Introduction
The increasing rate of obesity is directly
associated with specific factors, including a sedentary lifestyle,
overeating and genetic disorders (1). Furthermore, environmental chemicals
that disrupt metabolic function have been linked to obesity
(2). Individuals are exposed to
various chemicals through numerous sources (1,2).
Currently, 2,2-bis(4-hydroxyphenyl) or bisphenol (BP) A is one of
the most abundant environmental chemicals to which individuals are
widely exposed worldwide (3-5).
BPA is an exogenous endocrine disruptor associated with human
health complications. BPA is a solid crystal, organic, colourless
compound with a phenolic odour under ambient conditions, with
chemical formula C15H16O2 and
molecular weight of 228.29 g/mol (3). BPA is highly soluble in lipids and
weakly soluble in water (2). BPA
is primarily used to synthesise plastic, epoxy resin, thermal
paper, dental sealant, flame retardant, baby bottles and lacquer
coatings for metal products (4).
Numerous studies have indicated that BPA easily contaminates food
and drinks under certain conditions, such as heating (6,7).
During the fabrication, degradation and treatment of BPA-containing
materials, BPA is released into the environment (7). It easily enters the human body
through the respiratory and digestive tracts and is absorbed via
the skin (6). BPA release from
food products is increased by heating, contact with acid and
alkaline substances, exposure to microwaves and repeated use of
plastic containers containing BPA (8,9). BPA
is also a xenobiotic endocrine disruptor chemical. In mammary
glands, xenobiotic-metabolising enzymes metabolise BPA via two
pathways: Glucuronidation and sulfation (8). Cytochrome P450 (CYP) enzymes are key
enzymes in the liver that mediate the metabolism of BPA into
BPA-o-quinone and BPA-semiquinone (10,11).
Several studies have indicated that BPA also mimics hormones, such
as oestrogens and androgens, and may promote health complications,
such as hepatotoxicity, cardiotoxicity, type 2 diabetes, obesity
and cancer (1,12).
The present study investigated the effects of BPA on
human adult THLE-2 cells. Analysis of the peroxisome
proliferator-activated receptor (PPAR)-gamma expression profile in
tumour samples and paired normal tissue was performed using Gene
Expression Profiling Interactive Analysis (GEPIA). Then, THLE-2
cell viability following BPA treatment was assessed and gene
expression of clusterin of PPAR genes (lipid sensor) in BPA-treated
THLE-2 cells was measured by reverse transcription-quantitative PCR
(RT-qPCR). Subsequently, validation of gene expression data and
overall survival analysis were performed using The Cancer Genome
Atlas (TCGA) data in GEPIA. The present study assessed cytoplasmic
lipid accumulation in BPA-treated THLE-2 cells and performed
histopathological examination of liver tissue of rats fed normal or
high-fat diet with or without BPA. Finally, CYP gene expression in
BPA-treated THLE-2 cells was investigated by RT-qPCR. The present
results are important to raise awareness among the public and
policy-makers on general BPA use in the food and beverage industry
and to provide a foundation for development of strategies to curb
the high prevalence of obesity.
Materials and methods
Analysis of PPARγ expression profile
in tumour samples and paired normal tissue
The PPARγ expression profile in tumour samples and
paired normal tissue was investigated using GEPIA. This web server
extracts data from TCGA data portal and Genotype-Tissue Expression
(GTEx) database of normal tissue (gepia.cancer-pku.cn) (13). A total of 30 types of human tumour
and paired normal tissue was used to investigate gene expression.
These tumours included the following: Adrenocortical carcinoma,
bladder urothelial carcinoma, breast invasive carcinoma (BRCA),
cervical squamous cell carcinoma and endocervical adenocarcinoma
(CESC), cholangiocarcinoma, colon adenocarcinoma (COAD), lymphoid
neoplasm diffuse large B cell lymphoma, oesophageal carcinoma,
glioblastoma multiforme, head and neck squamous cell carcinoma
(HNSC), kidney chromophobe, kidney renal clear cell carcinoma,
kidney renal papillary cell carcinoma, acute myeloid leukaemia,
brain lower grade glioma, liver hepatocellular carcinoma (LIHC),
lung adenocarcinoma, lung squamous cell carcinoma (LUSC), ovarian
serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD),
pheochromocytoma and paraganglioma, prostate adenocarcinoma, rectum
adenocarcinoma (READ), sarcoma, skin cutaneous melanoma (SKCM),
stomach adenocarcinoma, testicular germ cell tumour, thyroid
carcinoma (THCA), thymoma and uterine corpus endometrial carcinoma
(UCEC). One-way analysis of variance (ANOVA) followed by Tukey's
post hoc test was used for comparing differential gene expression
in tumour vs. paired normal samples and log2(transcripts per
million+1) was used for log-scale plotting. The |log2 fold-change|
cut-off was set to 1 and Q-value cut-off of 0.01 was used for dot
and box plots. The overexpressed genes were marked in red;
underexpressed genes were marked in green.
Evaluation of BPA-treated THLE-2 cell
viability
Before evaluating PPARγ expression in THLE-2 cell
lines, the half maximal inhibitory concentration and the effect of
the carcinogen BPA on cell viability were investigated using MTT
assay, according to the manufacturer's instructions (Sigma-Aldrich;
Merck KGaA). The human adult liver THLE-2 cell line was obtained
from the American Type Culture Collection and BPA (>99% purity)
was purchased from Merck KGaA. THLE-2 cells were cultured with
bronchial epithelial growth medium (Thermo Fisher Scientific, Inc.)
in T25 culture flasks at 37˚C in a humidified atmosphere with 5%
CO2 for at least 2-3 days or until the cells reached 80%
confluence before subjecting to experiments. The growth medium was
supplemented with frozen additives, including 10% foetal bovine
serum (Thermo Fisher Scientific, Inc.), 0.4 ml phosphoethanolamine
(100 µg/ml stock; cat. no. P0503; Sigma-Aldrich; Merck KGaA) and
0.3 ml human recombinant epithelial growth factor (EGF; 10 µg/ml
stock; cat. no. 354052; Corning, Inc.). Cells were cultured until
~80% confluence. Then, cells were trypsinized and seeded in three
96-well plates at a density of 5x103 per well in growth
medium. The cells were incubated at 37˚C in a humidified atmosphere
with 5% CO2 for 24 h. Following incubation, the medium
in each well was removed and 100 µl fresh medium containing
different concentrations of BPA (7.81, 15.63, 31.25, 62.50, 125.00,
250.00, 500.00, 1,000.00 µg/ml) was added to each well. The 100
mg/ml BPA stock solution was prepared by dissolving 500 mg BPA in 1
ml DMSO. Then, the stock solution was aliquoted and stored at
-20˚C. The growth medium was used to adjust BPA solutions to the
concentrations needed for cell treatment. Treated cells were
incubated at 37̊C for 24, 48 and 72 h. After each incubation
period, 24 µl 2.5 mg/ml MTT reagent was added to each well. The
plate was incubated at 37˚C in 5% CO2 for 4 h. Finally,
100 µl DMSO was used to dissolve the purple formazan crystals. A
TECAN Sunrise™ microplate reader (Tecan Group, Ltd.) was used to
measure the absorbance of the samples at 570 nm. The percentage of
cytotoxicity was calculated as described previously (14). Each experiment was performed in
triplicate and ≥3 independent experiments were performed.
Assessment of clusterin, PPAR and CYP
gene expression in BPA-treated THLE-2 cells
The study performed assessment of clusterin, PPAR
and CYP gene expression in BPA-treated THLE-2 cells by culturing
THLE-2 cells as aforementioned. Cells were then trypsinised and
sub-cultured with fresh growth medium in 6-well plates at a density
of 2x105 cells/well and subjected to 35 µl/ml BPA
treatment at 37̊C for 24, 48 and 72 h. Total RNA was extracted from
BPA-treated THLE-2 cells using TRIzol® Total RNA
Isolation Reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
extracted total RNA concentration was determined using a NanoDrop™
(Thermo Fisher Scientific, Inc.) and the high purity of the
extracted RNA was preserved by storage at -80˚C until further use.
The extracted RNA integrity was checked using a 1% (w/v) agarose
gel with an electrophoresis system (Bio-Rad Laboratories, Inc.).
The extracted total RNA was reverse-transcribed into cDNA using a
Tetro cDNA Synthesis kit (Bioline; Meridian Bioscience) at room
temperature. The entire process took approximately one hour to
accomplish, according to the manufacturer's instructions. The
following primer pairs were used for qPCR: β-actin forward,
5'-CATTGCCGACAGGATGCA-3' and reverse, 5'-CCGATCCACACGGAGTACTTG-3';
clusterin forward, 5'-TCCACCGCCGGTTTATATGA-3' and reverse,
5'-GCCCAGCTATGGTTCAGACTAAAA-3'; PPARα forward,
5'-GCCAGTATTGTCGATTTCACAAG-3' and reverse,
5'-CTCCTTGTTCTGGATGCCATT-3'; PPARγ forward,
5'-CTTTATGGAGCCCAAGTTTGAG-3' and reverse,
5'-GCTTCACATTCAGCAAACCTG-3'; cytochrome P450 family 1 subfamily A
member 1 (CYP1A1) forward, 5'-TCAGGAGAAGCAGCTGGATGA-3' and reverse,
5'-GAGGTCCAAGACGATGTTAATGATC-3'; CYP1 subfamily B member 1 (CYP1B1)
forward, 5'-ATCAGGTGAGGTGTGCTCCAT-3' and reverse,
5'-TCTCCCAGAAGCTCCTGCATA-3'; and CYP family 2 subfamily S member 1
(CYP2S1) forward, 5'-GACAGGGTTAATGTCTCCAGAGTGT-3' and reverse,
5'-GGACAGACTCCGGAAAACAACT-3'. All primer pairs were designed using
Primer Express Software v.3.0.1 (Thermo Fisher Scientific, Inc.).
