiology of AOA1 in terms of causative
mutations.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RA, AA and SB designed the study. AA, GA and BA
collected the clinical data. AA, GA, BA and RA analysed and
interpreted the data. RA, AA, GA, BA and SB drafted the manuscript.
RA and SB confirmed the authenticity of the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was carried out in accordance with the
code of international and local Ethics (Declaration of Helsinki).
This study was reviewed and approved by the local ethics committee
of the King Fahad Specialist Hospital Dammam (Dammam, Saudi
Arabia).
Patient consent for publication
The parents of the patient provided written informed
consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Coutinho P, Barbot C and Coutinho P:
Ataxia with oculomotor apraxia type 1. In: Adam MP, Everman DB,
Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A,
editors. GeneReviews® [Internet]. Seattle (WA): University of
Washington, Seattle;. 1993(2022)2002.
|
|
2
|
Sandford E and Burmeister M: Genes and
genetic testing in hereditary ataxias. Genes (Basel). 5:586–603.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Inlora J, Sailani MR, Khodadadi H,
Teymurinezhad A, Takahashi S, Bernstein JA, Garshasbi M and Snyder
MP: Identification of a novel mutation in the APTX gene associated
with ataxia-oculomotor apraxia. Cold Spring Harb Mol Case Stud.
3(a002014)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Moreira MC, Barbot C, Tachi N, Kozuka N,
Uchida E, Gibson T, Mendonça P, Costa M, Barros J, Yanagisawa T, et
al: The gene mutated in ataxia-ocular apraxia 1 encodes the new
HIT/Zn-finger protein aprataxin. Nat Genet. 29:189–193.
2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sun Y, Zheng L, Yang Y, Qian X, Fu T, Li
X, Yang Z, Yan H, Cui C and Tan W: Metal-Organic framework
nanocarriers for drug delivery in biomedical applications.
Nanomicro Lett. 12(103)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ahel I, Rass U, El-Khamisy SF, Katyal S,
Clements PM, McKinnon PJ, Caldecott KW and West SC: The
neurodegenerative disease protein aprataxin resolves abortive DNA
ligation intermediates. Nature. 443:713–716. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen R, Im H and Snyder M: Whole-Exome
enrichment with the agilent sureselect human all exon platform.
Cold Spring Harb Protoc. 2015:626–633. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Koboldt DC: Best practices for variant
calling in clinical sequencing. Genome Med. 12(91)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Date H, Onodera O, Tanaka H, Iwabuchi K,
Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, et al:
Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is
caused by mutations in a new HIT superfamily gene. Nat Genet.
29:184–188. 2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sekijima Y, Hashimoto T, Onodera O, Date
H, Okano T, Naito K, Tsuji S and Ikeda S: Severe generalized
dystonia as a presentation of a patient with aprataxin gene
mutation. Mov Disord. 18:1198–1200. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zheng J, Croteau DL, Bohr VA and Akbari M:
Diminished OPA1 expression and impaired mitochondrial morphology
and homeostasis in Aprataxin-deficient cells. Nucleic Acids Res.
47:4086–4110. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Le Ber I, Moreira MC, Rivaud-Péchoux S,
Chamayou C, Ochsner F, Kuntzer T, Tardieu M, Saïd G, Habert MO,
Demarquay G, et al: Cerebellar ataxia with oculomotor apraxia type
1: Clinical and genetic studies. Brain. 126 (Pt 12):2761–2772.
2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Castellotti B, Mariotti C, Rimoldi M,
Fancellu R, Plumari M, Caimi S, Uziel G, Nardocci N, Moroni I,
Zorzi G, et al: Ataxia with oculomotor apraxia type1 (AOA1): Novel
and recurrent aprataxin mutations, coenzyme Q10 analyses, and
clinical findings in Italian patients. Neurogenetics. 12:193–201.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shimazaki H, Takiyama Y, Sakoe K, Ikeguchi
K, Niijima K, Kaneko J, Namekawa M, Ogawa T, Date H, Tsuji S, et
al: Early-onset ataxia with ocular motor apraxia and
hypoalbuminemia: The aprataxin gene mutations. Neurology.
59:590–595. 2002.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tranchant C, Fleury M, Moreira MC, Koenig
M and Warter JM: Phenotypic variability of aprataxin gene
mutations. Neurology. 60:868–870. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Habeck M, Zühlke C, Bentele KH, Unkelbach
S, Kress W, Bürk K, Schwinger E and Hellenbroich Y: Aprataxin
mutations are a rare cause of early onset ataxia in Germany. J
Neurol. 251:591–594. 2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Amouri R, Moreira MC, Zouari M, El Euch G,
Barhoumi C, Kefi M, Belal S, Koenig M and Hentati F: Aprataxin gene
mutations in Tunisian families. Neurology. 63:928–929.