All oligonucleotide primers were obtained from Integrated DNA
Technologies, Inc. The primer stock was packed in desalted
lyophilised form and dissolved in RNase-free water (Sigma-Aldric;
Merck KGaA) to generate a final 100 µM stock of each primer
solution. The primer stock and working solutions were stored at
-20˚C until further use. RT-qPCR was performed using an Agilent
AriaMx Real-Time PCR System (Agilent Technologies, Inc.). The PCR
cocktail was prepared by adding 10.0 µl Applied Biosystems PowerUp™
SYBR® Green Master Mix (Thermo Fisher Scientific, Inc.),
0.8 µl 10.0 µM forward and reverse primers solutions for the gene
of interest (GOI), as well as 50.0 ng cDNA diluted in nuclease-free
water to a total volume of 20.0 µl in each well. All solutions were
prepared in 0.1 ml/8-tube quantitative PCR strips (AITbiotech Pte,
Ltd.). The thermocycling conditions were as follows: Hot start at
95˚C for 30 sec followed by 40 cycles of amplification at 95˚C for
15 sec as the denaturation step and 60˚C for 1 min as the annealing
and extension steps. The reactions were continued for one cycle of
95˚C for 15 sec followed by 60˚C for 1 min, 95˚C for 30 sec and
60˚C for 15 sec to generate a melting curve to ensure that the
specific target region of GOI was amplified. The expression of GOI
was normalised to β-actin. The fold-change used to determine the
association between the normalised GOI in treated and untreated
(control) samples was calculated using the 2-ΔΔCq method
(15). The fold-change value for
the vehicle control was set to 1. Expression change >1
represented upregulation and <1 represented downregulation.
Gene expression validation and overall
survival analysis using TCGA data in GEPIA
The gene expression validation presented in the box
plot based on GEPIA dynamically shows the expression profiles of
the following genes: Clusterin, PPARγ and PPARα. The colour density
of each block represents the median expression value of a gene in
LIHC normalised by the maximum median expression value across all
blocks. The plot modification parameters, such as width, jitter
size and group colours provided by GEPIA, can be accessed in
do-it-yourself expression sub-features. Additionally, GEPIA
performed an overall survival analysis based on the identified gene
expression levels in LIHC for the overall or disease-free survival
analysis. GEPIA uses a log-rank test for hypothesis evaluation,
also known as the Mantel-Cox test (13). The study also included the Cox
proportional hazard ratio and the survival plot 95% confidence
interval (CI). In the present study, plots for high and low gene
expression levels were analysed using the two-stage hazard rate
comparison method for Kaplan-Meier plots in the case of crossing
curves (16). The null
(h0) and alternative (h1) hypotheses were set
as the survival hazard ratio of subjects over high and low
expression, respectively. P-value ranked the gene expression of
overall survival analysis in LIHC as significant when the plots run
parallel patterns (h0). P-value ranked the gene
expression of overall survival analysis in LIHC as not significant
when the plots crossed (h1).
Examination of cytoplasmic lipid
accumulation in BPA-treated THLE-2 cells
Oil Red O staining was performed to examine
cytoplasmic lipid accumulation in BPA-treated THLE-2 cells. THLE-2
cells were trypsinised and sub-cultured at 37̊C for 24 h with fresh
growth medium in 24-well plates at a density of 2x105
cells/well. Subsequently, THLE-2 cells were treated with 35 µg/ml
BPA at 37̊C for 24, 48 and 72 h. The BPA-treated cells were fixed
with cold 4% paraformaldehyde in PBS (pH 7.4) for 10 min at room
temperature before being stained with Oil Red O solution (Sigma
Aldrich; Merck KGaA) at 37˚C for 2 h. The stained cells were
counterstained with Mayer haematoxylin solution at room temperature
for 5 min (Sigma Aldrich; Merck KGaA) and mounted with an aqueous
mounting medium. The morphology of the stained cells was examined
using an Eclipse TS100 Inverted Light Microscope (Nikon
Corporation) at 200x magnification and images were captured with a
Nikon COOLPIX digital camera (Nikon Corporation).
Histopathological examination
For histological examination, liver tissue of 96
Sprague Dawley rats (age, 5-6 weeks) weighing 150-180 g were
obtained from Dr Yong Yoke Keong at Animal Research Centre of
Universiti Putra Malaysia (UPM), Serdang, Malaysia. Following the
guidelines, the study was conducted and approved by the Institution
of Animal Care and Use Committee at UPM (17). Briefly, the rats were housed in
plastic cages distilled water and standard pellets ad libitium
under housing condition 22̊C, with 12 h light/dark
cycles. Rats (n=18/group) were then fed normal diet + vehicle
(olive oil), normal diet + BPA (50 mg/kg/day), high-fat diet
without vehicle, high-fat diet + vehicle or high-fat diet + BPA (50
mg/kg/day). After 8 weeks, all rats were fasted for 12 h and
administered with a lethal dose of sodium thiopental (150 mg/kg)
intraperitoneally, which caused a quick and painless death by
acting on the central nervous system to induce a cardiopulmonary
arrest (18). The rats were
confirmed dead when lacking a heartbeat, respiration or corneal
reflex. The rats were dissected, and liver tissue was collected for
histopathological examination. The tissue samples were fixed in 10%
neutral formalin for 12 h at 25˚C. The tissue pieces were
dehydrated using ascending concentrations of ethyl alcohol and
embedded in paraffin at room temperature for 2 h to form blocks.
The blocks were trimmed and cut into 6 µm thick sections. The
sections were dewaxed in an oven at 60̊C overnight. The section was
dipped in xylene twice for 1-2 min. Next, the sections were gone
through a series of washing and staining steps, starting with
dipping the sections with absolute alcohol twice for 1-2 min, 95%
alcohol twice for 1-2 min, haematoxylin for 2 min, 1% acid alcohol
for 1 min, then washing with running tap water for 2 min. The
sections were then incubated with 1% ammonia for 1 min, washed with
running tap water for 2 min, 95% alcohol for 1-2 min, eosin for 3
min, 95% alcohol for 1-2 min, followed by absolute alcohol twice.
Lastly, immersed the sections in xylene twice before mounting. All
the procedures were performed at room temperature. The stained
tissue sections were examined under a light microscope to assess
the histological changes. Qualitative analysis of the sections was
performed using ToupView digital imaging software (BX41; Olympus
Corporation) at 100x magnification and ImageJ version 1.46r
(National Institutes of Health).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 9.4.0 (GraphPad Software, Inc.). The data are
expressed as the mean ± standard deviation. Data were compared
using one-way ANOVA with Tukey's post hoc test or two-way ANOVA
with Bonferroni's post hoc test. One-way ANOVA was used for lipid
accumulation analysis whereas two-way ANOVA was used for gene mRNA
expression analyses. Each sample was measured in ≥3 replications
and ≥3 independent experiments were performed. P<0.05 was
considered to indicate a statistically significant difference.
Results
Increased PPARγ expression profile in
tumour samples associated with digestive system
The present analysis compared PPARγ expression
levels between tumour and normal tissue. Data extracted from TCGA
database revealed that PPARγ expression was notably different in
only 11 types of tumours compared with matched normal tissue using
RNA-sequencing data from GTEx. Of these, only three showed
increased PPARγ expression and eight showed decreased expression
(Fig. 1A). The number of samples
for these three tumours was as follows: COAD, PAAD and READ. Number
of samples for the eight tumours that showed decreased expression
was as follows: BRCA, CESC, HNSC, LUSC, OV, SKCM, THCA and UCEC
(Fig. 1B). PPARγ showed increased
expression in cancers associated with the digestive system (COAD,
PAAD and READ), compared with normal tissue using GEPIA. Therefore,
carcinogens that are associated with colorectal carcinogenesis or
digestive system, including the liver, may also be associated with
PPARγ expression.
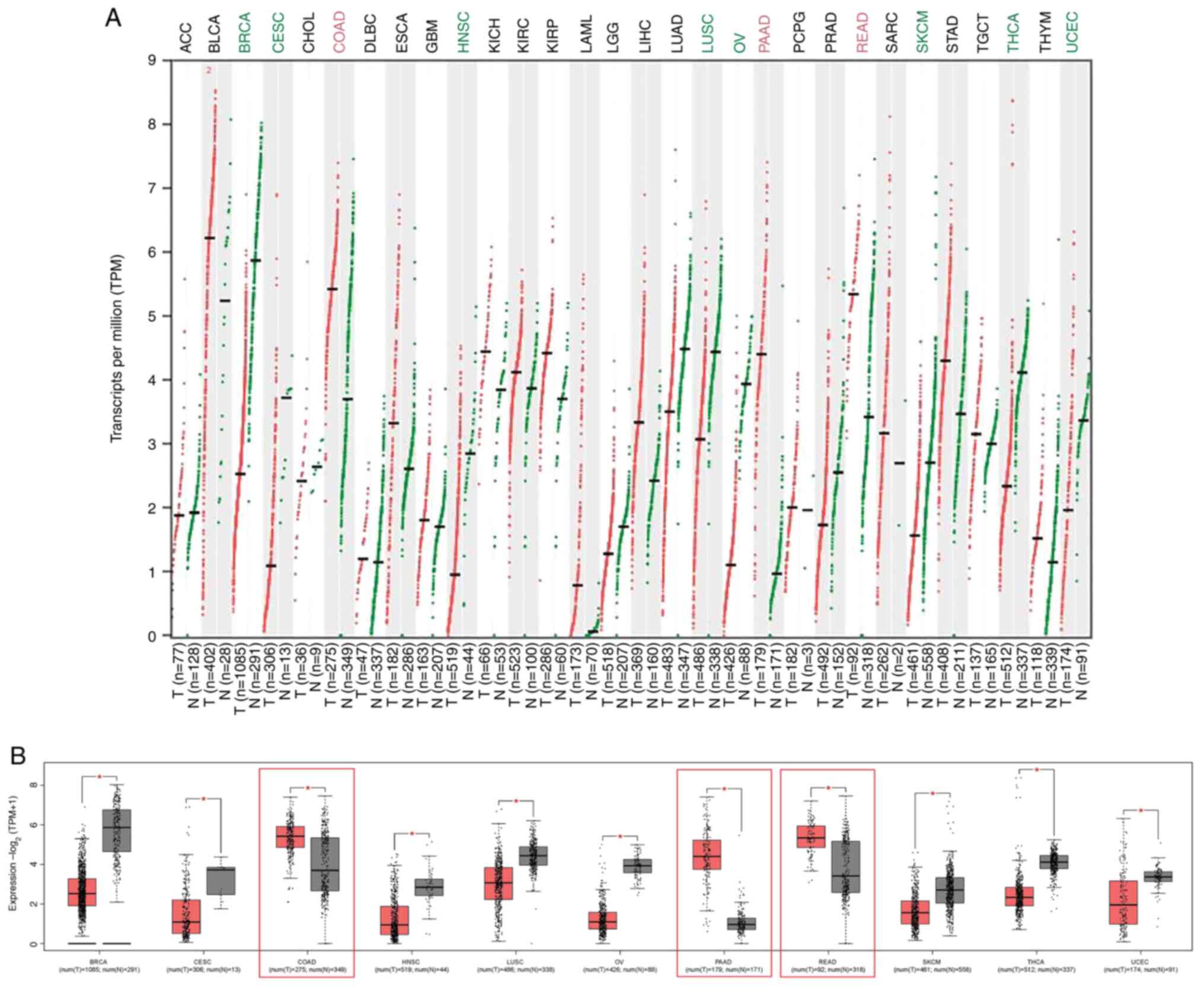 | Figure 1PPARγ expression in cancer. PPARγ
expression levels in 30 TCGA T (tumour; red) and matched N (normal;
green) samples with GTEx data using Gene Expression Profiling
Interactive Analysis webserver. (A) Dot and (B) box plots showing
upregulation of PPARγ in three types of cancer.
*P<0.05. TPM, transcripts per million; ACC,
adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA,
breast invasive carcinoma; CESC, cervical squamous cell carcinoma
and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD,
colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B cell
lymphoma; ESCA, oesophageal carcinoma; GBM, glioblastoma
multiforme; HNSC, head and neck squamous cell carcinoma; KICH,
kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP,
kidney renal papillary cell carcinoma; AML, acute myeloid
leukaemia; LGG, lower grade glioma; LIHC, liver hepatocellular
carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell
carcinoma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic
adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD,
prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC,
sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach
adenocarcinoma; TGCT, testicular germ cell tumour; THCA, thyroid
carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial
carcinoma; TCGA, The Cancer Genome Atlas; GTEx, Genotype-Tissue
Expression; T, tumour; N, normal; PPAR, peroxisome
proliferator-activated receptor. |
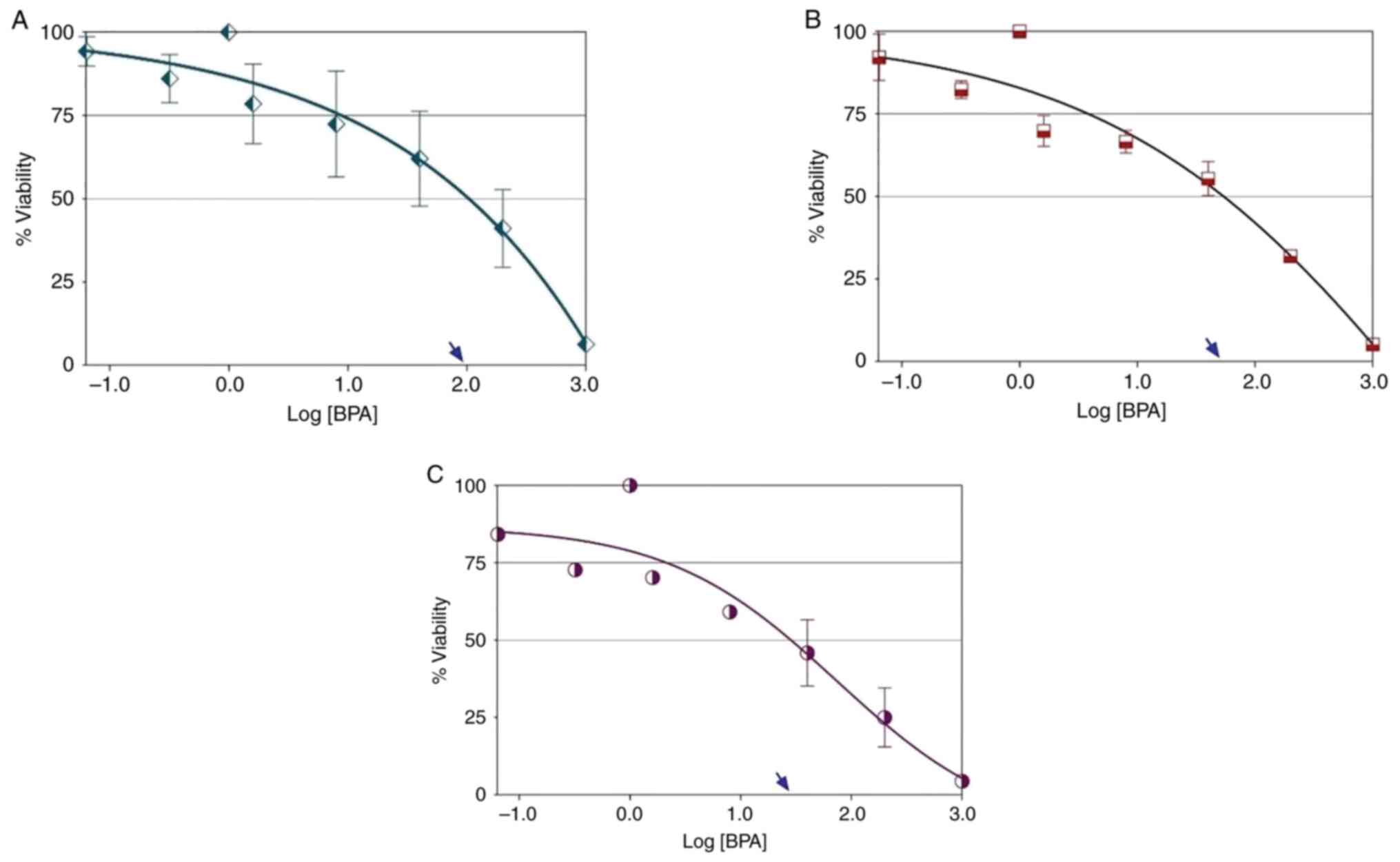
BPA induced chemosensitivity of THLE-2
cells
MTT assay analysis showed that treating THLE-2 cells
with BPA for 24 h did not produce an ideal dose-response (Fig. 2A). This phenomenon was also
observed in THLE-2 cells following BPA treatment for 48 h (Fig. 2B). The curves of the descending
hyperbola represent a growing drug resistance in the test cells.
The hyperbolic curve shows that BPA did not exhibit ideal
dose-response cell death at all concentrations. By contrast, BPA
was toxic to cells. The ideal dose-response curve (logistic or
sigmoidal shape) of BPA was observed exclusively in THLE-2 cells
following treatment with BPA for 72 h (Fig. 2C). The best-fit value of the
hillslope at the curves following 72 h treatment was -0.568,
indicating that the effect of BPA increased with treatment
duration. The maximal response following BPA treatment of THLE-2
cells for 72 h was ≤25%. The EC50 value of BPA in THLE-2
cells following 72 h treatment was ~33.70 µg/ml, indicating that
the BPA distribution of chemosensitivity occurred at lower
concentrations and increased with treatment duration. Therefore,
subsequent experiments used a concentration of 35 µg/ml BPA.
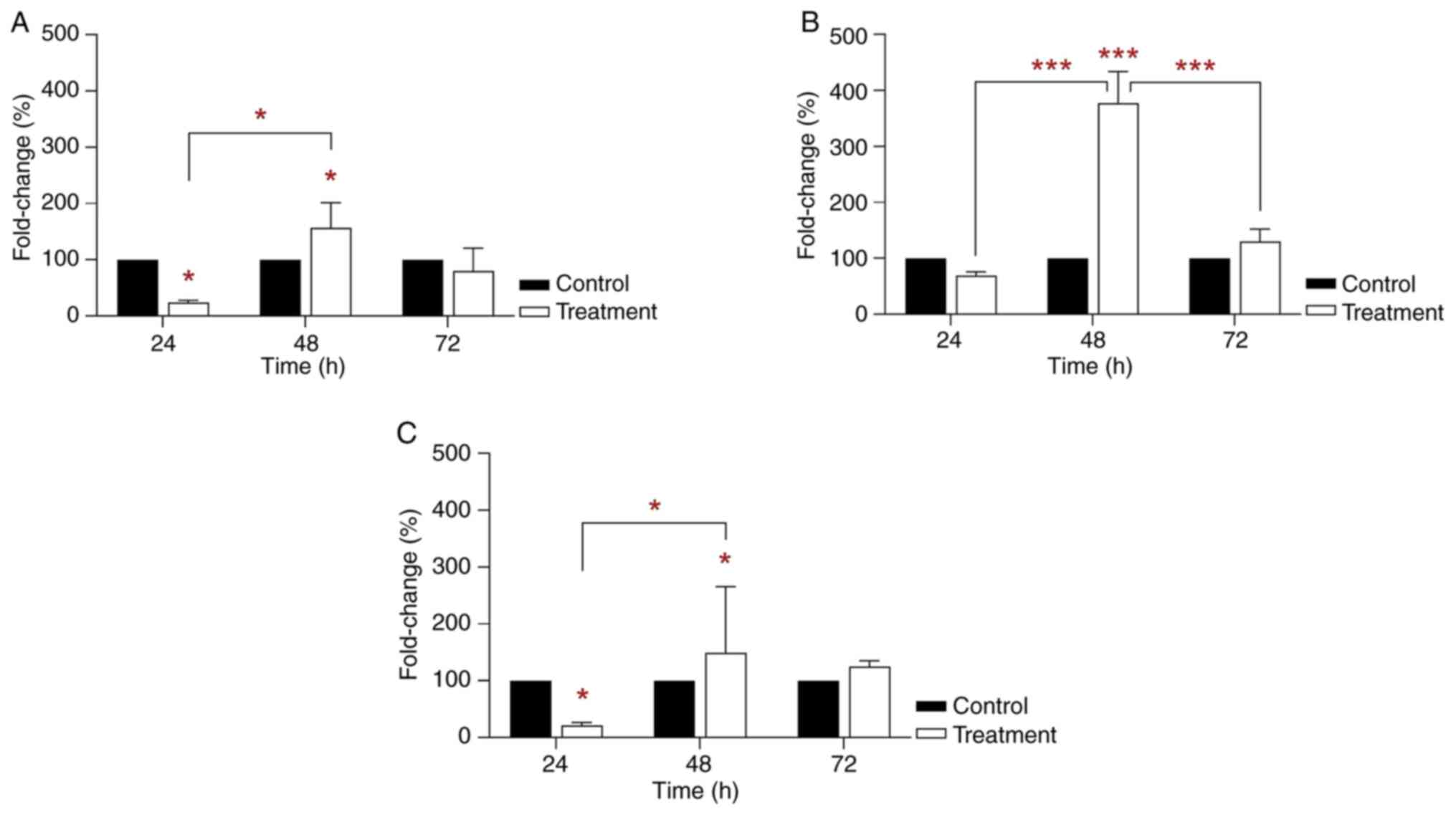
Increased gene clusterin and PPAR mRNA
expression in BPA-treated THLE-2 cells
RT-qPCR analysis showed an inhibitory effect on
clusterin mRNA expression in THLE-2 cells following 24 h 35 µg/ml
BPA treatment (24.58% vs. control; P<0.05; Fig. 3A). However, mRNA clusterin
expression was significantly increased in THLE-2 cells following 48
h BPA treatment (156.54% vs. control; P<0.05; Fig. 3A). Clusterin mRNA expression was
decreased in THLE-2 cells following 72 h BPA treatment. RT-qPCR
analysis also indicated that treatment of THLE-2 cells with 35
µg/ml BPA for 48 h significantly increased PPARγ mRNA expression
(377.11% vs. control; P<0.001; Fig.