2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yan J, Qiao L, Peng H, Liu A, Wu J and
Huang J: A novel missense pathogenic variant in NEFH causing rare
Charcot-Marie-Tooth neuropathy type 2CC. Neurol Sci. 42:757–763.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Pedroso JL, Vale TC, da Costa SCG, Santos
M, Alonso I and Barsottini OGP: Complex movement disorders in
ataxia with oculomotor apraxia type 1: Beyond the cerebellar
syndrome. Tremor Other Hyperkinet Mov (N Y). 10(39)2020.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Ababneh NA, Ali D, Al-Kurdi B, Sallam M,
Alzibdeh AM, Salah B, Ryalat AT, Azab B, Sharrack B and Awidi A:
Identification of APTX disease-causing mutation in two unrelated
Jordanian families with cerebellar ataxia and sensitivity to DNA
damaging agents. PLoS One. 15(e0236808)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hengel H, Buchert R, Sturm M, Haack TB,
Schelling Y, Mahajnah M, Sharkia R, Azem A, Balousha G, Ghanem Z,
et al: First-line exome sequencing in Palestinian and Israeli Arabs
with neurological disorders is efficient and facilitates disease
gene discovery. Eur J Hum Genet. 28:1034–1043. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Renaud M, Moreira MC, Ben Monga B,
Rodriguez D, Debs R, Charles P, Chaouch M, Ferrat F, Laurencin C,
Vercueil L, et al: Clinical, biomarker, and molecular delineations
and genotype-phenotype correlations of ataxia with oculomotor
apraxia type 1. JAMA Neurol. 75:495–502. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Hirano M, Matsumura R, Nakamura Y, Saigoh
K, Sakamoto H, Ueno S, Inoue H and Kusunoki S: Unexpectedly mild
phenotype in an ataxic family with a two-base deletion in the APTX
gene. J Neurol Sci. 378:75–79. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Karimzadeh P, Khayatzadeh Kakhki S, Esmail
Nejad SS, Houshmand M and Ghofrani M: Ataxia oculomotor apraxia
type 1 in the siblings of a family: A novel mutation. Iran J Child
Neurol. 11:78–81. 2017.PubMed/NCBI
|
|
26
|
van Minkelen R, Guitart M, Escofet C, Yoon
G, Elfferich P, Bolman GM, van der Helm R, van de Graaf R and van
den Ouweland AM: Complete APTX deletion in a patient with ataxia
with oculomotor apraxia type 1. BMC Med Genet.
16(61)2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Paucar M, Alonso I, Eriksson M, Beniaminov
S, Coutinho P and Svenningsson P: Novel APTX mutation in a hispanic
subject affected by ataxia with oculomotor apraxia type 1. Mov
Disord Clin Pract. 2:90–92. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nouri N, Nouri N, Aryani O, Kamalidehghan
B, Sedghi M and Houshmand M: A novel mutation in the aprataxin
(APTX) gene in an Iranian individual suffering early-onset ataxia
with oculomotor apraxia type 1(AOA1) disease. Iran Biomed J.
16:223–225. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yoon G, Westmacott R, Macmillan L, Quercia
N, Koutsou P, Georghiou A, Christodoulou K and Banwell B: Complete
deletion of the aprataxin gene: Ataxia with oculomotor apraxia type
1 with severe phenotype and cognitive deficit. BMJ Case Rep.
2009(bcr08.2008.0688)2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ferrarini M, Squintani G, Cavallaro T,
Ferrari S, Rizzuto N and Fabrizi GM: A novel mutation of aprataxin
associated with ataxia ocular apraxia type 1: Phenotypical and
genotypical characterization. J Neurol Sci. 260:219–224.
2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hirano M, Asai H, Kiriyama T, Furiya Y,
Iwamoto T, Nishiwaki T, Yamamoto A, Mori T and Ueno S: Short
half-lives of ataxia-associated aprataxin proteins in neuronal
cells. Neurosci Lett. 419:184–187. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ceyhan-Birsoy O, Murry JB, Machini K, Lebo
MS, Yu TW, Fayer S, Genetti CA, Schwartz TS, Agrawal PB, Parad RB,
et al: Interpretation of genomic sequencing results in healthy and
Ill Newborns: Results from the babyseq project. Am J Hum Genet.
104:76–93. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Manzoor H, Bukhari I, Wajid M, Zhang Y,
Zhang H, Brüggemann N, Klein C, Shi Q and Naz S: A Novel APTX
variant and ataxia with oculomotor apraxia type 1. J Clin Neurol.
13:303–305. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Yokoseki A, Ishihara T, Koyama A, Shiga A,
Yamada M, Suzuki C, Sekijima Y, Maruta K, Tsuchiya M, Date H, et
al: Genotype-phenotype correlations in early onset ataxia with
ocular motor apraxia and hypoalbuminaemia. Brain. 134 (Pt
5):1387–1399. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Backenroth D, Homsy J, Murillo LR,
Glessner J, Lin E, Brueckner M, Lifton R, Goldmuntz E, Chung WK and
Shen Y: CANOES: Detecting rare copy number variants from whole
exome sequencing data. Nucleic Acids Res. 42(e97)2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gordeeva V, Sharova E, Babalyan K,
Sultanov R, Govorun VM and Arapidi G: Benchmarking germline CNV
calling tools from exome sequencing data. Sci Rep.
11(14416)2021.PubMed/NCBI View Article : Google Scholar
|