3B). On the other hand, PPARα mRNA expression was significantly
decreased in THLE-2 cells treated with 35 µg/ml BPA for 24 h
(22.18%-fold change; P<0.05; Fig.
3C) compared with the control. However, the greatest PPARα mRNA
expression was observed following 48 h treatment (167.42% vs.
control; P<0.05; Fig. 3C).
However, these levels were lower than those of PPARγ. The
semi-ideal bell-shaped curve was observed for clusterin and PPARα
mRNA expression and an ideal bell-shaped curve was observed across
the treatment time points for PPARγ mRNA expression. The results
revealed the greatest PPARγ mRNA expression at 48 h treatment. In
addition, clusterin and PPARα mRNA expression may also serve an
important role in cell signaling.
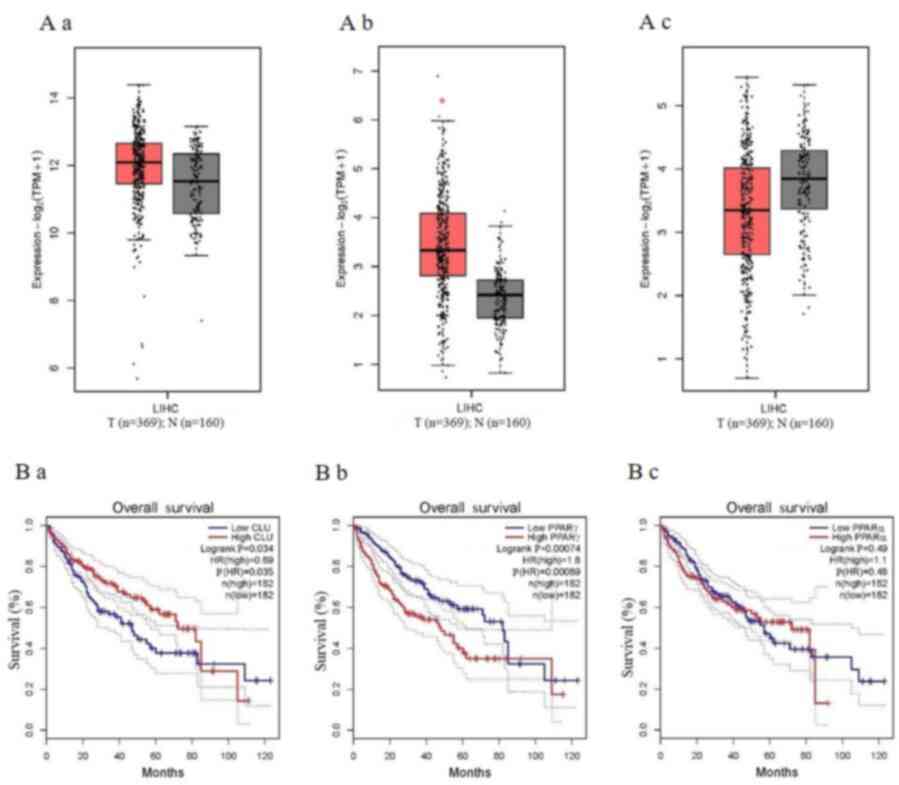
Association of PPARγ expression with
poorer overall survival in LIHC
Gene expression validation of the box plots for
clusterin, PPARγ and PPARα in LIHC was performed using TCGA data in
GEPIA. Clusterin (Fig 4A-a) and
PPARα (Fig. 4A-c) did not
significantly differ between tumour and paired normal tissue, while
only PPARγ showed significant differences between tumour and paired
normal tissue (P<0.05; Fig.
4A-b). As tumour progression affects prognosis, the analysis
also validated the identified genes by investigating their role in
LIHC overall survival time (19).
Higher clusterin expression possessed a significantly longer
overall survival time compared with lower clusterin expression
(log-rank P<0.05; Fig. 4B-a).
However, a shorter overall survival time was associated with higher
PPARγ expression of LIHC, for a mean survival time of <80 months
(log-rank P<0.05; Fig. 4B-b),
which demonstrated higher PPARγ expression was associated with
poorer overall survival in LIHC.
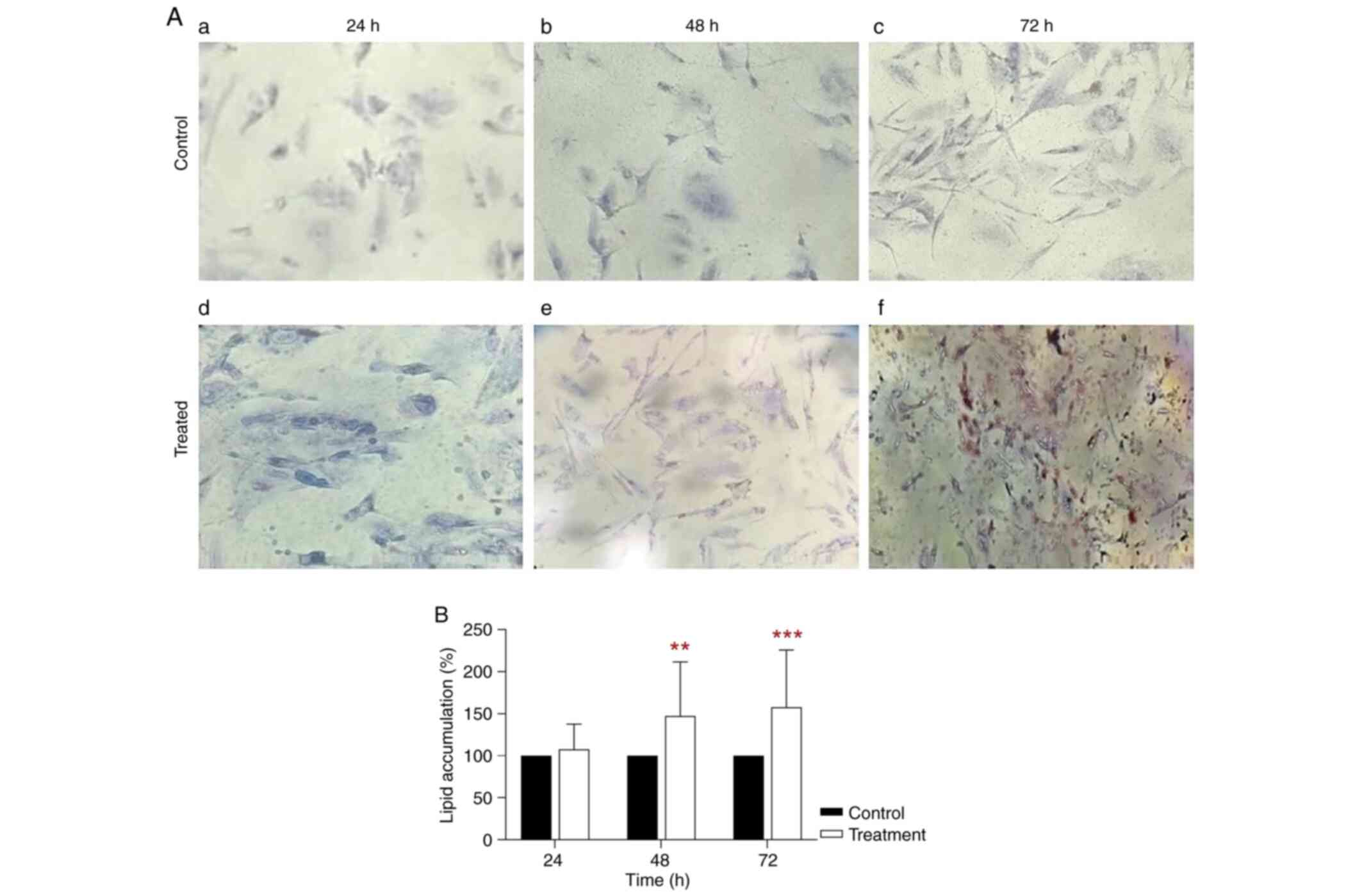
Increased lipid accumulation in
BPA-treated THLE-2 cells
Oil Red O staining showed high cytoplasmic lipid
accumulation in THLE-2 cells treated with 35 µg/ml BPA for 72 h
(Fig. 5A). Statistical analysis
revealed that lipid accumulation in THLE-2 cells treated with BPA
for 48 h was 156.20% of that in control (P<0.01; Fig. 5B). Following 72 h treatment, lipid
accumulation remained in the BPA-treated THLE-2 cells was 166.24%
of that in control (P<0.001; Fig.
5B). This finding indicates that THLE-2 cells exposed to BPA
showed differentiation due to increased lipid accumulation.
Increased lipid accumulation and
inflammatory features in rat liver tissue
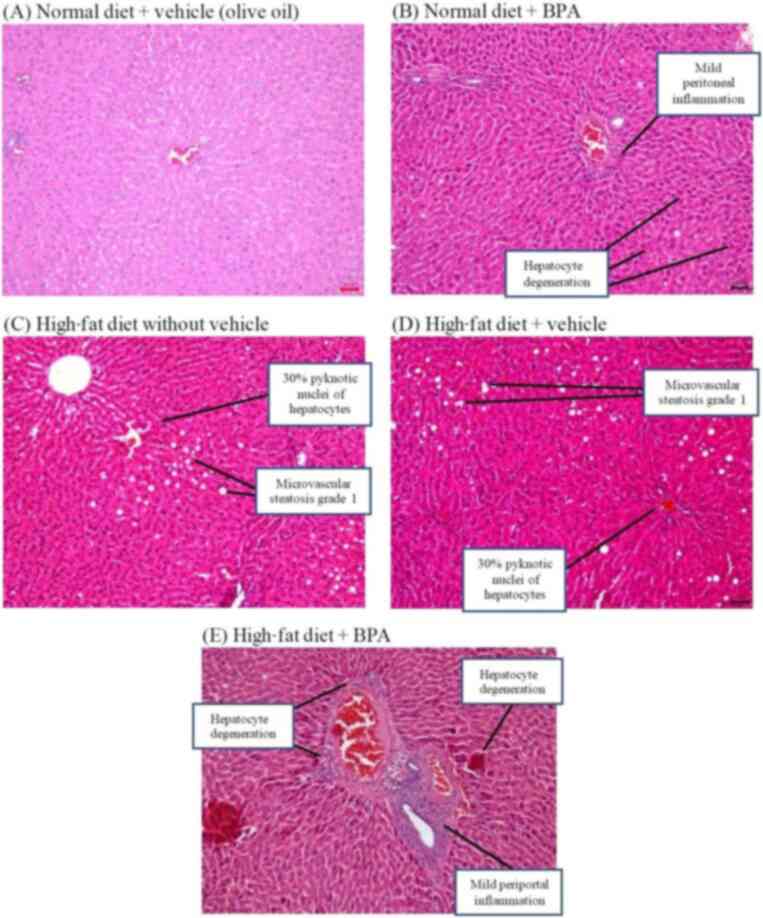
Haematoxylin and eosin staining showed that the rats
fed with a normal diet (and vehicle; olive oil) displayed normal
liver architecture had a classic arrangement with central nuclei,
sharp nuclear membranes and red-stained cytoplasm (Fig. 6A). The liver tissue of rats fed
normal diet + BPA showed deformed hepatocytes, potentially due to
inflammation (Fig. 6B). The
histopathology also revealed ballooning degeneration of hepatocytes
with minimal congestion. On the other hand, liver tissue of rats
fed high-fat diet with and without vehicle showed grade 1
microvesicular steatosis, indicating small lipid droplets in the
tissue (Fig. 6C and D). The analysis also found clusters
(aggregates) of inflammatory cells in the hepatocytes. The liver
tissue of rats fed high-fat diet + BPA showed periportal
inflammation, which is a sign of sepsis (Fig. 6E). The tissue also showed lipid
droplets, ballooning degeneration of hepatocytes and fibrous tissue
proliferation around hepatic lobules. The results revealed that BPA
exposure induced abnormal liver architecture, lipid accumulation
and inflammation.
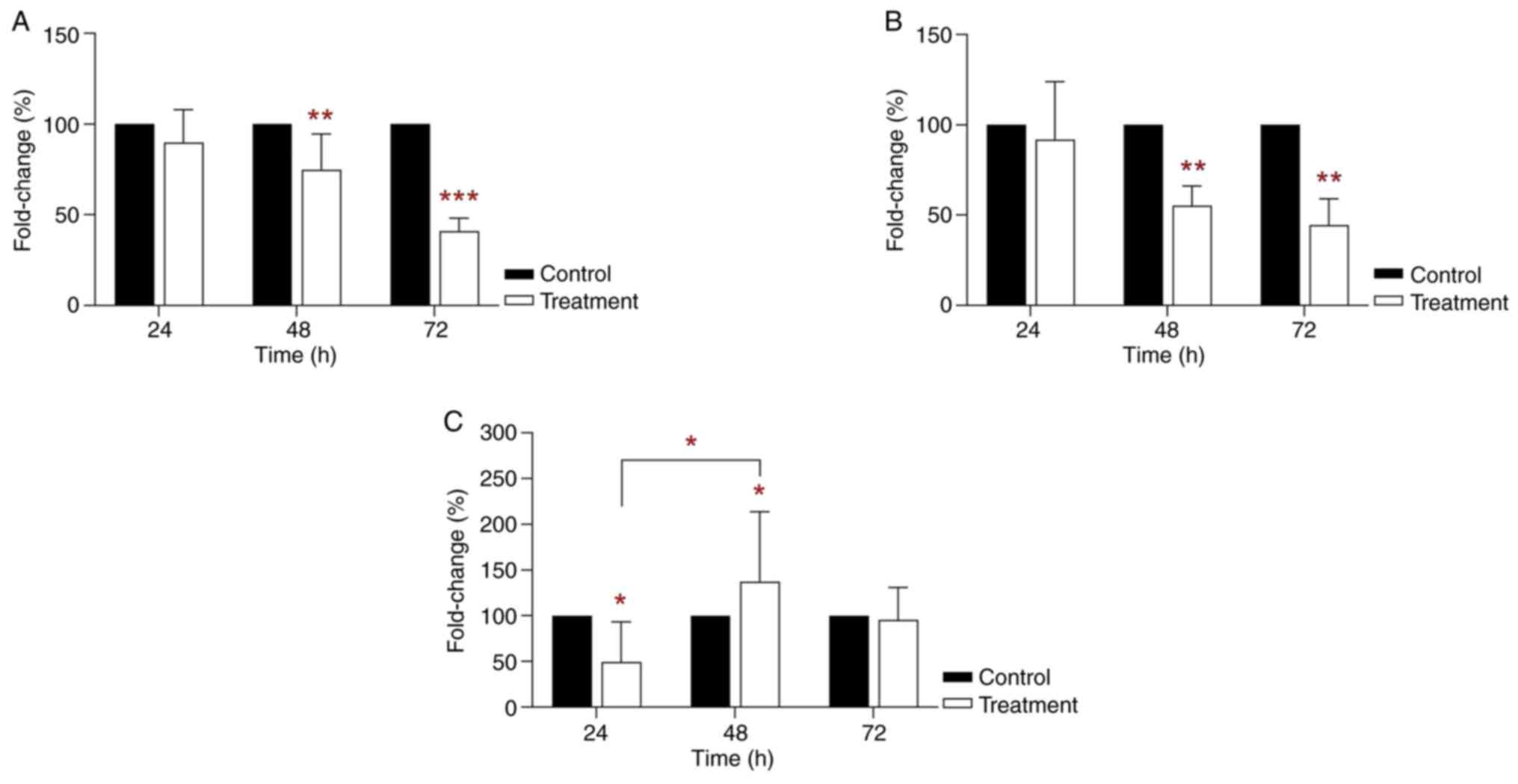
Changed CYP mRNA expression in
BPA-treated THLE-2 cells
RT-qPCR analysis revealed altered mRNA expression of
CYP1A1, CYP1B1 and CYP2S1, which are involved in cell metabolism
(20), in THLE-2 cells following
treatment with 35 µg/ml BPA for 72 h. Compared with the control, 35
µg/ml BPA significantly decreased CYP1A1 mRNA expression to 76.24
(P<0.01) and 41.39% (P<0.001; Fig. 7A) following 48 and 72 h treatment,
respectively. Compared with control, 35 µg/ml BPA significantly
inhibited CYP1B1 expression levels to 55.82 (P<0.01; Fig. 7B) and 45.67% (P<0.01; Fig. 7B) following 48 and 72 h treatment,
respectively. On the other hand, CYP2S1 mRNA expression was
significantly decreased to 56.16% after 24 h treatment compared
with the control (P<0.05; Fig.
7C). However, CYP2S1 mRNA expression was significantly
increased to 146.36% following 48 h treatment compared with the
control (P<0.05; Fig. 7C).
Semi-ideal bell-shaped curve for CYP2S1 mRNA expression was
observed across the treatment time points. This suggested that BPA
decreased capacity for carcinogen metabolism, while exerting
minimal cytotoxicity in THLE-2 cells.
Discussion
TCGA database revealed that PPARγ showed increasing
levels in cancers associated with the digestive system. PPARγ mRNA
was expressed in THLE-2 cells exposed to BPA with an
EC50 value of 35 µg/ml. In addition, 48 h treatment may
have been the optimal time point to signal cellular differentiation
in treated THLE-2 cells. BPA is a colorectal cancer carcinogen that
caused increased expression of lipid sensors, such as PPARγ, and
decreased metabolism in cytoplasm of THLE-2 cells (12,21).
Higher PPARγ expression was significantly associated with ~80
months overall survival of LIHC in the TCGA database. PPARγ may
have been the primary signalling molecule that induces lipid
accumulation in THLE-2 cells following BPA treatment, which was
observed in the present study. The liver tissue of rats fed
high-fat diet + BPA showed signs of periportal inflammation, lipid
droplets, ballooning degeneration of hepatocytes and fibrous tissue
proliferation around hepatic lobules. In THLE-2 cells, BPA
treatment also decreased the capacity for carcinogen metabolism and
increased cytotoxicity. This indicated that PPARγ expression and
BPA exposure were associated with colorectal cancer and may induce
liver disease.
PPARγ is a master regulator of adipogenesis and fat
storage (22). PPARγ regulates
adipocyte differentiation and insulin sensitivity in adipose tissue
(22). Several reports have
highlighted the anti-proliferative and pro-differentiative actions
of PPARγ ligands in cancer cell lines and animal models of human
neoplastic disease (22,23). Previous studies illustrating the
tumour-promoting effects of PPARγ in colon and breast cancer models
raise concerns about the practicability and safety of PPARγ ligands
as anticancer agents (22,24,25).
Consistently, increased PPARγ expression was found in cancer
associated with the digestive system, including urothelial bladder
carcinoma and colon, pancreatic and rectum adenocarcinoma. A
potential role for PPARγ as an inducer of differentiation of cancer
stem cells has been explored. PPARγ protein levels in tumour
specimens serve as a key prognostic marker (26). The PPARγ signalling pathway may
explain discrepancies regarding the dual role of PPARγ, whether it
acts as an anti-proliferative or a tumour-promoting action in
gastrointestinal cancer (22,27).
PPARγ acts as an antiproliferative agent, but PPARγ also acts as a
tumour-promoting molecule (22).
PPARγ mRNA expression was also increased in liver cells exposed to
BPA.
BPA is a ubiquitous ingredient in epoxy resin and
polycarbonate plastic commonly used in manufacturing water bottles
and food containers (28). BPA is
also a synthetic oestrogen associated with increased risk of type 2
diabetes mellitus and obesity (29). Despite an official ban against the
use of BPA in baby bottles in Malaysia since 2012, BPA remains
present in other food containers and beverage packaging (30). Additionally, BPA leaches into foods
from these containers when placed in microwaves for extended
periods or exposed to vegetable oil and sodium chloride solution
(1). The present study
investigated the potential role of BPA in cancers. Several studies
have suggested that the degree of BPA toxicity to human and animal
health depends on the dose, duration of exposure, age of the
exposed individual and frequency of exposure (3,31,32).
Liver and kidney are the organs with the highest bioaccumulation of
BPA (33); therefore, these organs
should be checked for BPA accumulation. Several studies have
reported that BPA interacts with nuclear receptors, including
oestrogen receptors, PPARs, retinoid and thyroid receptors
(34). BPA induces lipid
accumulation by stimulating these liver receptors (3,35).
BPA has been shown to activate PPARγ, thereby inducing expression
of PPARγ and its target genes in a mouse RAW264.7 macrophage cell
line (36,37). These results indicated that BPA
disrupts lipid metabolism by regulating PPARγ (38). To the best of our knowledge, only
one study has used the human liver HL-7702 cells as an in
vitro model to study the effect of BPA on PPARs (39). The study used human liver HL-7702
cells as an in vitro model to study the effect of BPA on
PPARs, whereas our study used the cells obtained from human adult
hepatocytes, THLE-2. Because the cell sources are different, both
studies imply two different mechanistic studies induced by BPA. In
the present study, BPA induced toxic effect in THLE-2 cells during
early treatment, such as reduced cell viability and increased
cytoplasmic lipid accumulation after 48 h of treatment.
BPA exhibits obesogenic properties that alter PPARγ
mRNA expression in THLE-2 cells. The obesogenic properties include
adipogenesis, stimulating lipid accumulation in adipose tissue and
liver, and perturbs obesity-related cytokines levels (40,41).
The expression of this adipogenesis gene or lipid sensor indicates
an increase in lipid accumulation and adipogenesis in the liver in
response to BPA exposure. The liver is the key organ that controls
lipid metabolism via numerous signalling pathways (42). The present study showed that
cytoplasmic lipid accumulation in THLE-2 cells exposed to 35 µg/ml
BPA for 72 h was significantly increased due to the effect of gene
expression on hepatic lipid accumulation. A previous study showed
that BPA concentrations of 10-100 ng/ml are representative of
occupational exposure (43). Based
on the magnitude value analysis, humans are exposed to 0.4-4.2
µg/kg body weight/day BPA (44).
These BPA concentrations are high, especially considering BPA
levels associated with long-term environmental exposure (45,46).
Therefore, for an in vitro study, treatment of THLE-2 cells
with 35 µg/ml BPA for 72 h and feeding experimental rats 50 mg/kg
BPA was equivalent to the predicted concentrations, which is
~10-fold higher than the actual situation. Numerous studies have
also demonstrated that BPA has a specific role in hepatic lipid
accumulation (47,48). For example, lipid accumulation
increases in HepG2 cells treated with 10 µM BPA for 24 h (47). Furthermore, an in vivo study
illustrated that long-term BPA exposure serves a crucial role in
lipid accumulation in the liver of mice (48). In the present study, PPARα, PPARγ
and clusterin mRNA expression levels were increased in the
BPA-treated THLE-2 cells. PPARγ gene exhibited the most significant
increase in expression in THLE-2 cells treated with 35 µg/ml BPA
for 72 h. Consistently, BPA treatment enhances the expression of
PPARγ (3,47,49).
A recent study demonstrated that HePG2 cells treated with 10 µM BPA
also exhibit increased PPARα expression (47). In addition, BPA upregulates PPARα
expression levels in the liver of offspring rats (50). However, the increase in PPARα
expression was not as high as that for PPARγ following BPA
treatment in the present study. Therefore, only PPARγ expression is
significantly upregulated in BPA-treated THLE-2 cells.
Haematoxylin and eosin staining showed that exposure
to BPA caused notable disruption of liver architecture, resulting
in cellular infiltration, large cytoplasmic vacuoles and hepatic
sinusoids, and an increased number of Kupffer cells in liver tissue
(51). The present analysis showed
deformed hepatocytes due to inflammation in the liver tissue of
rats fed normal diet + BPA. Tissue histopathology also revealed
hepatocyte balloon degeneration with minimal congestion lesions
compared with rats fed normal diet. Liver tissue of rats fed
high-fat diet with and without vehicle showed grade 1
microvesicular steatosis, which is a sign of small lipid droplets
on the tissue. Further analysis found inflammatory cell aggregates
in hepatocytes of rats fed high-fat diet + BPA. A previous study
demonstrated that liver tissue exposed to BPA show vacuolar
degeneration, sinusoidal dilatation, vascular congestion and
glycogen depletion that increases with exposure (52). The hepatic lobular structure
obtained from liver tissue of rats and pigs exposed to BPA are
close to physiological levels observed in the human liver (52). BPA causes liver damage in
developing children prone to the common neural, psychological and
metabolic disorders (52).
Children continuously exposed to BP compounds for several years may
experience a significantly increased risk of liver damage and
altered metabolism (52). BPA also
causes hepatic oxidative stress and steatosis, impairing secretory
function and liver integrity (7).
Moreover, BPA increases lipid peroxidation and decreases
antioxidant defence enzymes in rat liver (53). BPA reacts with oxygen radicals,
decomposing the radicals to reactive metabolites with potent
oxidant activity (54). These
metabolites increase production of reactive oxygen species (ROS),
inhibit activity of antioxidant enzymes and increase the reactivity
of H2O2 and thiobarbituric acid in the liver
(54). BPA-mediated increase in
ROS content enhances peptide chain cleavage and amino acid
cross-linking in enzymes, leading to change or loss of liver enzyme
activity (55). Therefore, the
mechanism of oxidative damage caused by BPA may involve inhibition
of the antioxidant enzyme system. BPA also causes dyslipidaemia,
increases accumulation of triglycerides (steatosis) and
cholesterol, and induces intracellular accumulation of fat droplets
in a dose-dependent manner (56).
Furthermore, BPA exposure results in upregulation of genes
associated with de novo lipogenesis, such as ATP citrate
lyase, and cholesterol synthesis, such as mevalonate (diphospho)
decarboxylase (Mvd) (57). BPA
induced hepatic lipid accumulation may arise from increased
expression of transcription factor sterol regulatory
element-binding protein-1, which increases the quantity and
activity of the enzymes that catalyse lipogenesis, triggering
hepatic lipid accumulation (56).
Hepatic lipid accumulation and oxidative stress followed by liver
injury and inflammation are pathogenic manifestations of
steatohepatitis not caused by consumption of alcohol (58). Oxidative stress induced by BPA and
lipid peroxidation enhances hepatic damage and disrupts cellular
membrane integrity leading to leakage of cytoplasmic liver enzymes
(59). Additionally, patients with
liver disease exhibit higher levels of BPA than healthy
individuals, suggesting a link between BPA exposure and liver
health status (60).
The effect of BPA on THLE-2 cells may also be
associated with CYP gene expression. The present study illustrated
significant inhibition of CYP1A1 and CYP1B1 mRNA expression in
THLE-2 cells treated with BPA for 72 h compared with the untreated
group. Several studies have suggested that CYPs are strongly
associated with the toxicity and metabolism of BPA in the mammalian
body via enhancement or inhibition of activity (3,9). As
observed in mice, in utero exposure to BPA disrupts CYP1A1
mRNA expression levels in the embryo (61). Other studies have shown
downregulation of CYP1A1 mRNA expression in mouse Hepa-1c1c7 cells
treated with 10 and 50 µM BPA for 18 h (62). Various nuclear receptors, including
PPARs and oestrogen-associated receptors, regulate
sulphotransferase expression, such as CYP1B1, thereby altering
xenobiotic disposition (20).
Inhibition of CYP1B1 mRNA expression has been found in human foetal
liver tissue treated with 35.4-56.1 ng/g BPA (63). Previous studies have demonstrated
that BPA upregulates CYP1B1 expression, which induces oxidative
stress (64,65). For example, exposure to 100 µM BPA
for 24 h increases CYP1B1 mRNA expression in human foetal lung
fibroblasts (65). CYP1A1 and
CYP1B1 enzymes serve a role in catalysing oxidation of
procarcinogens to carcinogenic reactive metabolites (66). Therefore, downregulation of CYP1A1
and CYP1B1 expression inhibits the cells from catalysing oxidation
of procarcinogens to carcinogenic reactive metabolites.
CYP2S1 mRNA was significantly overexpressed in the
present study. The present study also indicated that 48 h was the
optimum treatment time point for THLE-2 cells to metabolise BPA via
CYP2S1 expression. Higher CYP2S1 indicates better BPA metabolism.
CYPs are enzymes predominantly secreted by the liver and involved
in the metabolism or detoxification of xenobiotics and endogenous
compounds during phase one reactions (66). CYP enzymes cause molecules to be
hydrophilic, less toxic and easily excreted from the body based on
the presence of polar functional groups in their structure
(20). Human CYP2S1 is a complex
and most prominent family of CYP genes that is located at the end
of a cluster on chromosome 19q of CYP2 family members (67). Previous studies have revealed that
frequent xenobiotic exposure induces the highest CYP2S1 mRNA
expression in epithelial mouse and human tissues (68-73).
Another study demonstrated that oxaliplatin induces CYP2S1
expression in HCT116 cells and subsequently inhibits cell
proliferation. Moreover, increased survival was observed in HCT116
cells with CYP2S1 knockdown (66,74).
This phenomenon implies that cell growth is inhibited if CYP2S1 is
expressed at high levels.
In conclusion, PPARγ expression is associated with
digestive system cancer (18,75).
THLE-2 cells showed decreased viability and cytoplasmic lipid
accumulation following 48 h exposure to BPA. Liver-specific PPARγ
gene expression was significantly associated with overall survival.
Haematoxylin and eosin staining revealed notable disruption of the
liver architecture in rat liver tissue exposed to BPA. The observed
downregulation of CYP1A1 and CYP1B1 mRNA expression indicated that
BPA-treated THLE-2 cells did not catalyse the carcinogen to
reactive metabolites. By contrast, CYP2S1 mRNA expression was
associated with decreased proliferation of cells treated with
BPA.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Malaysia Research
University Network (MRUN) Program Grant of Higher Education
Ministry of Malaysian (Grant no. USM/203/CIPPM/6720020).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MA, TJ, HB, YK and KY conceived and designed the
study. LI and AA performed experiments. KY and YK confirm the
authenticity of all the raw data. LI and KY interpreted the data,
drafted and revised the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Experiments using rat liver tissue were approved by
the Institution of Animal Care and Use Committee at University
Putra Malaysia (approval no. UPM/IACUC/AUP-R068/2019).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Khoo Boon Yin, ORCID no. 0000-0003-1915-6606
References
|
1
|
Rochester JR: Bisphenol A and human
health: A review of the literature. Reprod Toxicol. 42:132–155.
2013.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ma Y, Liu H, Wu J, Yuan L, Wang Y, Du X,
Wang R, Marwa PW, Petlulu P, Chen X, et al: The adverse health
effects of Bisphenol A and related toxicity mechanisms. Environ
Res. 176(108575)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Quesnot N, Bucher S, Fromenty B and Robin
MA: Modulation of metabolizing enzymes by Bisphenol A in human and
animal models. Chem Res Toxicol. 27:1463–1473. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bertoli S, Leone A and Battezzati A: Human
Bisphenol A exposure and the diabesity phenotype. Dose Response.
13(1559325815599173)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Salehi A, Loganathan N and Belsham DD:
Bisphenol A induces Pomc gene expression through neuroinflammatory
and PPARγ nuclear receptor-mediated mechanisms in POMC-expressing
hypothalamic neuronal models. Mol Cell Endocrinol. 479:12–19.
2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Peyre L, Rouimi P, de Sousa G,
Héliès-Toussaint C, Carré B, Barcellini S, Chagnon MC and Rahmani
R: Comparative study of Bisphenol A and its analogue bisphenol S on
human hepatic cells: A focus on their potential involvement in
non-alcoholic fatty liver disease. Food Chem Toxicol. 70:9–18.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Eweda SM, Newairy ASA, Abdou HM and Gaber
AS: Bisphenol A-induced oxidative damage in the hepatic and cardiac
tissues of rats: The modulatory role of sesame lignans. Exp Ther
Med. 19:33–44. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jalal N, Surendranath AR, Pathak JL, Yu S
and Chung CY: Bisphenol a (BPA) the mighty and the mutagenic.
Toxicol Rep. 5:76–84. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ohore OE and Songhe Z: Endocrine
disrupting effects of Bisphenol A exposure and recent advances on
its removal by water treatment systems. A review. Sci Afr.
5(e00135)2019.
|
|
10
|
Kourouma A, Quan C, Duan P, Qi S, Yu T,
Wang Y and Yang K: Bisphenol A induces apoptosis in liver cells
through Induction of ROS. Adv Toxicol. 2015(901983)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kang JH, Katayama Y and Kondo F:
Biodegradation or metabolism of Bisphenol A: From microorganisms to
mammals. Toxicology. 217:81–90. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen ZJ, Yang XL, Liu H, Wei W, Zhang KS,
Huang HB, Giesy JP, Liu HL, Du J and Wang HS: Bisphenol A modulates
colorectal cancer protein profile and promotes the metastasis via
induction of epithelial to mesenchymal transitions. Arch Toxicol.
89:1371–1381. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Abdalkareem EA, Ong CY, Lim BH and Khoo
BY: Neutralizing FGF4 protein in conditioned medium of
IL-21-silenced HCT116 cells restores the migratory activity of the
colorectal cancer cells. Cytotechnology. 70:1363–1374.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10(e0116774)2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Institutional Animal Care and Use
Committee (IACUC): Universiti Putra Malaysia Code of Practice for
the Care and Use of Animals for Scientific Purposes. University
Putra Malaysia, Seri Kembangan, 2020. http://www.tncpi.upm.edu.my/upload/dokumen/20180522092738IACUC_-_UPM_Code_of_Practice.pdf.
|
|
18
|
Jucá MJ, Bandeira BC, Carvalho DS and Leal
AT: Comparative study of 1,2-dimethylhydrazine and azoxymethane on
the induction of colorectal cancer in rats. J Coloproctology.
34:167–173. 2014.
|
|
19
|
Wee Y, Liu Y, Lu J, Li X and Zhao M:
Identification of novel prognosis-related genes associated with
cancer using integrative network analysis. Sci Rep.
8(3233)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Esteves F, Rueff J and Kranendonk M: The
central role of cytochrome P450 in xenobiotic metabolism-a brief
review on a fascinating enzyme family. J Xenobiot. 11:94–114.
2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hafezi SA and Abdel-Rahman WM: The
Endocrine disruptor Bisphenol A (BPA) exerts a wide range of
effects in carcinogenesis and response to therapy. Curr Mol
Pharmacol. 12:230–238. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pazienza V, Vinciguerra M and Mazzoccoli
G: PPARs Signaling and cancer in the gastrointestinal system. PPAR
Res. 2012(560846)2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Eibl G: The role of PPAR-gamma and its
interaction with COX-2 in pancreatic cancer. PPAR Res.
2008(326915)2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Burstein HJ, Demetri GD, Mueller E, Sarraf
P, Spiegelman BM and Winer EP: Use of the peroxisome
proliferator-activated receptor (PPAR) gamma ligand troglitazone as
treatment for refractory breast cancer: A phase II study. Br Cancer
Res Treat. 79:391–397. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Augimeri G, Giordano C, Gelsomino L,
Plastina P, Barone I, Catalano S, Andò S and Bonofiglio D: The role
of PPARγ ligands in breast cancer: From basic research to clinical
studies. Cancers (Basel). 12(2623)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Robbins GT and Nie D: PPAR gamma,
bioactive lipids and cancer progression. Front Biosci (Landmark
Ed). 17:1816–1834. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Dai Y, Qiao L, Chan KW, Yang M, Ye J, Ma
J, Zou B, Gu Q, Wang J, Pang R, et al: Peroxisome
proliferator-activated receptor-gamma contributes to the inhibitory
effects of Embelin on colon carcinogenesis. Cancer Res.
69:4776–4783. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vandenberg LN, Hauser R, Marcus M, Olea N
and Welshons WV: Human exposure to Bisphenol A (BPA). Reprod
Toxicol. 24:139–177. 2007.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Vom Saal FS, Nagel SC, Coe BL, Angle BM
and Taylor JA: The estrogenic endocrine disrupting chemical
Bisphenol A (BPA) and obesity. Mol Cell Endocrinol. 354:74–84.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mahamuni D and Shrinithivihahshini ND:
Need for regulatory policies in India, on the use of Bisphenol A in
food contact plastic containers. Curr Sci. 113:861–868. 2017.
|
|
31
|
Bhandari R, Xiao J and Shankar A: Urinary
Bisphenol A and obesity in U.S. children. Am J Epidemiol.
177:1263–1270. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pereira-Fernandes A, Demaegdt H,
Vandermeiren K, Hectors TL, Jorens PG, Blust R and Vanparys C:
Evaluation of a screening system for obesogenic compounds:
Screening of endocrine disrupting compounds and evaluation of the
PPAR dependency of the effect. PLoS One. 8(e77481)2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Moreno-Gómez-Toledano R, Arenas MI,
Sánchez-Esteban S, Cook A, Saura M and Bosch RJ: Critical analysis
of human exposure to Bisphenol A and its novel implications on
renal, cardiovascular and hypertensive diseases. In: Hot Topics in
Endocrinology and Metabolism [Internet]. Heshmati HM (ed).
IntechOpen, London, 2021.
|
|
34
|
Filardi T, Panimolle F, Lenzi A and Morano
S: Bisphenol A and phthalates in diet: An emerging link with
pregnancy complications. Nutrients. 12(525)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ahmed S and Atlas E: Bisphenol S- and
Bisphenol A-induced adipogenesis of murine preadipocytes occurs
through direct peroxisome proliferator-activated receptor gamma
activation. Int J Obes (Lond). 40:1566–1573. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Rogers JA, Metz L and Yong VW: Review:
Endocrine disrupting chemicals and immune responses: A focus on
bisphenol-A and its potential mechanisms. Mol Immunol. 53:421–430.
2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
MacKay H and Abizaid A: A plurality of
molecular targets: The receptor ecosystem for bisph++enol-A (BPA).
Horm Behav. 101:59–67. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gao P, Wang L, Yang N, Wen J, Zhao M, Su
G, Zhang J and Weng D: Peroxisome proliferator-activated receptor
gamma (PPARγ) activation and metabolism disturbance induced by
Bisphenol A and its replacement analog bisphenol S using in vitro
macrophages and in vivo mouse models. Environ Int.
134(105328)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li CH, Zhang DH, Jiang LD, Qi Y and Guo
LH: Binding and activity of Bisphenol Analogues to human peroxisome
proliferator-activated receptor β/δ. Ecotoxicol Environ Saf.
226(112849)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Cimmino I, Fiory F, Perruolo G, Miele C,
Beguinot F, Formisano P and Oriente F: Potential mechanisms of
Bisphenol A (BPA) contributing to human disease. Int J Mol Sci.
21(5761)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Oliviero F, Marmugi A, Viguié C, Gayrard
V, Picard-Hagen N and Mselli-Lakhal L: Are BPA substitutes as
obesogenic as BPA? Int J Mol Sci. 23(4238)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Alves-Bezerra M and Cohen DE: Triglyceride
metabolism in the liver. Compr Physiol. 8:1–8. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ribeiro E, Ladeira C and Viegas S:
Occupational exposure to Bisphenol A (BPA): A reality that still
needs to be unveiled. Toxics. 5(22)2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Geens T, Aerts D, Berthot C, Bourguignon
JP, Goeyens L, Lecomte P, Maghuin-Rogister G, Pironnet AM,
Pussemier L, Scippo ML, et al: A review of dietary and non-dietary
exposure to bisphenol-A. Food Chem Toxicol. 50:3725–3740.
2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Valentino R, D'Esposito V, Ariemma F,
Cimmino I, Beguinot F and Formisano P: Bisphenol A environmental
exposure and the detrimental effects on human metabolic health: Is
it necessary to revise the risk assessment in vulnerable
population? J Endocrinol Invest. 39:259–263. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ribeiro E, Delgadinho M and Brito M:
Environmentally relevant concentrations of Bisphenol A interact
with doxorubicin transcriptional effects in human cell lines.
Toxics. 7(43)2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Liu Q, Shao W, Weng Z, Zhang X, Ding G, Xu
C, Xu J, Jiang Z and Gu A: In vitro evaluation of the hepatic lipid
accumulation of Bisphenol Analogs: A high-content screening assay.
Toxicol In Vitro. 68(104959)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ke ZH, Pan JX, Jin LY, Xu HY, Yu TT, Ullah
K, Rahman TU, Ren J, Cheng Y, Dong XY, et al: Bisphenol A exposure
may induce hepatic lipid accumulation via reprogramming the DNA
methylation patterns of genes involved in lipid metabolism. Sci
Rep. 6(31331)2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wang J, Sun B, Hou M, Pan X and Li X: The
environmental obesogen Bisphenol A promotes adipogenesis by
increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in
the adipose tissue of children. Int J Obes (Lond). 37:999–1005.
2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Xia W, Jiang Y, Li Y, Wan Y, Liu J, Ma Y,
Mao Z, Chang H, Li G, Xu B, et al: Early-life exposure to Bisphenol
A induces liver injury in rats involvement of mitochondria-mediated
apoptosis. PLoS One. 9(e90443)2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hussein RM and Eid JI: Pathological
mechanisms of liver injury caused by oral administration of
Bisphenol A. Life Sci J. 10:663–673. 2013.
|
|
52
|
Thoene M, Rytel L, Dzika E, Włodarczyk A,
Kruminis-Kaszkiel E, Konrad P and Wojtkiewicz J: Bisphenol A causes
liver damage and selectively alters the neurochemical coding of
intrahepatic parasympathetic nerves in juvenile porcine models
under physiological conditions. Int J Mol Sci.
18(2726)2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Abdel-Wahab WM: Thymoquinone attenuates
toxicity and oxidative stress induced by Bisphenol A in liver of
male rats. Pak J Biol Sci. 17:1152–1160. 2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Hassani FV, Abnous K, Mehri S, Jafarian A,
Birner-Gruenberger R, Yazdian Robati R and Hosseinzadeh H:
Proteomics and phosphoproteomics analysis of liver in male rats
exposed to Bisphenol A: Mechanism of hepatotoxicity and biomarker
discovery. Food Chem Toxicol. 112:26–38. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ke C, Liu X, Zuo H, Zhao J, Yang X and
Yuan J: The oxidative damage of Bisphenol A on the organs of the
mice. Sci Res. 5:1190–1194. 2013.
|
|
56
|
Lin Y, Ding D, Huang Q, Liu Q, Lu H, Lu Y,
Chi Y, Sun X, Ye G, Zhu H, et al: Downregulation of miR-192 causes
hepatic steatosis and lipid accumulation by inducing SREBF1: Novel
mechanism for Bisphenol A-triggered non-alcoholic fatty liver
disease. Biochim Biophys Acta Mol Cell Biol Lipids. 1862:869–882.
2017.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Marmugi A, Ducheix S, Lasserre F, Polizzi
A, Paris A, Priymenko N, Bertrand-Michel J, Pineau T, Guillou H,
Martin PG, et al: Low doses of Bisphenol A induce gene expression
related to lipid synthesis and trigger triglyceride accumulation in
adult mouse liver. Hepatology. 55:395–407. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Periasamy S, Chien SP, Chang PC, Hsu DZ
and Liu MY: Sesame oil mitigates nutritional steatohepatitis via
attenuation of oxidative stress and inflammation: A tale of two-hit
hypothesis. J Nutr Biochem. 25:232–240. 2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Rönn M, Kullberg J, Karlsson H, Berglund
J, Malmberg F, Orberg J, Lind L, Ahlström H and Lind PM: Bisphenol
A exposure increases liver fat in juvenile fructose-fed Fischer 344
rats. Toxicology. 303:125–132. 2013.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Nicolucci C, Errico S, Federico A, Dallio
M, Loguercio C and Diano N: Human exposure to Bisphenol A and liver
health status: Quantification of urinary and circulating levels by
LC-MS/MS. J Pharm Biomed Anal. 140:105–112. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Nishizawa H, Imanishi S and Manabe N:
Effects of exposure in utero to Bisphenol A on the expression of
aryl hydrocarbon receptor, related factors and xenobiotic
metabolizing enzymes in murine embryos. J Reprod Dev. 51:593–605.
2005.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Jeong HG, Kimand JY and Choi CY:
Down-regulation of murine Cyp1a-1 in mouse hepatoma Hepa-1c1c7
cells by Bisphenol A. Biochem Biophys Res Commun. 277:594–598.
2000.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Nahar MS, Kim JH, Sartor MA and Dolinoy
DC: Bisphenol A-associated alterations in the expression and
epigenetic regulation of genes encoding xenobiotic metabolizing
enzymes in human fetal liver. Environ Mol Mutagen. 55:184–195.
2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Meli R, Monnolo A, Annunziata C, Pirozzi C
and Ferrante MC: Oxidative stress and BPA toxicity: An antioxidant
approach for male and female reproductive dysfunction. Antioxidants
(Basel). 9(405)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Mahemuti L, Chen Q, Coughlan MC, Qiao C,
Chepelev NL, Florian M, Dong D, Woodworth RG, Yan J, Cao XL, et al:
Bisphenol A induces DSB-ATM-p53 signaling leading to cell cycle
arrest, senescence, autophagy, stress response and estrogen release
in human fetal lung fibroblasts. Arch Toxicol. 92:1453–1469.
2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Khor CY and Khoo BY: PPARα plays an
important role in the migration activity and the expression of
CYP2S1 and CYP1B1 in Chrysin-treated HCT116 cells. Biotechnol Lett.
42:1581–1595. 2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hoffman SMG, Nelson DR and Keeney DS:
Organization, structure and evolution of the CYP2 gene cluster on
human chromosome 19. Pharmacogenetics. 11:687–698. 2001.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Rylander T, Neve EP, Ingelman-Sundberg M
and Oscarson M: Identification and tissue distribution of the novel
human cytochrome P450 2S1 (CYP2S1). Biochem Biophys Res Commun.
281:529–535. 2001.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Rivera SP, Saarikoski ST and Hankinson O:
Identification of a novel dioxin-inducible cytochrome P450. Mol
Pharmacol. 61:255–259. 2002.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Choudhary D, Jansson I, Schenkman JB,
Sarfarazi M and Stoilov I: Comparative expression profiling of 40
mouse cytochrome P450 genes in embryonic and adult tissues. Arch
Biochem Biophys. 414:91–100. 2003.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Smith G, Wolf CR, Deeni YY, Dawe RS, Evans
AT, Comrie MM, Ferguson J and Ibbotson SH: Cutaneous expression of
cytochrome P450 CYP2S1: Individuality in regulation by therapeutic
agents for psoriasis and other skin diseases. Lancet.
361:1336–1343. 2003.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Saarikoski ST, Rivera SP, Hankinson O and
Husgafvel-Pursiainen K: CYP2S1: A short review. Toxicol Appl
Pharmacol. 207 (Suppl 2):S62–S69. 2005.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Saarikoski ST, Wikman HA, Smith G, Wolff
CH and Husgafvel-Pursiainen K: Localization of cytochrome P450
CYP2S1 expression in human tissues by in situ hybridization and
immunohistochemistry. J Histochem Cytochem. 53:549–556.
2005.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Yang C, Zhou Q, Li M, Tong X, Sun J, Qing
Y, Sun L, Yang X, Hu X, Jiang J, et al: Upregulation of CYP2S1 by
oxaliplatin is associated with p53 status in colorectal cancer cell
lines. Sci Rep. 6(33078)2016.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Peters JM, Shah YM and Gonzalez FJ: The
role of peroxisome proliferator-activated receptors in
carcinogenesis and chemoprevention. Nat Rev Cancer. 12:181–195.
2012.PubMed/NCBI View Article : Google Scholar
|